

Abstract
In the present work, carboxyl terminated PAMAM G-3.5 was covalently attached to SN38 via glycine and β-alanine spacers. The conjugates were stable at pH 7.4 and moderately hydrolyzed in cell culture media and rat plasma. Similar to SN38 but to a lesser extent, both conjugates inhibited proliferation of human colorectal cancer HCT-116 cells, arrested the cell cycle in the G2/M phase and led to nuclear fragmentation. However activity of the conjugate with glycine spacer (IC50 = 129 nM) was higher compared to the β-alanine linked conjugate (IC50 = 387 nM). These PAMAM-SN38 conjugates have potential for targeted therapy of colorectal carcinoma.
Polymeric drug carriers have emerged as promising alternatives to small molecule therapeutics due to their many advantages over parent drugs, which include enhanced aqueous solubility, reduced systemic toxicity and improved efficacy (1, 2). In this context, dendrimers have emerged as promising candidates for targeted delivery of chemotherapeutics. They are nanometer sized, have multiple surface groups and low polydispersity. The large number of functionalizable sites at the periphery of the dendrimers, allow facile attachment of multiple drugs, targeting moieties and imaging agents. Pharmaceutical compounds can also be physically encapsulated in the dendrimer interior to enhance water solubility and reduce toxicity (3, 4).
7-ethyl-10-hydroxy camptothecin (SN38) is a chemotherapeutic agent belonging to the camptothecin family of highly potent topoisomerase I inhibitors (5, 6). It has extremely low water solubility and is not used directly for clinical applications. CPT-11 (Irinotecan) is the water soluble, prodrug of SN38 which is currently approved for colorectal cancer. However it is 100–1000 times less active than SN38, causes gastrointestinal toxicity, and similar to SN38 has poor oral bioavailability (7, 8). Additionally, the pharmacologically important lactone ring in irinotecan is converted to the inactive carboxylate form in the presence of human plasma albumin (9). A promising approach to increase the solubility of SN38 and facilitate delivery to the site of action is attachment to water soluble polymeric carriers (10, 11) or antibodies (12). Recently, we demonstrated that complexation of SN38 with amine terminated generation 4 PAMAM (poly (amido amine)) dendrimers (G4-NH2) enhance water solubility of the drug, increases cellular uptake and its transepithelial transport across Caco-2 cells (13). While such complexes show promise in oral drug delivery, covalent conjugation of SN38 to dendrimers can impart greater stability, prevent premature release and facilitate targeted delivery (14). In addition, we have previously demonstrated in vitro that concentration, generation of dendrimer, and incubation time influence the toxicity observed with cationic PAMAM dendrimers whereas under similar conditions carboxyl terminated anionic dendrimers have shown less toxicity and yet maintained comparable transepithelial transport (15, 16). These observations, along with the inherent advantages of polymeric drug delivery systems, suggest that development of anionic PAMAM-SN38 conjugates can increase the bioavailability of the drug, its efficacy and reduce toxicity. Here we report the synthesis, characterization and in vitro stability and activity of G3.5-SN38 conjugates in a colorectal cancer cell model.
Experimental Procedures
PAMAM G3.5 dendrimer, N-(tert-butoxycarbonyl)glycine, N-(tert-butoxycarbonyl)beta alanine, N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide (EDC), N-hydroxy succinimide (NHS), 4-(dimethylamino)pyridine (DMAP), N,N diisopropylethylamine (DIPEA) and di-tert-butyl dicarbonate (Boc2O) were obtained from Aldrich, (St. Louis, MO, USA). 7-ethyl-10-hydroxy camptothecin (SN38) was obtained from AK Scientific Company (California, USA) and its purity was determined by 1H NMR and HPLC. Anyhdrous dichloromethane (DCM) and dimethylsulphoxide (DMSO) were purchased and used without further purification.
1H NMR of intermediates and conjugates were recorded on a Bruker 400 or Varian Unity 500 MHz spectrophotometer. Size exclusion chromatography (SEC) for characterization and purification of G3.5-drug conjugates were performed on an Akta Fast Protein Liquid Chromatography (FPLC) system, using a Superose12 analytical and Hiload 16/60 Superdex 75 preparative grade columns (GE, USA) respectively. The dendrimer peak was analyzed to determine the hydrodynamic radius using a Wyatt Quasi-Elastic Light Scattering Detector (QELS) and the calculations were performed using the Astra 5.3.4 software. All samples were run in triplicate. Reverse phase, High Performance Liquid Chromatography (HPLC) was performed on Hewlet Packard series 1100 using an agilent C18 column, 250 mm × 4.5 mm.
Synthesis of SN38-gly(3)
The overall synthetic strategy is depicted in Scheme 1. SN38 derivatized at the 20-OH with a glycine spacer was synthesized according to a reported protocol with modifications (11). Briefly, SN38 (0.8g, 2.04 mmol) was stirred for 12h with di-tert-butyl dicarbonate (0.579g, 2.65 mmol) and anhydrous pyridine (4 mL, 42 mmol) in 40 ml of CH2Cl2 to obtain compound 1 (Scheme 1) in 90% yield. To a solution of 1 (0.5g, 1.0 mmol) in CH2Cl2(10 ml), N-(tert-butoxycarbonyl)glycine (0.5g, 3.0 mmol), EDC (0.9 mL, 5.0 mmol) and DMAP (0.350g, 3.0 mmol) were added and stirred for 2h. Subsequently the reaction mixture was washed with 0.5 % NaHCO3 and 0.1 N HCl. The crude product obtained was purified by column chromatography using 1–3% MeOH/CHCl3 to obtain compound 2 (Scheme 1) in 63 % yield. Deprotection of 2 (0.25g, 0.3 mmol) in 30% TFA/DCM gave SN38-gly (compound 3, 0.24g, 95 % yield).1H NMR (400 MHz, CD3OD):δ 1.07 (t, 3H, J = 5.8 Hz), 1.34 (t, 3H, J = 6.2 Hz), 2.18–2.30 (m, 2H), 3.07–3.12 (m, 2H), 4.17 (d,1H, J = 14.4 Hz), 4.27 (d,1H, J = 14.0 Hz), 5.21 (s, 2H), 5.48 (d,1H, J = 13.2 Hz), 5.63 (d,1H, J = 13.2 Hz), 7.31(s, 1H), 7.40–7.42 (m, 2H), 7.96 (d, 1H, J = 7.2 Hz). ESI-TOF MS: m/z Calc. for C24H23N3O6 + H+: 450.17; Obsd. 450.12.
Scheme 1.

Synthesis of SN38 derivatives, SN38-gly with glycine spacer (3) and SN38 with β-alanine spacer (5) at the 20-OH position.
Synthesis of SN38-βala (5)
In an analogous manner, compound 1 (0.4g, 0.8 mmoles) was reacted with N-(tert-butoxycarbonyl)beta alanine (0.28g, 2.4 mmol) to obtain compound 4 (0.38g, 70% yield) which upon deprotection with 30% TFA/CH2Cl2 gave compound 5 in 90% yield (Scheme 1).1H NMR (400 MHz, CD3SOCD3): δ 0.91(t, 3H, J = 5.6 Hz), 1.28 (t, 3H, J = 5.8 Hz), 2.16–2.18 (m, 2H), 2.87–3.1 (m, 6H), 5.30 (s, 2H), 5.49 (s, 2H), 7.03 (s, 1H), 7.41 (s, 2H), 7.80 (bs, 3H) 8.0 (d, 1H, J = 7.6 Hz). ESI-TOF MS: m/z Calc. for C24H25N3O6 + H+ : 464.18; Obsd. 464.18.
Synthesis of G3.5-SN38 conjugates
The methanolic solution of PAMAM G3.5 carboxylate equivalent to 250 mg (0.02 mmoles) was evaporated under vacuum. The residue was dissolved in water, pH adjusted to 3.0 and lyophilized. To the acidified G3.5 in DMSO (15 ml) was added EDC (0.22 ml, 1.28 mmol) and NHS (0.28 g, 2.56 mmol) and the reaction mixture stirred for 10 mins. To this solution, compound 3 or 5 (0.32 mmol) was added followed by DIPEA (0.07 ml, 0.32 mmol) (Scheme 2). The reaction mixture was stirred at room temperature for 8h. DMSO was removed under high vaccum at 40 °C and residue redissolved in water, dialyzed using 3500 cut off membrane and sample lyophilized. The obtained product was further purified by SEC to remove low molecular weight impurities, unreacted dendrimer and possible crosslinked impurities. The conjugates were obtained in ~ 65 % yield and characterized by SEC and 1H NMR.
Scheme 2.

Synthesis of G3.5-SN38 conjugates
In vitro stability of conjugates
Solutions of G3.5-gly-SN38 (0.75 mg/ml) and G3.5-βala-SN38 (0.5 mg/ml) in PBS, pH 7.4, Mcyoy's 5A media (ATCC, Manassa, VA, USA) with 10% FBS, or 50% rat plasma (Rat plasma was prepared by centrifuging rat whole blood for 10 mins at 1000 rpm and the clear supernatant isolated as plasma) in PBS were incubated at 37 °C with shaking. At periodic intervals, samples (0.1 ml) were withdrawn, 0.1 ml acetonitrile was added, vortexed for 30 secs, and centrifuged at 1600 rpm to precipitate the proteins. The clear supernatant was then extracted with 0.1 ml of chloroform and the procedure was repeated in triplicate. The chloroform extracts were pooled, evaporated under nitrogen, redissolved in 80 ul of 1:1 DMSO/0.1 N HCl and analyzed by reverse phase HPLC using a UV detector for SN38 detection. A gradient elution method of methanol/water in 0.1% TFA at a flow rate of 1ml/min was used. The concentration of SN38 from each sample was determined from the peak areas observed, based on a calibration curve (n = 3) for SN38 in the concentration range 0.1– 60 ug/ml. The calibration curves were plots of peak area as a function of SN38 concentration. For accurate determination of SN38 concentration in extracted samples, extraction efficiencies for SN38 using the above described method were determined in each of the media, which were 100%, 103% and 62% respectively for PBS, cell culture media and 50 % rat plasma respectively.
Evaluation of cell proliferation and viability
HCT-116, human colorectal cancer cell line was obtained from American Type Culture Collection (ATCC, Manassas, VA). The cells were cultured in Mcyoy's 5A medium (ATCC) containing 10% fetal bovine serum (FBS) at 37 °C. Cells were grown at 37 °C in a humidified atmosphere of 5% CO2 (v/v) in air. Cell growth kinetic and viability were assessed by utilizing a highly water-soluble tetrazolium salt, WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt], as a component of Cell Counting Kit-8 from Dojindo Molecular Technologies, Inc. (Gaithersburg, MA). Cells were plated in 96 well plates (4000 cells/well) and incubated for 24 h, media was then gently aspirated and cells were treated with different concentrations (serially diluted) of SN38, SN38-gly, SN38-βala, G3.5, G3.5-gly-SN38, or G3.5-βala-SN38 in 0.5% DMSO containing media. DMSO was used to ensure complete dissolution of SN38 in media.0.5% DMSO did not affect cell viability. After 48 h, the cells were washed with PBS, and CCK8 reagent was added following the manufacturer's protocol. The cells were then incubated for 90 mins and absorbance at 450 nm was measured using 630 nm as reference. Cell viability was determined as % absorbance relative to untreated control cells. Prism software (v 5.0, LaJolla, CA, USA) was used to generate the inhibition curves and IC50 determination.
Cell cycle analysis
Cell cycle progression was monitored by the flow cytometric measurement of DNA content (17). HCT-116 cells (0.25 × 106 cell/ml) were incubated in 6 well plates for 24h. The cells were then exposed to free SN38 (32 nM), G3.5-gly-SN38 (240 nM) and G3.5-βala-SN38 (760 nM) for further 24h. Cells were then harvested, washed with 1 ml of ice-cold PBS, and then fixed with 1 ml of 70% ethanol (previously frozen at −20 C) overnight. The cells were then washed a second time with 1 ml of ice-cold PBS and treated with 0.2 ml PBS solution containing Propidium iodide (Sigma-Aldrich, USA) (25 ug/ml), Triton X-100 (Sigma-Aldrich, USA)(0.05%), and RNAse (Sigma-Aldrich, USA) (180 ug/ml). The solutions were incubated at room temperature in the dark for 30 min prior to analysis. Analysis of DNA content in cells stained with propidium iodide was performed using FACScan (Becton Dickinson, Mountain View, CA). The percentage of cells in each phase of the cell cycle was evaluated using the ModFit software (Verity Software House, Topsham, ME).
Nuclear fragmentation study
HCT-116 cells (0.1 × 106) were plated in 6 well plates containing coverslips and incubated for 24h. SN38 (5nM), G3.5-gly-SN38 (40nM) and G3.5-βala-SN38 (120 nM) were added and cells were incubated for 36h. Cells were then washed with PBS three times, and fixed with 4% paraformaldehyde for 10 min. Cells were again washed with PBS three times, and each coverslip was mounted on glass slide with a drop of DAPI containing mounting medium (Santacruz Biotechnology, CA, USA). Nuclear fragmentation was visualized in fixed cells stained with DAPI by a laser scanning confocal microscope Olympus FluoView® FV1000 (Olympus America Corp., Center Valley, PA). The objective specifications were 60× oil immersion and numerical aperture 1.42.
Statistical Analysis
Statistical analysis was carried out using student's t-test with a probability value p <0.05 considered statistically significant.
Results and Discussion
Synthesis and Characterization
Anionic PAMAM G3.5 was covalently conjugated to SN38 via a glycine or β–alanine spacer. Amino acid linkers particularly glycine has been successfully used to attach SN38 and other camptothecin analogues to polymers such as poly ethylene glycol (PEG), poly glutamic acid and pegylated poly (L-lysine) dendrimer with such conjugates showing increased aqueous solubility and good in vitro and in vivo efficacy (11, 18–20). β–alanine differs from glycine in the presence of an additional methylene group and could thus act as a relatively extended spacer. We chose these two spacers to compare the possible effect of linker length on the extent of drug loading, rate of drug release and activity of the conjugates. SN38 derivatives, modified at the 20-OH position via an ester linker with glycine and β–alanine respectively, were synthesized as described in the experimental section (11). Briefly, the phenolic hydroxyl group at 10 position of SN38 was first protected with Boc group, and the resulting compound 1 was then reacted with N-Boc glycine to yield compound 2. Removal of both the Boc groups under acidic conditions yielded SN38-gly (compound 3). In an analogous manner SN38-βala (compound 5) was also synthesized starting from SN38 (Scheme 1).
The terminal carboxyl groups of G3.5 PAMAM dendrimer were then activated using EDC/ NHS, and then reacted with the primary amines of SN38-gly and SN38-βala respectively in DMSO. Thus, the dendrimer was linked via an amide bond at one end of the spacer while the drug was attached to the same spacer via an ester bond at the other end (Scheme 2). The dendrimer was conjugated at the 20-OH end of SN38 with the aim of stabilizing the lactone form of the camptothecin derivative under physiological conditions which is essential for antitumor activity [9].
The conjugates were extensively purified by dialysis followed by preparative SEC to remove low molecular weight impurities. There was an increase in the elution volume in SEC for both conjugates relative to G3.5 with the order of elution being G3.5 < G3.5-gly-SN38 < G3.5-βala-SN38. The hydrodynamic radii of both conjugates as measured by dynamic light scattering was less than that of G3.5 (Table 1). These observations suggest that, upon attachment of SN38, the dendrimer may have a more compact form to avoid unfavorable interactions between the aqueous solvent and the hydrophobic drug. It is also possible, that the overall charge density on the surface of the conjugates is lowered relative to G3.5, reducing the extent of solvation in aqueous solution, which results in a lower hydrodynamic radius.
Table 1.
Characteristics of G3.5 PAMAM-SN38 conjugates.
| Compound | No. of SN38 molecules/ PAMAMa | Wt. of SN38/wt. of conjugate (%) | Molecular weightb | Elution volume (ml) | Hydrodynamic Radius (nm) |
|---|---|---|---|---|---|
| G3.5 | - | - | 12,931 | 13.0 | 1.76±0.11 |
| G3.5-gly-SN38 | 2.9 | 8.0 | 14,184 | 13.4 | 1.37±0.06* |
| G3.5-βala-SN38 | 4.0 | 10.0 | 14,715 | 14.0 | 1.33±0.05** |
Determined from 1H NMR.
Determined from NMR data and reported molecular weight of PAMAM.
(p < 0.01) and
(p < 0.01) denote significant differences of hydrodynamic radius from unconjugated G3.5.
The number of drug molecules attached to the dendrimer was determined using 1H NMR, by comparing the number of protons in the dendrimer to the methyl groups of SN38. The protons corresponding to PAMAM appeared between chemical shift, 2.1–3.8 ppm, while the methyl protons appeared as a broad peak between 0.9–1.1 ppm (Figure 1). 1H NMR was recorded at three different concentrations, 5 mg/ml, 10 mg/ml and 25 mg/ml and no change in integral values corresponding to protons from the dendrimer or methyl protons of SN38 were observed. This suggested the absence of aggregation at concentrations used for NMR analysis. The drug loading per mole of dendrimer was 2.9 and 4 for G3.5-gly-SN38 and G3.5-βala-SN38 respectively (Table1). Thus increasing the spacer length by one methylene unit increased the drug loading slightly. For synthesis of dendrimers with above drug loading, 16 moles of compounds gly-SN38 or βala-SN38 was used for conjugation to G3.5. Use of further excess drug did not improve loading, while lower stoichiometric ratios of the drug derivatives decreased loading onto PAMAM dendrimers. In order to further confirm that SN38 was covalently linked to the dendrimer, the following control experiment was performed. Acidified PAMAM G3.5 was mixed with compound gly-SN38 in DMSO and stirred under identical conditions as the conjugation reactions in the absence of the activating reagents EDC/NHS. The resulting mixture was then purified using the same method as described for the conjugates and the isolated compound analyzed by SEC and 1H NMR. The SEC profile and 1H NMR were same as for the native G3.5 PAMAM dendrimer. No peaks due to SN38 were observed in the 1H NMR spectrum suggesting absence of complexed or free drug (supplementary information). These observations further confirm that the conjugates synthesized are not complexes and the SN38 is covalently bound to the dendrimer.
Figure 1.

1H NMR spectra (500 MHz, D2O) of G3.5-gly-SN38.
The solubility of the conjugates in water was ~ 140 mg/ml which corresponds to ~10 mg/ml and ~14mg/ml of SN38 respectively in case of G3.5-gly-SN38 and G3.5-βala-SN38. SN38 has a very poor solubility of 7μg/ml and conjugation to the PAMAM dendrimer has considerably improved its aqueous solubility, which is higher than earlier reported PEG-SN38 derivatives (6.7 mg/ml) [11].
Stability of the conjugates
In vitro release characteristics of the conjugates was studied to assess their stability in the following physiologically relevant solutions, PBS, pH =7.4, cell culture media-Mcyoy's 5A medium with 10% FBS and 50% rat plasma (11, 21, 22). Stability in cell culture media was studied to determine whether potential activity of the conjugates would be due to extracellular and/or intracellular release of the drug. 50% plasma was used since in whole blood the plasma content is about ~55%. SN38 binds strongly to plasma proteins and the extraction efficiency in 100% rat plasma was poor (35%) after 1h incubation, while in 50% plasma it was much higher (62%) and hence the released drug could be estimated more accurately.
In PBS, both conjugates were stable with approximately 5% of the drug released in 72h. Thus these conjugates are more stable than the PEG-SN38 conjugates which were reported to have a t1/2 of only 14h in PBS [11]. However in cell culture media and 50% rat plasma the extent of release was relatively higher compared to PBS (Figure 2). This is expected as enzymes such as esterases are present in both plasma and 10% FBS containing media. Esterases can specifically cleave the ester bond between the spacer and the drug, whereas in PBS the release is only due to nonspecific hydrolysis. In case of G3.5-gly-SN38 the release in plasma was slightly higher than in cell culture medium (p<0.04) but no significant difference was observed in case of β-alanine linked conjugate. It has been reported that increasing the length of the spacer increases the rate of hydrolysis of the ester bond due to increased accessibility to the enzyme (22, 23). Upon comparing the release rates of both conjugates, we did not observe an overall, large difference in the release upon increasing the linker by one methylene unit. However, it is observed that G3.5-gly-SN38 shows slightly faster release of the drug in the first 24h than G3.5-βala-SN38 in both media and plasma (p < 0.03), but returns to similar rate by 48h (p > 0.1). It is possible that glycine being a natural amino acid is a better substrate for enzymes compared to β-alanine and hence the glycine linked conjugate shows initial faster release rates in media containing esterases. The conjugate with glycine linker also showed a higher release of drug at pH 5 after 72h compared to the conjugate with the β-alanine linker (see supplemental Figure 4). However, the dendrimer is likely to have too many different conformations in solutions such that some result in increased accessibility to the enzymes while others lead to increased steric hindrance to ester degradation. Hence the overall release kinetics is complex (24, 25). The fact that only ~20% (Figure 2) is released even after 72h for both conjugates in rat plasma, suggest that the covalent conjugates are quite stable and hold promise as delivery systems for controlled release of the drug.
Figure 2.

Stability of conjugates in different media at 37 °C. Top panel: SN38 release from G3.5-gly-SN38; Bottom panel: SN38 release from G3.5-βala-SN38. The time intervals were 0, 1, 5, 10, 24, 48 and 72h. Each graph represents average of two independent runs. (m ± s.d). PBS (◆), 50% rat plasma (▴), and cell culture media (∎).
Figure 4.
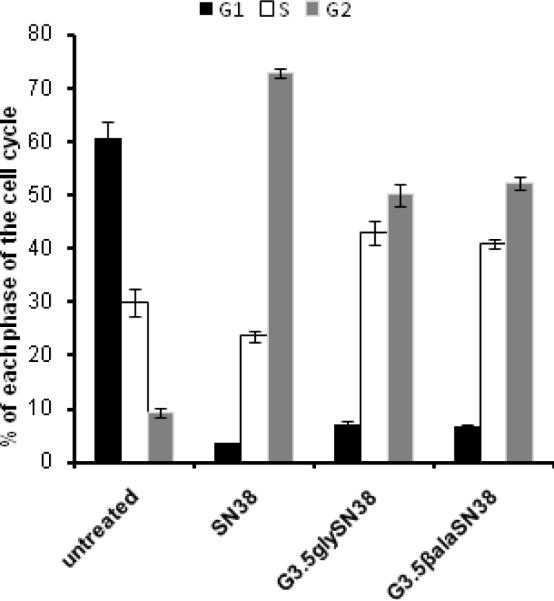
Effect of SN38 and G3.5-SN38 conjugates on cell cycle distribution of HCT-116 cells after 24h incubation. Values are representative of two independent experiments, each done in duplicate (m±s.d).
Inhibition of cell growth
The toxicity of the conjugates, linker modified SN38 derivatives (SN38-gly, SN38-βala), free SN38, and CPT-11 was compared upon 48h incubation with HCT-116 cells (Figure 3). SN38 was highly toxic to the cells with IC50 of approximately 16 nM. It was observed that both SN38-gly and SN38-βala had similar IC50 values and were slightly less cytotoxic than SN38 (p <0.02). This suggests that both SN38-gly and SN38-βala undergo rapid hydrolysis to release the free drug as the IC50 values were only about 3 times less than SN38. Both conjugates were far less cytotoxic with much higher IC50 values compared to the free drug. However their toxicity is still in the nanomolar range and are highly potent compared to CPT-11 (Table 2). PAMAM G3.5 was non-toxic in the concentration range used in case of the conjugates (inset, Figure 3). The lower activity of the PAMAM-SN38 conjugates compared to SN38 can be attributed to the slow release of the drug by hydrolysis of the linker from the sterically hindered dendrimer surface. G3.5-βala-SN38 exhibited lower activity as compared to G3.5-gly-SN38 (p <0.001) which could perhaps be due to the observed, lower initial release rate in cell culture media. There is also a possibility that the cellular uptake varies for the two different conjugates (26).
Figure 3.
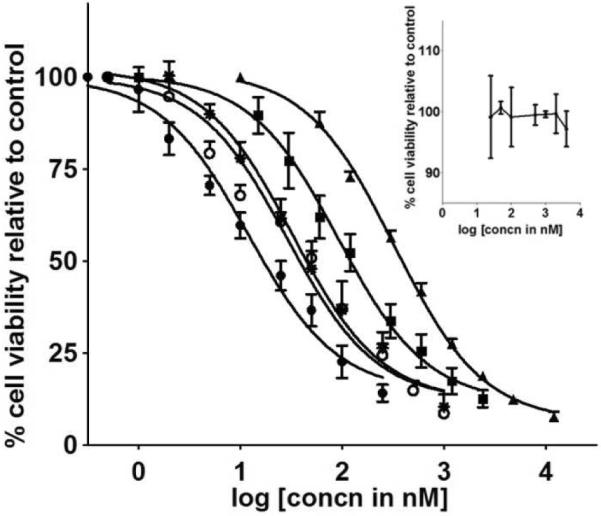
In vitro cytotoxicity of SN38 (●), SN38gly (◯), SN38βala (*), G3.5-gly-SN38 (∎), ▴G3.5- βala-SN38 in human colorectal cancer cell line (HCT-116) after 48h incubation. Each curve is representative of 3–5 independent experiments (m ± s.d). Inset: Inhibition curve for G3.5.
Table 2.
Cytotoxicity of different forms of SN38 toward human colorectal carcinoma HCT-116 cells. IC50 values presented as M±S.D. of 3–5 independent experiments.
| Compound | IC50 (nM) |
|---|---|
| SN38 | 16 ±3 |
| SN38-gly | 42 ±10 |
| SN38-βala | 45 ±6 |
| G3.5-gly-SN38 | 129 ±27* |
| G3.5-βala-SN38 | 387 ±20** |
| CPT-11 | 12 × 103 ± 3.8 × 103 |
(p <0.001) and
(p <0.001) denote significant differences of IC50 of G3.5-SN38 conjugates from SN38.
Induction of cell cycle arrest and nuclear fragmentation
As both PAMAM-SN38 conjugates inhibited cell proliferation of HCT-116 cells, we further investigated the mode of action of these conjugates. SN38 is a topoisomerase I inhibitor and is known to exhibit characteristic G2/M phase cell cycle arrest. The drug acts by covalent binding to the cleavable topoisomerase I-DNA complex, causing double strand DNA breaks and irreversible arrest of DNA replication. These events lead to arrest in the G2/M phase of the cell cycle (27, 28).
The progression of HCT-116 cells through the cell cycle after 24h treatment with 2 × IC-50 concentration of either conjugates or free drug was evaluated using flow cytometry. As expected, SN38, caused cell cycle arrest in the G2/M phase (73%) with considerable reduction in population of cells in the G0/G1 (4%) phase relative to untreated cells (61%). Upon exposure to G3.5-gly-SN38, a significant accumulation of cells in the G2/M phase (50 %) was observed with accompanied substantial decrease in the G0/G1 (7.0%). Similarly, in case of G3.5-βala-SN38, the accumulation of 52% cell population in the G2 phase was observed (Figure 4). These results suggest that the dendrimer-drug conjugates have a similar mechanism of action as SN38 and that the conjugates affect cells due to release of the free drug. However compared to both conjugates, SN38 used at equitoxic 2 × IC-50 concentration induced accumulation of larger number of cells in the G2/M phase of the cell cycle (p <0.001). This can be attributed to the slow release of drug from the conjugates.
In addition to inducing cell cycle arrest in cancer cells, anticancer drugs such as SN38 can kill cancer cells. The different modes of cell death include apoptosis, necrosis, autophagy or mitotic catastrophe with each associated with characteristic morphological changes and biochemical markers. Few studies have shown that SN38 causes cell death mainly via an apoptotic mechanism. The morphological changes that define apoptosis include chromatin condensation, nuclear fragmentation, plasma membrane blebbing and appearance of apoptotic bodies while biochemical markers include formation of DNA fragment `ladders'and caspase activation (28, 29).
Fluorescence microscopy was used to examine and compare the effect of conjugates and free drug on cell and nuclear morphology (Figure 5). HCT-116 cells were treated for 36h with either free SN38 or conjugates at 1/3 IC50, stained with the nuclear dye, DAPI and observed under the confocal fluorescence microscope. Untreated cells appeared mostly uniform with regularly shaped nuclei. In contrast, cells treated with either SN38 or the dendrimer-drug conjugates appeared heterogeneous. Nuclear fragments were distinctly visible, along with swollen and few condensed nuclei. Mitotic cells were also occasionally seen in the treated cells. Although, these observations are not conclusive evidence of apoptosis or necrosis, they do suggest that the free drug and conjugates have the same mode of action. The activity of the conjugates is due to the release of the free drug.
Figure 5.

Nuclear fragmentation in HCT-116 cells treated with drug/conjugates. Untreated cells (column 1); 5nM SN38 (column 2); 40 nM G3.5-gly-SN38 (column 3); 120 nM G3.5-βala-SN38 (column 4). Scale bar is 10 μm. Arrows indicate nuclear fragments. From bottom, 1st row: DIC (Differential interference contrast image) 2nd row: fluorescence image, 3rd row: overlay of DIC and fluorescence image.
Conclusions
PAMAM-SN38 conjugates with ester linkers which are stable at physiological pH and in plasma with low release of drug were synthesized. Conjugation of SN38 with the dendrimer significantly increased aqueous solubility. These dendrimer-drug conjugates are potent and have IC50 in the nanomolar range, and show activity due to slow release of SN38. Cell cycle analysis and observed nuclear fragmentation suggest that both free drug and the drug conjugated to the dendrimer have similar mechanism of action. These novel dendritic conjugates of SN38, coupled with our previous observations that PAMAM dendrimers are translocated across epithelial barrier of the gut, show potential to improve the oral bioavailability and targeted delivery of this potent drug.
Supplementary Material
Acknowledgements
Financial Support was provided by the NIH (R01 EB007470) and Utah Science Technology and Research (USTAR). The authors appreciate the assistance of Giridhar Thiagarajan with revision of the manuscript.
Footnotes
Supporting Information Available. Additional 1H NMR Spectra, representative histograms of cell cycle analysis and drug release data for SN38 at pH 5. This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- (1).Duncan R. Polymer conjugates as anticancer nanomedicines. Nat Rev Cancer. 2006;6:688–701. doi: 10.1038/nrc1958. [DOI] [PubMed] [Google Scholar]
- (2).Li C, Wallace S. Polymer-drug conjugates: recent development in clinical oncology. Adv Drug Deliv Rev. 2008;60:886–98. doi: 10.1016/j.addr.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Majoros IJ, Williams CR, Baker JR., Jr. Current dendrimer applications in cancer diagnosis and therapy. Curr Top Med Chem. 2008;8:1165–79. doi: 10.2174/156802608785849049. [DOI] [PubMed] [Google Scholar]
- (4).Duncan R, Izzo L. Dendrimer biocompatibility and toxicity. Adv Drug Deliv Rev. 2005;57:2215–37. doi: 10.1016/j.addr.2005.09.019. [DOI] [PubMed] [Google Scholar]
- (5).Pommier Y. DNA topoisomerase I inhibitors: chemistry, biology, and interfacial inhibition. Chem Rev. 2009;109:2894–902. doi: 10.1021/cr900097c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ulukan H, Swaan PW. Camptothecins: a review of their chemotherapeutic potential. Drugs. 2002;62:2039–57. doi: 10.2165/00003495-200262140-00004. [DOI] [PubMed] [Google Scholar]
- (7).Shimada Y, Rothenberg M, Hilsenbeck SG, Burris HA, 3rd, Degen D, Von Hoff DD. Activity of CPT-11 (irinotecan hydrochloride), a topoisomerase I inhibitor, against human tumor colony-forming units. Anticancer Drugs. 1994;5:202–6. doi: 10.1097/00001813-199404000-00011. [DOI] [PubMed] [Google Scholar]
- (8).Ulukan H, Muller MT, Swaan PW. Downregulation of topoisomerase I in differentiating human intestinal epithelial cells. Int J Cancer. 2001;94:200–7. doi: 10.1002/ijc.1463. [DOI] [PubMed] [Google Scholar]
- (9).Smith NF, Figg WD, Sparreboom A. Pharmacogenetics of irinotecan metabolism and transport: an update. Toxicol In Vitro. 2006;20:163–75. doi: 10.1016/j.tiv.2005.06.045. [DOI] [PubMed] [Google Scholar]
- (10).Caiolfa VR, Zamai M, Fiorino A, Frigerio E, Pellizzoni C, d'Argy R, Ghiglieri A, Castelli MG, Farao M, Pesenti E, Gigli M, Angelucci F, Suarato A. Polymer-bound camptothecin: initial biodistribution and antitumour activity studies. J Control Release. 2000;65:105–19. doi: 10.1016/s0168-3659(99)00243-6. [DOI] [PubMed] [Google Scholar]
- (11).Zhao H, Rubio B, Sapra P, Wu D, Reddy P, Sai P, Martinez A, Gao Y, Lozanguiez Y, Longley C, Greenberger LM, Horak ID. Novel prodrugs of SN38 using multiarm poly(ethylene glycol) linkers. Bioconjug Chem. 2008;19:849–59. doi: 10.1021/bc700333s. [DOI] [PubMed] [Google Scholar]
- (12).Moon SJ, Govindan SV, Cardillo TM, D'Souza CA, Hansen HJ, Goldenberg DM. Antibody conjugates of 7-ethyl-10-hydroxycamptothecin (SN-38) for targeted cancer chemotherapy. J Med Chem. 2008;51:6916–26. doi: 10.1021/jm800719t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kolhatkar RB, Swaan P, Ghandehari H. Potential oral delivery of 7-ethyl-10-hydroxy-camptothecin (SN-38) using poly(amidoamine) dendrimers. Pharm Res. 2008;25:1723–9. doi: 10.1007/s11095-008-9572-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Patri AK, Kukowska-Latallo JF, Baker JR., Jr. Targeted drug delivery with dendrimers: comparison of the release kinetics of covalently conjugated drug and non-covalent drug inclusion complex. Adv Drug Deliv Rev. 2005;57:2203–14. doi: 10.1016/j.addr.2005.09.014. [DOI] [PubMed] [Google Scholar]
- (15).Kolhatkar RB, Kitchens KM, Swaan PW, Ghandehari H. Surface acetylation of polyamidoamine (PAMAM) dendrimers decreases cytotoxicity while maintaining membrane permeability. Bioconjug Chem. 2007;18:2054–60. doi: 10.1021/bc0603889. [DOI] [PubMed] [Google Scholar]
- (16).Kitchens KM, Foraker AB, Kolhatkar RB, Swaan PW, Ghandehari H. Endocytosis and interaction of poly (amidoamine) dendrimers with Caco-2 cells. Pharm Res. 2007;24:2138–45. doi: 10.1007/s11095-007-9415-0. [DOI] [PubMed] [Google Scholar]
- (17).Shapiro HM. 1988. p. 353p. Alan R. Liss Inc, NewYork.
- (18).Singer JW, De Vries P, Bhatt R, Tulinsky J, Klein P, Li C, Milas L, Lewis RA, Wallace S. Conjugation of camptothecins to poly-(L-glutamic acid) Ann N Y Acad Sci. 2000;922:136–50. doi: 10.1111/j.1749-6632.2000.tb07032.x. [DOI] [PubMed] [Google Scholar]
- (19).Bhatt R, de Vries P, Tulinsky J, Bellamy G, Baker B, Singer JW, Klein P. Synthesis and in vivo antitumor activity of poly(l-glutamic acid) conjugates of 20S-camptothecin. J Med Chem. 2003;46:190–3. doi: 10.1021/jm020022r. [DOI] [PubMed] [Google Scholar]
- (20).Fox ME, Guillaudeu S, Frechet JM, Jerger K, Macaraeg N, Szoka FC. Synthesis and in vivo antitumor efficacy of PEGylated poly(l-lysine) dendrimercamptothecin conjugates. Mol Pharm. 2009;6:1562–72. doi: 10.1021/mp9001206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Chen X, McRae S, Parelkar S, Emrick T. Polymeric phosphorylcholinecamptothecin conjugates prepared by controlled free radical polymerization and click chemistry. Bioconjug Chem. 2009;20:2331–41. doi: 10.1021/bc900339x. [DOI] [PubMed] [Google Scholar]
- (22).Najlah M, Freeman S, Attwood D, D'Emanuele A. Synthesis, characterization and stability of dendrimer prodrugs. Int J Pharm. 2006;308:175–82. doi: 10.1016/j.ijpharm.2005.10.033. [DOI] [PubMed] [Google Scholar]
- (23).Ishikawa T, Otsuki M, Iwatsuki T. Enzymatic esterolysis of polymers containing 2-biphenylyl ester bonds in side chains. Bull Chem Soc Jpn. 1986;59 [Google Scholar]
- (24).Vicent MJ, Greco F, Nicholson RI, Paul A, Griffiths PC, Duncan R. Polymer therapeutics designed for a combination therapy of hormone-dependent cancer. Angew Chem Int Ed Engl. 2005;44:4061–6. doi: 10.1002/anie.200462960. [DOI] [PubMed] [Google Scholar]
- (25).Khandare JJ, Jayant S, Singh A, Chandna P, Wang Y, Vorsa N, Minko T. Dendrimer versus linear conjugate: Influence of polymeric architecture on the delivery and anticancer effect of paclitaxel. Bioconjug Chem. 2006;17:1464–72. doi: 10.1021/bc060240p. [DOI] [PubMed] [Google Scholar]
- (26).Perumal O, Khandare J, Kolhe P, Kannan S, Lieh-Lai M, Kannan RM. Effects of branching architecture and linker on the activity of hyperbranched polymer-drug conjugates. Bioconjug Chem. 2009;20:842–846. doi: 10.1021/bc800526z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Tsao YP, D'Arpa P, Liu LF. The involvement of active DNA synthesis in camptothecin-induced G2 arrest: altered regulation of p34cdc2/cyclin B. Cancer Res. 1992;52:1823–9. [PubMed] [Google Scholar]
- (28).Ueno M, Nonaka S, Yamazaki R, Deguchi N, Murai M. SN-38 induces cell cycle arrest and apoptosis in human testicular cancer. Eur Urol. 2002;42:390–7. doi: 10.1016/s0302-2838(02)00321-4. [DOI] [PubMed] [Google Scholar]
- (29).Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L, Kroemer G. Cell death modalities: classification and pathophysiological implications. Cell Death Differ. 2007;14:1237–43. doi: 10.1038/sj.cdd.4402148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.