

Abstract
The success of nucleoside reverse transcriptase inhibitors (NRTIs) in treating HIV-1 infection and reducing mother-to-child transmission of the virus during pregnancy is accompanied by evidence that NRTIs cause long-term health risks for cancer and mitochondrial disease. Thus, agents that mitigate toxicities of the current combination drug therapies are needed. Previous work had shown that the NRTI-drug pair zidovudine (AZT)–didanosine (ddI) was highly cytotoxic and mutagenic; thus, we conducted preliminary studies to investigate the ability of the active moiety of amifostine, WR1065, to protect against the deleterious effects of this NRTI-drug pair. In TK6 cells exposed to 100 μM AZT-ddI (equimolar) for 3 days with or without 150 μM WR1065, WR1065 enhanced long-term cell survival and significantly reduced AZT-ddI-induced mutations. Follow-up studies were conducted to determine if coexposure to AZT and WR1065 abrogated the antiretroviral efficacy of AZT. In human T-cell blasts infected with HIV-1 in culture, inhibition of p24 protein production was observed in cells treated with 10 μM AZT in the absence or presence of 5–1,000 μM WR1065. Surprisingly, WR1065 alone exhibited dose-related inhibition of HIV-1 p24 protein production. WR1065 also had antiviral efficacy against three species of adenovirus and influenza A and B. Intracellular levels of unbound WR1065 were measured following in vitro/in vivo drug exposure. These pilot study results indicate that WR1065, at low intracellular levels, has cytoprotective and antimutagenic activities against the most mutagenic pair of NRTIs and has broad spectrum anti-viral effects. These findings suggest that the activities have a possible common mode of action that merits further investigation.
Keywords: antimutagenesis, antiviral, amifostine, WR1065, cytoprotection
INTRODUCTION
Zidovudine (AZT or 3′-azido-2′,3′-dideoxythymidine) was the first nucleoside reverse transcriptase inhibitor (NRTI) used to treat HIV-1 infection and to reduce vertical transmission of the virus during pregnancy, and the largest body of data showing the potential for NRTIs to induce long-term side effects pertains to AZT (reviewed in [IARC, 2000; Wutzler and Thust, 2001; Dagan et al., 2002; Poirier et al., 2004; NTP, 2006; Kohler and Lewis, 2007; Walker and Poirier, 2007]). Phillips et al. [1991] reported that in vivo exposure of mice to AZT caused significant increases in micronucleated cells in bone marrow; since then additional studies have described the DNA damaging effects, clastogenicity, mutagenicity, and carcinogenicity of AZT and other NRTIs in animal models and humans [Olivero et al., 1997, 1999; Bonnet et al., 2004; Poirier et al., 2004; Brock et al. 2006; NTP, 2006; Escobar et al., 2007; Meng et al., 2007; Walker et al., 2007; Witt et al., 2007]. Previous work showed that in vitro and in vivo exposure of cells to AZT caused significant increases in mutations in reporter genes and in genes associated with neoplastic transformation (reviewed in [Walker and Poirier, 2007]). The combined use of AZT and a second NRTI enhanced cellular DNA incorporation of AZT and induction of mutations in the hypoxanthine-guanine phosphoribosyltransferase (HPRT) reporter gene compared with the responses from single NRTIs following in vitro exposure of human TK6 lymphoblastoid cells or in utero exposure of human infants [Meng et al., 2000, 2007; Escobar et al., 2007].
In spite of these potential toxicities, NRTI-based combinatorial drug therapy in the form of highly active antiretroviral therapy continues to be the most successful regimen for controlling HIV-1 infection and the progression of AIDS-associated symptoms, and reducing mother-to-child transmission of HIV-1 during pregnancy [Watts, 2006; Thorne and Newell, 2007]. Because NRTIs are likely to remain the backbone of HIV therapy for the foreseeable future, investigations of treatment options to minimize toxicities are a necessity [Foster and Lyall, 2008]. Potential options for mitigating the additive/synergistic cytotoxic and mutagenic effects of NRTI drug combinations are limited, and include the use of less toxic drug combinations, the addition of cytoprotective/anti-mutagenic agents to the currently used drug cocktails, or the development of alternative antiviral drugs with reduced toxicity [Walker and Poirier, 2007].
In the 1950s through the 1970s, a class of phosphorothioate drugs known informally as aminothiols was developed for use by the military as radioprotective compounds [Davidson et al., 1980]. Amifostine (WR2721) was one of the most successful drugs developed and tested; it is an organic thiophosphate that selectively protects normal tissues, but not most tumor cells, against the cytotoxicity of ionizing radiation. Amifostine has the chemical name S-2-(3-aminopropyl)-aminoethyl phosphorothioic acid and is a prodrug that is dephosphorylated to the active free thiol metabolite (WR1065) by alkaline phosphatase located in the cell membrane of most cells of many species including man [Capizzi, 1996; Grdina et al., 2000, 2002c; Khodarev et al., 2004; Kouvaris et al., 2007]. Following dephosphorylation, WR1065 is rapidly taken into the cell where it may be metabolized further to a symmetrical disulfide (WR33278) as well as other products [Grdina et al., 2000].
Investigations of the aminothiols have shown that their activity falls into several categories. Their radioprotective capabilities have been well documented in in vitro and in vivo studies demonstrating multifold protection against different types of radiation in a variety of model systems and species (reviewed in [Grdina et al., 2000, 2002a]). More recent studies have shown the capacity of amifostine, and some related compounds, to provide cytoprotection in patients undergoing chemotherapy for head and neck cancers and small cell lung cancer [reviewed in [Culy and Spencer, 2001; Block and Gyllenhaal, 2005]. Amifostine currently is FDA approved and marketed as Ethyol® for use as a cytoprotective agent during the treatment of these and a growing number of cancer types. Grdina et al. [1989a,b] and Kataoka et al. [1992] showed that amifostine also functions as an antimutagen, an anticancer agent, and an antimetastatic agent. Grdina et al. [1992] demonstrated that the aminothiol, WR151327, protected Hep2G cells against the DNA damaging effects of AZT; these findings were significant because they extended the range of the antimutagenic activity of the aminothiols from the DNA damage effects attributed to radiation, and some chemo-therapeutic agents, to the broader range of DNA lesions associated with exposure to AZT as a representative NRTI.
The use of less toxic antiretroviral drug combinations has been explored, and the narrow range in the relative mutagenic potencies exhibited by individual NRTIs, or combinations of NRTIs [Carter et al., 2007; Torres et al., 2007], led to current efforts to identify cytoprotective and antimutagenic agents that could be used along with antiretroviral agents to mitigate deleterious side effects of NRTI-based highly active antiretroviral therapy. During the course of the pilot studies reported here, we showed that WR1065 had both cytoprotective and antimutagenic activities against the synergistic toxicities of the most highly mutagenic pair of NRTIs, AZT, and didanosine (ddI or 2′,3′-dideoxyinosine), in cultured human lymphoblastoid cells. Extension of these studies to assess the potential impact of WR1065 on the therapeutic efficacy of AZT in phytohemagglutinin-stimulated primary human lymphocytes infected with HIV-1 in culture led to the discovery that WR1065 alone had anti-HIV activity; further work showed that WR1065 inhibited replication of three species of adenovirus and influenza A and B. The possibility of overlapping modes of action for WR1065-mediated cytoprotective, antimutagenic, and antiviral activities was considered in this report.
MATERIALS AND METHODS
Chemicals, Media, and Cells/Cell Lines
AZT and ddI were obtained from Sigma-Aldrich (St. Louis, MO) or Byron Chemical Co. (Long Island City, NY), and were dissolved in sterile physiologically buffered saline (Sigma-Aldrich) for cell treatments. Amifostine was purchased from Sigma-Aldrich and WR1065 was acquired from the NCI Chemical Repository and was stored according to instructions provided. HEPES, 6-thioguanine, cytidine, hypoxanthine, aminopterin, thymidine, alkaline phosphatase, phytohemagglutinin (PHA), formaldehyde, Trypan blue, and crystal violet were purchased from Sigma-Aldrich. For in vitro studies of TK6 cells, cell culture components including RPMI 1640, L-glutamine, MEM nonessential amino acids, and penicillin–streptomycin were obtained from BioWhittaker (Walkersville, MD), and FBS was purchased from Biomeda (Foster City, CA). For in vitro studies of human peripheral blood mononuclear cells (PBMCs), cell culture components included RPMI 1640 medium (American Type Culture Collection, Manassas, VA), FBS (Hyclone, Logan, UT), penicillin/streptomycin/glutamine (Invitrogen, Gaithersburg, MD), and recombinant human IL-2 (BD Biosciences, San Jose, CA). For in vitro studies of adenovirus and influenza virus replication, cell culture components including minimum essential medium, newborn calf serum, penicillin–streptomycin, and L-glutamine were obtained from Invitrogen. Human TK6 lymphoblastoid cells were from an established cell line [Liber and Thilly, 1982] and were a kind gift from Dr. J. Patrick O’Neill (University of Vermont), whereas fresh human PBMCs were obtained from the NIH Transfusion Center (Bethesda, MD) under a NIH Human Studies Research Board approved protocol. A549 cells, MDCK cells, adenovirus species, influenza A, and influenza B were obtained from the American Type Culture Collection.
The absolute purity of the WR1065 used in the pilot studies presented below was uncertain. WR1065 is sensitive to oxidation (as well as other reactions) and can undergo reaction to forms that appear to lack activities associated with the free thiol [North et al., 2000]. Prior to the initiation of these pilot studies, WR1065 was weighed (mol wt. 134) and stored frozen in RPMI 1640 medium as a 10 mg/ml working solution. However, the vial of lyophilized WR1065 used in the experiments presented here had been opened briefly two or three times on previous occasions, with the possibility that minimal oxidation may have occurred. The HPLC apparatus used in one set of experiments was available on one occasion only, thus, determination of the purity of the WR1065 used in these experiments was not feasible. Accordingly, in the tables and figures in the Results section, the indicated concentrations of WR1065 represent the maximum amount of active compound that could be present; the true concentration of active compound might be less. Also, for concentrations of WR1065 of less than 100 μM, estimates of the true efficacy of the aminothiol are limited by the fact that a portion of the compound has been found to be inactivated by a variety of medium components [Grdina et al., 2002a].
Cloning Efficiencies and HPRT Mutant Frequencies in TK6 Cells Exposed for 3 Days to AZT-ddI With or Without WR1065
A T-cell cloning assay was performed with TK6 cells cultured in supplemented RPMI 1640 growth medium as described [Sussman et al., 1999]. Cells were grown and all assays were performed using culture conditions that optimized cell viability, based upon Trypan blue exclusion, to ensure that changes in cell viability or cloning efficiencies (CEs) were due to the treatments and not to suboptimal cell culture conditions [Carter et al., 2007]. TK6 cells were expanded in T-flasks, and 4 to 5 days before starting treatments, the cells were changed to growth medium that contained CHAT [cytidine (C), hypoxanthine (H), aminopterin (A), and thymidine (T)] for 48 hr, then to conditioned medium that contained THC for 24 hr to reduce the spontaneous background HPRT mutant frequency [Liber and Thilly, 1982]. Conditioned medium was prepared 24 hr before needed, as follows. TK6 cells in log-phase growth were diluted to 3 to 5 × 105 cells/ml with fresh medium. The next day, this medium was removed and an additional 10% FBS (above that already in the medium) was added. The conditioned medium was filter sterilized using a 0.45-μm filter to remove any cells and mixed with freshly prepared RPMI 1640 growth medium to achieve a 50:50 ratio. Following the exposures, cells were washed, resuspended, and subcultured in nonselective medium for 7 days. For CE measurements, a fraction of treated and vehicle-exposed cells from each flask was plated at an average of two viable cells per well, in the presence of 4 × 104 lethally irradiated TK6 “feeder” cells, in 96-well U-bottom microtiter dishes. Fourteen days later, the plates were scored for positive colonies, based on morphology, size, and density. CEs were calculated as described by Sussman et al. [1999], and expressed as the mean ± standard deviation (SD).
The ability of WR1065 to prevent AZT-ddI-induced mutations was tested following exposure of T-cells for 3 days to 0 or 100 μM AZT-ddI (equimolar), with or without 150 μM WR1065 (n = 6 replicates/group). After a 7-day expression period, cells from each flask were plated in 96-well U-bottom microtiter dishes in nonselective medium to measure CEs and selective medium containing 6-thioguanine (1 μg/ml medium) to measure HPRT mutant frequencies. Fourteen days after plating, the microtiter dishes were examined for colonies in selective and nonselective medium. Observed HPRT mutant frequencies were calculated as previously described [Sussman et al., 1999], and expressed as the mean ± SD. Experiments were performed in duplicate, and data were combined for presentation here.
Inhibition of HIV-1p24 Protein Production by AZTand/or WR1065
An initial experiment was conducted to compare p24 protein production inhibition by AZT with or without WR1065. Fresh human PBMCs were cultured in 96-well plates at a density of 5 × 105 cells for 48 hr in RPMI 1640 medium with 10% fetal bovine serum, 10 U/ml human IL-2, 1% penicillin/streptomycin/glutamine, and 20 μg/ml PHA as a mitogen. Proliferating T-cell blasts then were infected with HIV-1BZ-167 (S. Sharpe, New York University, New York, NY) for 2 hr [Perno et al., 1988], washed to remove excess virus, and incubated with 10 μM AZT, 1,000 μM WR1065, or 10 μM AZT plus 1,000 μM WR1065 for 72 hr with no further changes in medium. Control groups included HIV-1 infected and uninfected PHA-stimulated T-cell blasts that were untreated. After 72 hr, cells were harvested and the HIV-1 infection status was determined in the experimental and control groups by measuring p24 protein using an ELISA kit (RETRO-TEK HIV-1, p24 Extended Range ELISA) (ZeptoMetrix, Buffalo, NY).
Based on our finding of apparent antiretroviral activity by WR1065 in this first experiment, subsequent experiments were performed (using the above protocol) to define the dose-response relationship between the concentration of WR1065 and p24 levels in HIV-1-infected PHA-stimulated T-cell blasts. In the second experiment, cells were treated for 72 hr with 10 μM AZT or 100, 330, or 1,000 μM WR1065. In the third experiment, HIV-1-infected cells were treated for 72 hr with 10 μM AZT or 10, 33, or 66 μM WR1065. Note that for experiments #1 and #2, aliquots of virus that had been stored previously at the NCI were used; for experiment #3, newly obtained HIV-1BZ-167 (from the same source as above) was used.
In parallel with these experiments, the degree of treatment-induced cytotoxicity was assessed by measuring relative levels of cell viability in human T-cell blasts treated with vehicle alone or drugs in the absence of HIV-1 infection. Cell viability was determined by comparing the numbers of viable and nonviable cells using a hemocytometer and Trypan blue exclusion.
Effects of WR1065 on the Replication of Human Adenoviruses or Influenza Viruses
Antiviral activities of WR1065 against adenoviruses were tested in A549 cells (alveolar type II, derived from a human lung carcinoma) and against influenza A or influenza B in MDCK cells (a canine kidney epithelial cell line) grown to near confluence in minimal essential medium in six-well plates, using standard culture media and growth conditions recommended for these cell lines (American Type Culture Collection). Aliquots of a given virus were prepared at an estimated concentration of 1,000 plaque forming units (PFUs) in 100 μL; cells were infected with serial dilutions of these aliquots and virus was absorbed to the cells for 1 hr at 37°C. After adsorption, cells were replenished with minimal essential medium containing up to 100 μM WR1065. Cultures were maintained at 37°C in 5% CO2 for 7 days for adenovirus assays and 3 days for influenza virus assays. Postinfection, infected cells were frozen and infectious virus yield titers in freeze-thawed preparations were determined by plaque assay. WR1065 antiviral activity was evaluated using two different dilutions of virus, which gave different infectious virus yields wherever possible.
To assess WR1065 effects upon A549 cell viability, uninfected A549 cells were grown in flat-bottomed six-well plates until near 100% confluence and then refed with freshly prepared medium with or without 150 μM WR1065. The cells were held for 5 to 7 days, and cell viability was assessed by gross and microscopic evaluation of wells for evidence of cell loss.
To estimate the amount of plaque reduction per unit dose of WR1065 for these antiviral analyses, the average reduction in PFUs per unit dose of WR1065 at each administered dose of WR1065 was calculated as follows: (average PFUs for vehicle-exposed cells − average PFUs at dose Y WR1065) ÷ (dose Y (μM) of WR1065) = average plaque reduction effect per unit dose WR1065.
Intracellular Concentrations of WR1065 in Lymphocytes Following In Vitro or In Vivo Treatment With Amifostine
The relationship between the dose of amifostine and the range of intracellular levels of WR1065 was examined following in vitro exposure of mouse, rat, and human lymphocytes and in vivo exposure of mice. Peripheral blood samples were collected from two of the authors using vacutainer tubes containing sodium heparin, and PBMCs were separated on ficol solution, washed, and cultured in RPMI 1640 medium including PHA as described earlier. Previously described techniques were used for isolating lymphocytes and culturing T-cells from mice and rats [Torres et al., 2007]. Briefly, the animals were euthanized by CO2 asphyxiation, spleens were removed aseptically and macerated, single cell suspensions were layered onto ficol solution, and washed cells (i) were cultured in RPMI 1640 medium in the presence of growth factor and mitogen (concanavalin A) for in vitro exposure to amifostine or (ii) were frozen following in vivo exposure of mice to amifostine. Animals were housed under standard conditions [Torres et al., 2007] and the procedures using rodents were approved by the Institutional Animal Care and Use Committee of Lovelace Respiratory Research Institute; procedures using human lymphocytes were approved by an Institutional Review Board.
For the in vitro exposures, primary mouse, rat, and human lymphocytes, maintained in T-25 flasks containing supplemented RPMI 1640 medium, were exposed for 1 or 2 hr with 0, 10, 50, or 100 μM amifostine that was dephosphorylated by preincubation with alkaline phosphatase (n = 2–4 flasks/time point and concentration) [Livesey et al., 1988]. Dephosphorylated amifostine was used in these experiments, as opposed to WR1065, because fresh WR1065 was not available. After treatment, cells were washed, concentrated, snap frozen, and stored at −80°C until analysis. For in vivo exposures of mice, individual animals were treated by subcutaneous injection with 0, 15, or 30 mg/kg amifostine dissolved in sterile saline (n = 3–4 mice/group), and animals were necropsied 2 hr later for isolation and freezing of splenic lymphocytes as described.
On the day of analysis, the frozen cells were homogenized in water (20% w/v), precipitated with an equal volume of perchloric acid–EDTA and centrifuged at 4°C, as described by Pamujula et al. [2004]. The analysis of WR1065 was performed using a high-pressure liquid chromatography method with electrochemical detection and previously described chromatographic conditions [Srinivasan et al., 2002; Pamujula et al., 2004], except that the signal was monitored on four channels and the 400 mV channel (which was the most sensitive) was used for generating a standard curve. For each sample, the concentration of WR1065 per million cells was determined by interpolating the peak areas to a standard curve for WR1065 and analyses were performed in duplicate.
Statistical Analyses
Differences between individual treatment groups vs. controls, or paired sets of samples, were determined by the Student’s t-test or Mann-Whitney U-statistic. In examining the antiviral activities of WR1065 and/or AZT, the χ2 test was used to assess differences in the proportions of HIV p24 protein production or infectious virus yields, and a multiple linear regression was used to test trends in antiviral activity. Statistical analyses were performed using SigmaStat (SSPSSC, Chicago, IL); P-values < 0.05 were considered significant.
RESULTS
Effect of 3-Day Exposures to AZT-ddI, With or Without WR1065, on CEs and HPRT Mutant Frequencies in TK6 Cells
Experiments were conducted to determine the potential impact of exposure to WR1065 (150 μM) on cytotoxic and mutagenic effects of AZT-ddI (100 μM each), the most mutagenic pair of NRTIs tested to date [Carter et al., 2007], following 3-day treatments of TK6 cells (Table I). No difference in average CEs between vehicle-exposed vs. WR1065-exposed cells was observed, although there was a small nonsignificant decrease in the viability of WR1065-treated cells (P = 0.3). In contrast, significant differences in CEs were observed between vehicle- and AZT-ddI-exposed cells, with the relative cell survival for AZT-ddI-exposed cells reduced to only 34% of control cells (P < 0.001), a finding consistent with the reported cytotoxicity of this NRTI drug combination [Meng et al., 2000; Carter et al., 2007]. Exposure of TK6 cells to the combined drug treatment of AZT-ddI plus WR1065 increased the relative cell survival to 52% of vehicle-exposed cells, a result that fell just short of a significant increase in relative cell survival compared with treatment with AZT-ddI without WR1065 (P = 0.06). These results are compatible with the reported activity of WR1065 as a cytoprotective agent.
TABLE I.
Observed Cloning Efficiencies at 21 Days Following 3-Day Exposures of TK6 Cells to AZT-ddI (Equimolar) and/or WR1065a
| Treatment group | CE (relative cell survival)b | P-valuec |
|---|---|---|
| Vehicle-control | 79 ± 10.9% (NA)d | – |
| WR1065 (150 μM) | 69.7 ± 5.3% (88%) | 0.3 |
| AZT-ddI (100 μM) | 27.1 ± 11.0% (34%) | <0.001 |
| AZT-ddI (100 μM) + WR1065 (150 μM) | 40.7 ± 13.0% (52%) | 0.001 |
TK6 human lymphoblastoid cells were exposed to vehicle or the given concentrations of AZT-ddI and/or WR1065 for 3 days, and then washed, diluted, and plated after a 7-day expression period under nonselection conditions to measure colony outgrowth as described in the Materials and Methods. Plates were scored 14 days after plating to determine average cloning efficiency (CE).
Adjusted relative cell survival values, in parentheses, represent the ratio of observed CEs in drug-exposed cells versus the average control value.
Mann-Whitney test.
Not applicable.
The ability of WR1065 to mitigate AZT-ddI-induced HPRT mutations was assessed in the same pilot study. A minimal nonsignificant increase in mean HPRT mutant frequencies was observed in cells exposed only to WR1065 (mutant frequency = 3.6 × 10−6) compared with vehicle-exposed cells (mutant frequency = 2.8 × 10−6). AZT-ddI-exposed cells had significantly increased mean HPRT mutant frequencies (mutant frequency = 12.1 × 10−6) compared with all other groups (P < 0.05). Coexposure to AZT-ddI plus WR1065 (mutant frequency = 4.2 × 10−6) abrogated the synergistic mutagenic effects of the NRTIs by at least 85% (P < 0.05) (Fig. 1). Notably, the increase in HPRT mutant frequencies observed in cells exposed to AZT-ddI plus WR1065 was not significantly different from the background HPRT mutant frequencies found in vehicle-exposed cells (P > 0.05), and approximated closely the mutant frequencies of cells exposed to WR1065 alone. These findings demonstrate the substantial efficacy of WR1065 as a protectant against AZT-ddI-associated mutagenic effects.
Fig. 1.

Comparison of the induced mutant frequency at the HPRT locus of human TK6 lymphoblastoid cells exposed for 3 days to vehicle alone, 150 μM WR1065, 100 μM AZT-ddI (equimolar), or 100 μM AZT-ddI (equimolar) plus 150 μM WR1065. Drug-induced mutant frequencies were determined by subtracting the mean mutant frequency measured in the vehicle-exposed group (mutant frequency = 2.8 × 10−6) from the mean observed mutant frequency measured in individual treatment groups. An asterisk (*) designates that the mean observed mutant frequency in AZT-ddI-exposed cells was significantly increased over the mean (vehicle-exposed) control cell value (P < 0.05), whereas the double asterisk (**) indicates that the mean observed mutant frequency in cells exposed to AZT-ddI plus WR1065 was significantly reduced compared with cells exposed to AZT-ddI only (P < 0.05) but was not significantly different from the mean observed value for vehicle-exposed cells. WR1065 reduced the mutagenic effects of AZT-ddI by at least 85%. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Inhibition of HIV-1Replication by AZTand/or WR1065 Following Infection of Human T-Cell Lymphocytes With HIV-1
The aforementioned findings led to investigation of the potential impact of WR1065 on the antiretroviral efficacy of AZT. The production of HIV-1 p24 protein was examined in fresh human PBMCs cultured with PHA to produce T-cell blasts, infected with HIV-1BZ-167 for 2 hr, washed, and then treated for 72 hr with AZT alone (10 μM), WR1065 alone (1,000 μM), or AZT plus WR1065 at the same drug concentrations (Table II, Experiment 1). Uninfected T-cell blasts served as controls for evaluation of drug toxicity (see later). PHA-stimulated blasts uninfected with HIV-1, and cells that were infected but untreated, served as control groups. Assessment of HIV-1 replication showed that 10 μM AZT resulted in 100% inhibition of p24 protein production as determined by ELISA in the presence or absence of WR1065. This experiment also revealed that 1,000 μM WR1065 provided ~100% inhibition of p24 protein production when applied alone to HIV-1-infected T-cell blasts.
TABLE II.
Inhibition of HIV-1 Replication as Assessed by Inhibition of p24 Protein Production in PHA-Stimulated Human T-Cell Blasts by AZT and/or WR1065a
| Exposure groups | p24 protein (pg/ml) | Estimated % inhibition |
|---|---|---|
| Experiment #1 | ||
| Cells (uninfected) | 0.28 | NA |
| HIV-1-infected cells | 498.62 | NA |
| HIV-1-infected cells + 10 μM AZT | 0.13b | ~100% |
| HIV-1-infected cells + 1,000 μM WR1065 | 0.13b | ~100% |
| HIV-1-infected cells + 10 μM AZT + 1,000 μM WR1065 | 0.13b | ~100% |
| Experiment #2 | ||
| Cells alone (uninfected) | 8.99 | NA |
| HIV-1-infected cells | 176.45 | NA |
| HIV-1-infected cells + 10 μM AZT | 9.07b | 94.9% |
| HIV-1-infected cells + 1,000 μM WR1065 | 8.92b | 94.9% |
| HIV-1-infected cells + 330 μM WR1065 | 9.31b | 94.7% |
| HIV-1-infected cells + 100 μM WR1065 | 8.92b | 94.9% |
| Experiment #3 | ||
| Cells alone (uninfected) | 3.0 | NA |
| HIV-1-infected cells | 44,455.8 | NA |
| HIV-1-infected cells + 10 μM AZT | 2.9b | ~100% |
| HIV-1-infected cells + 66 μM WR1065 | 33.2b | 99.9% |
| HIV-1-infected cells + 33 μM WR1065 | 503.2b | 98.9% |
| HIV-1-infected cells + 10 μM WR1065 | 911.2b | 98.0% |
| HIV-1-infected cells + 5 μM WR1065 | 10,293.7b | 76.8% |
NA, not applicable.
Primary human lymphocytes were stimulated with PHA, and resulting T-cell blasts were infected with HIV-1BZ-167 for 2 hr, washed, and treated with drug(s) for 72 hr, and then assessed for HIV p24 protein production using an ELISA kit as described in the Materials and Methods.
P < 0.001 (χ2 test), compared with p24 production in untreated HIV-infected cells.
Additional experiments were designed to assess the relationship between the concentration of WR1065 and p24 protein levels in HIV-1-infected PHA-stimulated human PBMCs. In a second experiment, the estimated % inhibition of p24 production in cells treated with 100, 330, or 1,000 μM WR1065 was high across all three aminothiol concentrations (~95%) and was similar to that provided by 10 μM AZT (Table II, Experiment 2). In a third experiment, 10 μM AZT provided ~100% inhibition of HIV-1 replication, whereas, 5, 10, 33, or 66 μM WR1065 exhibited dose-related inhibition ranging from ~77% at 5 μM to nearly 100% at 66 μM (Table II, Experiment 3). As noted earlier, different aliquots of HIV-1 virus (obtained from the same source) were used in experiments #1 and #2 vs. experiment #3. Elevated p24 protein production in infected, untreated cells was observed in experiment #3 compared with that of the previous two experiments (see Table II). Thus, comparisons between the percent reductions in p24 protein production within each experiment can be made with confidence, but comparisons between experiments should be made with caution. Yet, these combined experiments showed that WR1065 significantly reduced p24 production even though a broad range of WR1065 concentrations was tested, and the replicative capacity of the virus varied between experiments. These findings support the conclusion that the aminothiol alone inhibited HIV-1 replication. A companion report further examines the activity of WR1065 against HIV-1 in vitro and simian immunodeficiency virus ex vivo [Poirier et al., submitted].
To determine if some of the inhibition of virus replication might be due to WR1065-mediated cytotoxicity, HIV-1-uninfected fresh PBMCs were cultured in 0, 1, 5, 10, 50, 100, 500, or 1,000 μM WR1065 for 72 hr, and cell viability was determined using Trypan blue exclusion. These experiments showed that cell survival ranged from ~90% at 10 or 50 μM WR1065 down to 73% at 500 μM WR1065 and 30% at 1,000 μM WR1065. The data indicated that doses ≥100 μM showed cytotoxicity that may have been associated with some HIV-1 growth inhibition, but at doses of 1–50 μM WR1065, there was no significant contribution of cytotoxicity to the antiretroviral effects observed. Additional experiments confirming and extending these observations are presented in a companion report [Poirier et al., submitted].
WR1065-Mediated Inhibition of Adenovirus Replication in A549 Cells or Inhibition of Influenza A and B Replication in MDCK Cells
The aforementioned findings led to investigations to determine the ability of WR1065 to inhibit replication of viruses from other virus families including adenoviruses representing species B, C, and E, and influenza A and B. The impact of WR1065 treatment upon the replication of adenovirus type 7 of species B, adenovirus type 5 of species C, and adenovirus type 4 of species E in A549 cell monolayers is presented in Table III. Ad7p (strain Gomen) was used as a representative species B adenovirus associated with respiratory infections in pediatric and military populations; Ad5p (strain Adenoid 75) was used as representative of species C adenoviruses associated with upper respiratory infections in immunocompromised patients; Ad4p (strain RI-67) was used as representative of species E adenoviruses associated with conjunctivitis and respiratory infections in military populations. The results of these studies demonstrated that WR1065 reduced infectious virus yields for the three evaluated species of adenovirus. In these pilot studies, exposure to 100 μM WR1065 resulted in a significant reduction in PFUs in experiments where infected, untreated cells had infectious virus yields greater than 50 × 106 PFU/ml. For experiments where infected, untreated cells had infectious viral yields greater than 500 × 106 PFU/ml, exposure to 33 μM WR1065 also had a significant antiviral effect. Nonsignificant reductions in PFUs were observed in all other experiments and/or when lower doses of WR1065 were used (e.g., see Fig. 2). These findings support the conclusion that WR1065 had antiviral activity against these species of adenovirus.
TABLE III.
Impact of WR1065 Treatment on the Replication of Prototype Adenovirus Strains of Serotypes 4, 5, and 7 in A549 Cellsa
| Virus | Species | Experiment number | Mean infectious virus yields (PFUs × 106/ml) 96-hr postinfection in the presence of WR1065
|
|||
|---|---|---|---|---|---|---|
| 0 μM WR1065 | 10 μM WR1065 | 33 μM WR1065 | 100 μM WR1065 | |||
| Ad7p (Gomen) | B | 1b | 3.5 | 1.85 (47%) | 0.85 (76%) | 1.15 (67%) |
| 2b | 58.2 | – | – | 30.5 (48%)e,d | ||
| Ad5p (Adenoid 75) | C | 1b | 8.9 | 6.2 (30%) | 5.7 (36%) | 2.2 (75%)e |
| 2e | 665 | – | 600 (9%)e | 500 (25%)f | ||
| 3c | 1,920 | – | 1,660 (14%)e | – | ||
| Ad4p (RI-67) | E | 1b | 1.6 | 0.35 (78%) | – | – |
| 2b | 22.5 | – | – | 9.5 (58%) | ||
| 3b | 150 | – | – | 40 (73%)f | ||
| 4c | 1,900 | – | 1,420 (25%)e | 980 (48%)f | ||
Infectious virus yields were determined in A549 cells infected with adenovirus and treated with 0 or up to 100 μM WR1065. Cells in six-well plates were infected with a given species of adenovirus. After 1 hr adsorption, cells were replenished with minimum essential medium containing 0 or up to 100 μM WR1065. At 7 days postinfection, infected cells were frozen and infectious virus yields in freeze-thawed preparations were titered by plaque assay.
Infected with 103 PFUs, adsorption for 1 hr.
Infected with 104 PFUs, adsorption for 1 hr.
Two additional experiments using 100 μM WR1065 yielded similar results.
P < 0.05 (χ2 test), compared with untreated infected A549 cells.
P < 0.001 (χ2 test), compared with untreated infected A549 cells.
Fig. 2.
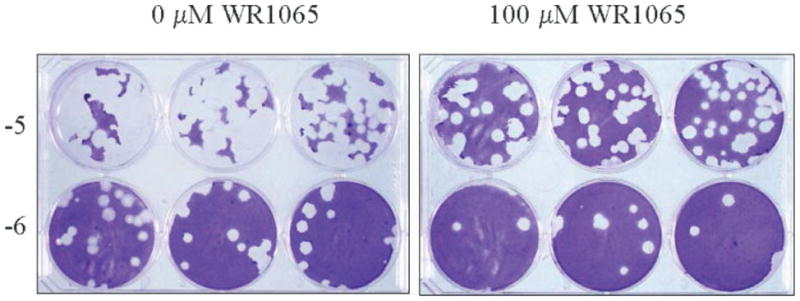
Titration of Ad4p infectious virus yields by plaque assay in A549 cell monolayers. Cells in six-well plates were infected with 1000 PFU of Ad4p (RI-67) and treated with 0 or 100 μM WR1065. After 1 hr adsorption, cells were replenished with minimum essential medium containing 0 or 100 μM WR1065. At 7 days postinfection, infected cells were frozen and freeze-thawed preparations were titered by standard plaque assay. Titers shown are from Table III, experiment 1 except the mean virus yields are expressed as PFUs × 106/ml.
To assess WR1065-mediated effects upon A549 cell viability, uninfected A549 cells were incubated with 0, 10, 30, and 100 μM WR1065 to determine the effects of the drug upon cell viability. A549 cells formed uniform monolayers with no evidence of grossly or microscopically visible focal areas of cell death when incubated with any of the above WR1065 concentrations for up to 7 days. These results demonstrate that these exposure concentrations of WR1065 lacked cytotoxic effects upon A549 cells in the absence of viral infection.
Subsequently, WR1065 was evaluated for its ability to inhibit viral replication and development of cytopathic effects in influenza A- or influenza B-infected MDCK cell monolayers. Increasing doses of WR1065 resulted in significant trends in the reduction of infectious virus yields for both influenza A and B virus strains (Table IV) (P < 0.05), but pairwise comparisons between infectious virus yields in cells treated with a given dose of WR1065 and infected, untreated control cells were not significant in these pilot experiments. Efficacy appeared to be dependent upon both the viral strain being tested and the drug dose administered. These findings support the conclusion that WR1065 had antiviral efficacy against influenza A and influenza B.
TABLE IV.
Impact of WR1065 Treatment on Replication of Influenza A/HKx31 (H3N2) or Influenza B/Lee/40 in MDCK Cell Monolayersa
| Virus | Mean plaque number (% reduction)
|
P-valueb | |||
|---|---|---|---|---|---|
| 0 μM WR1065 | 10 μM WR1065 | 33 μM WR1065 | 100 μM WR1065 | ||
| A/HKx31(H3N2) | 66.3 | 59.3 (11%) | 57.0 (14%) | 47.7 (28%) | <0.05 |
| Influenza B/Lee/40 | 26.7 | 24.0 (10%) | 20.3 (24%) | 18.6 (30%) | <0.05 |
Infectious virus yields were determined in MDCK cells infected with virus and treated with 0 or up to 100 μM WR1065. Cells in six-well plates were infected with a given species of influenza virus. After 1 hr adsorption, cells were replenished with minimum essential medium containing 0 or up to 100 μM WR1065. At 3 days postinfection, infected cells were frozen and freeze-thawed preparations were titered by plaque assay.
Multiple linear regression.
As part of efforts to understand the effect of WR1065 on the inhibition of viral replication, we estimated the plaque reduction efficacy per unit dose of WR1065 using data from representative experiments performed with adenoviral- and influenza-infected cells (see Table V). Five viruses were tested in two different epithelial cell types; in each experiment the effect per unit dose of WR1065 was higher at the lowest dose tested and diminished as the applied dose increased. The trend was observed across all experiments where multiple dose levels were evaluated. Data for similar evaluations performed for HIV-infected cells are not presented due to the fact that these pilot studies included relatively high doses of WR1065; however, trends in WR1065-associated effects per unit dose similar to those above (Table V) were observed in some low-dose experiments of HIV-1 replication inhibition (data not shown).
TABLE V.
Average Infectious Virus Yield Reduction Per Unit Dose of WR1065 in Adenovirus or Influenza-Infected Cells
| Virus tested | Cell type used in the plaque reduction assay | Dose of WR1065 administered (μM) | Average # of PFUs reduced by exposure to WR1065 (×106) | PFUs reduced per unit dose of WR1065 (×106) |
|---|---|---|---|---|
| AD7-P | A549 | 0 | (3.5)a | NA |
| 10 | 1.65 | 0.17 | ||
| 33 | 2.65 | 0.08 | ||
| 100 | 2.35 | 0.02 | ||
| AD5-P | A549 | 0 | (8.9)a | NA |
| 10 | 2.7 | 0.27 | ||
| 33 | 3.2 | 0.10 | ||
| 100 | 6.7 | 0.07 | ||
| AD4-P | A549 | 0 | (1,900)a | NA |
| 33 | 480 | 14.55 | ||
| 100 | 920 | 9.20 | ||
| Influenza A/HKx31 (H3N2) | MDCK | 0 | (66.3)a | NA |
| 10 | 7 | 0.70 | ||
| 33 | 9.3 | 0.28 | ||
| 100 | 18.3 | 0.18 | ||
| Influenza B/Lee/40 | MDCK | 0 | (26.7)a | NA |
| 10 | 2.7 | 0.27 | ||
| 33 | 6.4 | 0.19 | ||
| 100 | 8.1 | 0.08 |
Average PFUs in vehicle-exposed cells.
NA, not applicable.
As was noted the results for the HIV-1 experiments, comparisons of drug efficacy between experiments using adenoviruses or influenza viruses should be made with caution. Although every effort was made to ensure that each aliquot of the evaluated viruses had equal replicative capacity, variations between aliquots were observed. Thus, comparisons of drug inhibitory effects upon viral replication within one experiment can be done with confidence, but comparisons between experiments should be made with caution because of the potential for differences in viral replication capacity between experiments. Additional work will be necessary to determine how well these in vitro experimental results predict in vivo drug efficacy.
Intracellular Levels of Free WR1065 in Cytoplasm of Exposed Primary Lymphocytes
An important issue was the range in intracellular levels of WR1065 that occurred following in vitro or in vivo treatment with WR1065, the free thiol of amifostine, that offered cytoprotective, antimutagenic, and/or antiviral activities under the exposure conditions used in the studies described earlier. To address this issue, pilot studies were performed to measure intracellular levels of unbound WR1065 in primary mouse, rat, and human lymphocytes at treatment levels of amifostine in cell culture medium previously found to provide significant protection against mutations induced by AZT-ddI and significant reduction in viral load in human cells in vitro. It should be noted that amifostine that had been dephosphorylated via alkaline phosphatase immediately prior to use was applied in these studies because fresh WR1065 was not available. Amounts of free WR1065 also were measured in splenic lymphocytes isolated after subcutaneous injection of mice with amifostine. These in vitro studies found an average concentration of free WR1065 of 32 pmol/106 cells in mouse and rat lymphocytes treated in culture with amifostine (Fig. 3), levels that are well below the accumulated intracellular concentration of ~30 nmol WR1065/106 cells found by Calabro-Jones et al. [1998] to produce significant cytotoxicity in multiple types of cultured cells. Similar levels of unbound WR1065 were measured in primary human lymphocytes exposed to dephosphorylated amifostine (data not shown), but the amount of WR1065 was more variable than that found in similarly treated rodent lymphocytes. In amifostine-exposed mice, dose-related amounts of free WR1065 were measured in splenic lymphocytes, with 56 ± 1 pmol/106 cells and 82 ± 17 pmol/106 cells found 2 hr after subcutaneous injection with 15 or 30 mg amifostine/kg. WR1065 was not detected in cells following in vitro or in vivo exposure to vehicle alone.
Fig. 3.

Dose response for intracellular concentration of WR1065 following treatment of primary mouse (A) or rat (B) lymphocytes for 1 or 2 hr with 0, 10, 50, or 100 μM amifostine converted to WR1065 (n = 2–4/species for each exposure group). Amifostine was dephosphorylated for 30 min with alkaline phosphatase to yield WR1065 for cell treatment. A high-pressure liquid chromatography method with electrochemical detection was used to measure WR1065, and the concentration of WR1065 per million cells in each sample was determined by interpolating the peak areas to a standard curve for WR1065 [Pamujula et al., 2004]. (A) Data from WR1065-exposed mouse lymphocytes; (B) data from WR1065-exposed rat lymphocytes.
These studies also showed a marked reduction in the accumulation of intracellular free thiol at exogenous doses above ~50 μM. These results are consistent with the observation that transport of WR1065 into cells occurs by two processes, one of which is active and operates at doses below ~50 μM, the second of which is passive and operates at WR1065 levels that exceed 50 μM [Grdina et al., 1995].
DISCUSSION
The initial goal of this work was to investigate the ability of the aminothiol, WR1065, to protect human cells against cytotoxic and mutagenic effects of NRTIs in vitro. These pilot studies showed that exposure of human TK6 lymphoblastoid cells to AZT-ddI plus WR1065 for 3 days had antimutagenic effects and long-term cytoprotective effects as measured using CEs, even though the doses of WR1065 used in these experiments were below those previously shown to be cytoprotective [Grdina et al., 2002b]. Extension of this work revealed that WR1065 had antiviral effects against HIV-1, three species of adenovirus, and influenza A and influenza B. The antimutagenic and antiviral activities were associated with low intracellular levels of WR1065, suggesting a common mode of action.
In assessing cytoprotective activity of WR1065, we chose to evaluate CEs instead of assessing cell viability by Trypan blue exclusion because measurements of CE provide a quantitative indication of the impact of drug exposure upon the functionality of genes involved in the ability of cells to survive mild physical stress and to plate down, to reenter the growth cycle, and to proliferate for a 14-day period. These studies showed that WR1065 provided long-term protection against AZT-ddI-induced cyto-toxicity and mutagenicity, findings that are in agreement with the reported ability of WR1065 to protect cells/tissues from the cytotoxic and mutagenic effects of radiation and chemotherapeutic agents [Grdina et al., 2000, 2002c], and that extend the range of known lesions against which WR1065 has efficacy as a protective agent.
A comparison of the degree of WR1065-mediated protection against mutagenesis found in our pilot study compared with that reported previously shows that the ~85% reduction in AZT-ddI-induced mutant frequencies in TK6 cells coexposed to WR1065 is similar to the mutant frequency reductions found in lymphocytes of mice given amifostine before in vivo exposure to gamma radiation or cyclophosphamide (i.e., 63 or 78% reduction in observed Hprt mutant frequencies, respectively) [Grdina et al., 1985, 1999; Kataoka et al., 1996]. The results of our AZT-ddI experiments support an earlier hypothesis that the effects of WR1065 on cell turnover rates led to enhanced DNA repair [Khodarev et al., 2004]; improved repair would have several effects: (i) improved survival for a subset of cells because of the prevention of lethal mutations, (ii) decreased cell viability for another subset because of the loss of cells that could not repair the NRTI-induced damage successfully, and (iii) a significant reduction in mutant frequencies.
The finding that WR1065 significantly reduced the mutagenicity of AZT-ddI in TK6 cells led to follow-up studies of the potential impact of WR1065 on the antiretroviral efficacy of AZT and the discovery that WR1065 alone reduced HIV-1 replication. The experiments reported here did not allow us to draw conclusions about potential interactions between AZT and WR1065 because, at the doses used, both drugs had substantial anti-HIV activity, and it was not possible to determine if one drug affected the efficacy of the other. The issue of potential drug interactions is addressed in a companion report [Poirier et al., submitted]. Cell viability studies conducted in parallel assays showed that WR1065-related toxicity increased with escalating dose, but cytotoxicity was not sufficient to account for the observed inhibition of viral p24 protein production. Optimal cell survival was achieved at doses of 10 and 50 μM, levels at which active transport of the drug into cells has been hypothesized to occur [Grdina et al., 1995].
These antiviral studies then were extended to distinctly different viruses. Under a current viral classification scheme (http://www.virology.net/Big_Virology/BVFamilyGenome.html), HIV-1 is classified in the family Retroviridae, part of the group of DNA and RNA reverse transcribing viruses. To investigate further the potential antiviral activity of WR1065, we examined its effects upon viral replication of adenovirus serotypes representative of species B, C, and E within the genus Mastadenovirus in the family Adenoviridae, part of the group of DNA double-stranded viruses, nucleic acid-type double-stranded DNA virus, and influenza A and B in the family Ortho-myxoviridae, part of the group of RNA viruses, nucleic acid type (−) single-stranded RNA viruses. WR1065, at concentrations of 100 μM and lower, reduced the replication of the tested species of adenovirus, and also reduced the replication of influenza A and B, with WR1065 having no observable toxicity over the course of treatment in A549 cells. In evaluating the results of these studies, it should be noted that the antiviral efficacy of WR1065 was tested at concentrations of virus with differing infectious virus yields. Significant antiviral efficacy was observed consistently across all experiments only for the higher infectious virus yields and for the highest concentrations of WR1065 tested. The inability to detect consistent antiviral efficacy at lower concentrations of virus and/or WR1065 may reflect the small sample size used in these pilot studies and additional work will be necessary to investigate these effects further.
As part of our efforts to understand the activity of WR1065 as an antiviral agent in greater detail, we determined the effect per unit dose and also measured levels of free drug in primary lymphocytes exposed in culture. Surprisingly, we found an inverse relationship between the dose of WR1065 used in the studies of inhibition of adenovirus and influenza virus replication and the effect per unit dose of WR1065 (Table V). Subsequent studies showed that these antiviral effects were achieved at low levels of intracellular free drug. It should be noted that the levels of bound drug were not measured; because bound drug may be the active form, these latter studies are more informative for the ability to achieve therapeutic intracellular drug levels than for the level of intracellular drug with antiviral and antimutagenic activity.
The findings of both antimutagenic and antiviral activity of WR1065 support the hypothesis that these effects involve a common drug mechanism. The antimutagenic mode of action of the aminothiols including WR1065 has been investigated previously, and another body of literature discusses potential modes of action for poly-amines, polyamine analogs, and thiol-containing compounds as antiviral agents. A thorough review of these studies is beyond the scope of this report, but some findings are informative for the pilot studies presented here. WR1065 has been shown to have both high-dose and low-dose effects [Grdina et al., 2000, 2002c]. Cytoprotection is associated with high-dose exposures, and it is considered to be optimal at doses near 4 mM and is attributed to WR1065’s activity as a free-radical scavenger, whereas the drug’s antimutagenic effects are associated with low-dose exposures around 100 μM or lower where changes in transcription factor binding, protein phosphorylation levels, and gene expression have been described [Grdina, 2002c]. Interestingly, low intracellular levels of WR1065 have been shown to affect the phosphorylation of topoisomerase II alpha, thereby, affecting its activity [Murley et al., 1997]. This enzyme is involved in host cell DNA replication and replication of HIV-1 [Freire et al., 2001] and effects upon its activity also could alter mutation induction by delaying cell cycle progression to allow more time for DNA repair [Kondapi et al., 2005].
Both polyamines and thiol-containing compounds have been shown to have antiviral activity. Ames and Dubin [1960] reported that polyamines inhibited replication of T4 bacteriophage. Mitchell et al. [1998] showed that WR1065 increased intracellular spermidine levels in cells exposed in vitro. Such a change has been suggested to have antimutagenic effects because of the DNA protective capacity of spermidine, and also could result in antiviral effects because of changes in intracellular polyamine levels [Bachrach and Rosenkovitch, 1972; Bachrach, 2007]. The precise mechanism(s) by which polyamines induce DNA protective, antimutagenic, and antiviral effects are uncertain. Some studies showed that polyamine metabolism by amine oxidases resulted in the formation of aldehydes, aminoaldehydes, and oxidized polyamines that had broad spectrum antiviral activity [Bachrach, 2007], and oxidation has been shown to reduce the activity of HIV-1 proteases [Davis et al., 1999]. It should be noted that WR1065 is metabolized in vitro to cysteamine, acrolein, other aldehydes, and hydrogen peroxide by copper-dependent amine oxidases present in serum [Meier and Issels, 1995]; however, oxidation metabolites of WR1065 do not appear to be sufficient to explain the inhibitory effects of WR1065 upon adenoviruses as shown in these pilot studies, because adenoviruses are resistant to many virucidal compounds including many aldehydes that are effective inhibitors of other viruses [Sauerbrei et al., 2007].
Several investigators have reported evidence that thiol-containing compounds have activity against HIV-1. In studies of the antiretroviral activity of WR151327, a derivative of amifostine, in chronically infected promonocytic cells, Kalebic and Schein [1994] reported drug-associated inhibitory effects upon the viral reverse transcriptase. Bergamini et al. [1994] studied the anti-HIV effects of cysteamine and its symmetrical diamine cystamine, two compounds that are structurally related to WR1065; however, they failed to find evidence of direct drug-associated effects upon the viral reverse transcriptase. Their studies suggested that these two agents inhibited several stages of HIV replication including assembly of infectious viral particles [Bergamini et al., 1994, 1996]. These thiol-containing compounds also have been demonstrated to protect DNA from radiation-induced damage [Allegra et al., 2002]; DNA protective effects could be related to an antimutagenic mode of action. Taken together, these studies suggest that polyamines, polyamine analogs, and thiol-containing compounds have a diverse range of activities. Although comparisons between studies must be made with caution because different model systems were used to evaluate drug-associated effects, it is noteworthy that no one mode of action described to date appears to be adequate to explain the diversity of these effects.
Amifostine and its active form WR1065 have been studied for decades as radioprotectants, and amifostine has been used clinically as a cytoprotectant since the mid-1990s. The range of activities identified for this small compound include scavenging of free radicals, cytoprotection against an array of physical agents and chemical compounds, and low-dose effects that include changes in gene expression, and protection against mutagenesis, neoplastic transformation, and metastasis. The pilot studies presented here add to this diversity of effects by extending the range of its cytoprotective and antimutagenic effects and by demonstrating in vitro antiviral activity against viruses classified in three distinct groups, with diverse mechanisms involved in their replication, survival, and pathogenicity. These findings show novel activities for WR1065 and support the conclusion that the amino-thiols represent a new class of broad spectrum antiviral agents. These results indicate the need for additional investigations of the mode of action for the antimutagenic and antiviral activity of this drug.
Acknowledgments
Grant sponsor: The National Institute of Child Health and Human Development; Grant number: R01 HD033648; Grant sponsor: The National Cancer Institute; Grant number: R01 CA095741; Grant sponsor: The National Heart, Lung, and Blood Institute; Grant numbers: R01 HL072727, 1 F31 HL081928; Grant sponsor: The Intramural Research Program, National Cancer Institute, National Institute of Health.
The authors thank Steve Randock for assistance in preparing figures. The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. Patents pending: USA, NIH332.001VPC, April 2007, and USA, No. 41543-0602, April 2007.
Abbreviations
- AZT
zidovudine or 3′-azido-2′,3′-dideoxythymidine
- CE
cloning efficiency
- ddI
didanosine or 2′,3′-dideoxyinosine
- HPRT
hypoxanthine-guanine phosphoribosyltransferase
- NRTI
nucleoside reverse transcriptase inhibitor
- PBMCs
peripheral blood mononuclear cells
- PFUs
plaque forming units
- PHA
phytohemagglutinin
- SD
standard deviation
- WR1065
free thiol of amifostine
References
- Allegra P, Amodeo E, Colombatto S, Solinas SP. The ability of cystamine to bind DNA. Amino Acids. 2002;22:155–166. doi: 10.1007/s007260200004. [DOI] [PubMed] [Google Scholar]
- Ames BN, Dubin DT. The role of polyamines in the neutralization of bacteriophage deoxyribonucleic acid. J Biol Chem. 1960;235:769–775. [PubMed] [Google Scholar]
- Bachrach U. Antiviral activity of oxidized polyamines. Amino Acids. 2007;33:267–272. doi: 10.1007/s00726-007-0535-y. [DOI] [PubMed] [Google Scholar]
- Bachrach U, Rosenkovitch E. Effect of oxidized spermine and other aldehydes on the infectivity of vaccinia virus. Appl Microbiol. 1972;23:232–235. doi: 10.1128/am.23.2.232-235.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergamini A, Capozzi M, Ghibelli L, Dini L, Salanitro A, Milanese G, Wagner T, Beninati S, Pesce CD, Amici C, Rocchi G. Cystamine potently suppresses in vitro HIV replication in acutely and chronically infected human cells. J Clin Invest. 1994;93:2251–2257. doi: 10.1172/JCI117223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergamini A, Ventura L, Mancino G, Capozzi M, Placido R, Salanitro A, Cappannoli L, Faggioli E, Stoler A, Rocchi G. In vitro inhibition of the replication of human immunodeficiency virus type 1 by β-mercaptoethylamine (cysteamine) J Infect Dis. 1996;174:214–218. doi: 10.1093/infdis/174.1.214. [DOI] [PubMed] [Google Scholar]
- Block KI, Gyllenhaal C. Commentary: The pharmacological anti-oxidant amifostine—Implications of recent research for integrative cancer care. Integr Cancer Ther. 2005;4:329–351. doi: 10.1177/1534735405282842. [DOI] [PubMed] [Google Scholar]
- Bonnet F, Lewden C, May T, Heripret L, Jougla E, Bevilacqua S, Costagliola D, Salmon D, Chene G, Morlat P. Malignancy-related causes of death in human immunodeficiency virus-infected patients in the era of highly active antiretroviral therapy. Cancer. 2004;101:317–324. doi: 10.1002/cncr.20354. [DOI] [PubMed] [Google Scholar]
- Brock MV, Hooker CM, Engels EA, Moore RD, Gillison M, Alberg A, Keruly J, Yang SC, Heitmiller RF, Baylin SB, Herman JG, Brahmer J. Delayed diagnosis and elevated mortality in an urban population with HIV infection and lung cancer: Implications for patient care. J Acquir Immune Defic Syndr. 2006;43:47–55. doi: 10.1097/01.qai.0000232260.95288.93. [DOI] [PubMed] [Google Scholar]
- Calabro-Jones PM, Aguilera JA, Ward JF, Fahey RC. The limits to radioprotection of Chinese hamster V79 cells by WR-1065 under aerobic conditions. Radiat Res. 1998;149:550–559. [PubMed] [Google Scholar]
- Capizzi R. Amifostine: The preclinical basis for broad-spectrum selective cytoprotection of normal tissues from cytotoxic therapies. Semin Oncol. 1996;23 (Suppl 8):2–17. [PubMed] [Google Scholar]
- Carter MM, Torres SM, Cook DL, Jr, McCash CL, Yu M, Griegos J, Walker VE, Walker DM. Relative mutagenic potencies of several nucleoside analogs, alone or in drug pairs, at the HPRT and TK loci of human TK6 lymphoblastoid cells. Environ Mol Mutagen. 2007;48:239–247. doi: 10.1002/em.20282. [DOI] [PubMed] [Google Scholar]
- Culy CR, Spencer CM. Amifostine: An update on its clinical status as a cytoprotectant in patients with cancer receiving chemotherapy or radiotherapy and its potential therapeutic application in myelodysplastic syndrome. Drugs. 2001;61:641–684. doi: 10.2165/00003495-200161050-00012. [DOI] [PubMed] [Google Scholar]
- Dagan T, Sable C, Bray J, Gerschenson M. Mitochondrial dysfunction and antiretroviral nucleoside analog toxicities: What is the evidence? Mitochondrion. 2002;1:397–412. doi: 10.1016/s1567-7249(02)00003-x. [DOI] [PubMed] [Google Scholar]
- Davidson DE, Grenan MM, Sweeney TR. Biological characteristics of some improved radioprotectors. In: Brady LW, editor. Radiation Sensitizers; Their Use in the Clinical Management of Cancer. New York: Masson; 1980. pp. 309–320. [Google Scholar]
- Davis DA, Yusa K, Gillim LA, Newcomb FM, Mitsuya H, Yarchoan R. Conserved cysteines of the human immunodeficiency virus type 1 protease are involved in regulation of polyprotein processing and viral maturation of immature virions. J Virol. 1999;73:1156–1164. doi: 10.1128/jvi.73.2.1156-1164.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar P, Olivero OA, Wade NA, Abrams EJ, Nesel CJ, Ness RB, Day RD, Day BW, Meng Q, O’Neill JP, Walker DM, Poirier MC, Walker VE, Bigbee WL. Genotoxicity assessed by the comet and GPA assays following in vitro exposure of human lymphoblastoid cells (H9) or perinatal exposure of mother-child pairs to AZT or AZT-3TC. Environ Mol Mutagen. 2007;48:330–343. doi: 10.1002/em.20285. [DOI] [PubMed] [Google Scholar]
- Foster C, Lyall H. HIV and mitochondrial toxicity in children. J Antimicrob Chemother. 2008;61:8–12. doi: 10.1093/jac/dkm411. [DOI] [PubMed] [Google Scholar]
- Freire R, d’Adda Di Fagagna F, Wu L, Pedrazzi G, Stagljar I, Hickson ID, Jackson SP. Cleavage of the Bloom’s syndrome gene product during apoptosis by caspase-3 results in an impaired interaction with topoisomerase III α. Nucleic Acids Res. 2001;29:3172–3180. doi: 10.1093/nar/29.15.3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grdina DJ, Nagy B, Hill CK, Wells RL, Peraino C. The radioprotector WR-1065 reduces radiation-induced mutations at the hypoxanthine-guanine phosphoribosyl transferase locus in V79 cells. Carcinogenesis. 1985;6:929–931. doi: 10.1093/carcin/6.6.929. [DOI] [PubMed] [Google Scholar]
- Grdina DJ, Nagy B, Hill CK, Sigdestad CP. Protection against radiation-induced mutagenesis in V79 cells by 2-[(aminopropy-l)amino] ethanethiol under conditions of acute hypoxia. Radiat Res. 1989a;117:251–258. [PubMed] [Google Scholar]
- Grdina DJ, Sigdestad CP, Dale PJ, Perrin JM. The effect of 2-[(aminopropyl)amino] ethanethiol on fission-neutron-induced DNA damage and repair. Br J Cancer. 1989b;59:17–21. doi: 10.1038/bjc.1989.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grdina DJ, Dale P, Weichselbaum R. Protection against AZT-induced mutagenesis at the HGPRT locus in a human cell line by WR-151326. Int J Radiat Oncol Biol Phys. 1992;22:813–815. doi: 10.1016/0360-3016(92)90530-u. [DOI] [PubMed] [Google Scholar]
- Grdina DJ, Shigematsu N, Dale P, Newton GL, Aguilera JA, Fahey RC. Thiol and disulfide metabolites of the radiation protector and potential chemopreventive agent WR-2721 are linked to both its anti-cytotoxic and anti-mutagenic mechanisms of action. Carcinogenesis. 1995;16:767–774. doi: 10.1093/carcin/16.4.767. [DOI] [PubMed] [Google Scholar]
- Grdina DJ, Hunter N, Kataoka Y, Murley JS, Milas L. Chemopreventive doses of amifostine confer no cytoprotection to tumor nodules growing in the lungs of mice treated with cyclophosphamide. Semin Oncol. 1999;26 (2 Suppl 7):22–27. [PubMed] [Google Scholar]
- Grdina DJ, Kataoka Y, Murley JS. Amifostine: Mechanisms of action underlying cytoprotection and chemoprevention. Drug Metabol Drug Interact. 2000;16:237–279. doi: 10.1515/dmdi.2000.16.4.237. [DOI] [PubMed] [Google Scholar]
- Grdina DJ, Murley JS, Kataoka Y. Radioprotectants: Current status and new directions. Oncology. 2002a;63 (Suppl 2):2–10. doi: 10.1159/000067146. [DOI] [PubMed] [Google Scholar]
- Grdina DJ, Murley JS, Kataoka Y, Calvin DP. Differential activation of nuclear transcription factor κB, gene expression, and proteins by amifostine’s free thiol in human microvascular endothelial and glioma cells. Semin Radiat Oncol. 2002b;1 (Suppl 1):103–111. doi: 10.1053/srao.2002.31383. [DOI] [PubMed] [Google Scholar]
- Grdina DJ, Murley JS, Kataoka Y, Epperly W. Relationships between cytoprotection and mutation prevention by WR-1065. Mil Med. 2002c;167 (Suppl 2):51–53. [PubMed] [Google Scholar]
- IARC. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Some Antiviral and Antineoplastic Drugs, and Other Pharmaceutical Agents. 76. Lyon: IARC Scientific Publications; 2000. General remarks: And the Monograph on Zidovudine; pp. 32–35.pp. 73–127. [Google Scholar]
- Kalebic T, Schein PS. Organic thiophosphate WR-151327 suppresses expression of HIV in chronically infected cells. AIDS Res Hum Retroviruses. 1994;10:727–733. doi: 10.1089/aid.1994.10.727. [DOI] [PubMed] [Google Scholar]
- Kataoka Y, Basic I, Perrin J, Grdina DJ. Antimutagenic effects of radioprotector WR-2721 against fission-spectrum neurons and 60Co γ-rays in mice. Int J Radiat Biol. 1992;61:387–392. doi: 10.1080/09553009214551081. [DOI] [PubMed] [Google Scholar]
- Kataoka Y, Perrin J, Hunter N, Milas L, Grdina DJ. Antimutagenic effects of amifostine: Clinical implications. Semin Oncol. 1996;23 (Suppl 8):53–57. [PubMed] [Google Scholar]
- Khodarev NN, Kataoka Y, Murley JS, Weichselbaum RR, Grdina DJ. Interaction of amifostine and ionizing radiation on transcriptional patterns of apoptotic genes expressed in human micro-vascular endothelial cells (HMEC) Int J Radiat Oncol Biol Phys. 2004;60:553–563. doi: 10.1016/j.ijrobp.2004.04.060. [DOI] [PubMed] [Google Scholar]
- Kohler JJ, Lewis W. A brief overview of mechanisms of mitochondrial toxicity from NRTIs. Environ Mol Mutagen. 2007;48:166–172. doi: 10.1002/em.20223. [DOI] [PubMed] [Google Scholar]
- Kondapi AK, Padmaja G, Satyanarayana N, Mukhopadyaya R, Reitz MS. A biochemical analysis of topoisomerase II α and β kinase activity found in HIV-1 infected cells and virus. Arch Biochem Biophys. 2005;441:41–55. doi: 10.1016/j.abb.2005.06.021. [DOI] [PubMed] [Google Scholar]
- Kouvaris JR, Kouloulias VE, Vlahos LJ. Amifostine: The first selective-target and broad-spectrum radioprotector. Oncologist. 2007;12:738–747. doi: 10.1634/theoncologist.12-6-738. [DOI] [PubMed] [Google Scholar]
- Liber HL, Thilly WG. Mutation assay at the thymidine kinase locus in diploid human lymphoblasts. Mutat Res. 1982;94:467–485. doi: 10.1016/0027-5107(82)90308-6. [DOI] [PubMed] [Google Scholar]
- Livesey JC, Rasey JS, Vertrees S, Freeman LM, Magee S, Nelson NJ, Chin L, Grunbaum Z, Krohn KA. In vitro metabolism of the phosphorothioate radioprotectors WR-2721 and WR-3689. Pharmacol Ther. 1988;39:215–217. doi: 10.1016/0163-7258(88)90064-2. [DOI] [PubMed] [Google Scholar]
- Meier T, Issels RD. Degradation of 2-(3-aminopropylamino)-ethanethiol (WR-1065) by Cu-dependent amine oxidases and influence on glutathione status of Chinese hamster ovary cells. Biochem Pharmacol. 1995;50:489–496. doi: 10.1016/0006-2952(95)00164-u. [DOI] [PubMed] [Google Scholar]
- Meng Q, Walker DM, Olivero OA, Shi X, Antiochos BB, Poirier MC, Walker VE. Zidovudine-didanosine coexposure potentiates DNA incorporation of zidovudine and mutagenesis in human cells. Proc Natl Acad Sci USA. 2000;97:12667–12671. doi: 10.1073/pnas.220203197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Q, Olivero OA, Fasco MJ, Bellisario R, Kaminsky LS, Pass KA, Wade NA, Abrams EJ, Nessel CJ, Ness RB, Bigbee WL, O’Neill JP, Walker DM, Poirier MC, Walker VE Protocol team. Plasma and cellular markers of 3′-azido-3′dideoxythymidine (AZT) metabolism as indicators of DNA damage in cord blood mononuclear cells from infants receiving prepartum NRTIs. Environ Mol Mutagen. 2007;48:307–321. doi: 10.1002/em.20298. [DOI] [PubMed] [Google Scholar]
- Mitchell JL, Rupert J, Leyser A, Judd GG. Mammalian cell poly-amine homeostasis is altered by the radioprotector WR1065. Biochem J. 1998;335 (Part 2):329–334. doi: 10.1042/bj3350329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murley JS, Constantinou A, Kamath NS, Grdina DJ. WR-1065, an active metabolite of the cytoprotector amifostine, affects phosphorylation of topoisomerase II α leading to changes in enzyme activity and cell cycle progression in CHO AA8 cells. Cell Prolif. 1997;30:283–294. doi: 10.1046/j.1365-2184.1997.00092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North S, El-Ghissassi F, Pluquet O, Verhaegh G, Hainaut P. The cytoprotective aminothiol WR-1065 activates p2waf-1 and down regulates cell cycle progression through a p53-dependent pathway. Oncogene. 2000;19:1206–1214. doi: 10.1038/sj.onc.1203413. [DOI] [PubMed] [Google Scholar]
- NTP. NTP toxicology and carcinogenesis studies of transplacental 3′-azido-3′-dioxythymidine (AZT) (CAS No. 30516-87-1) in Swiss (CD-1(R)) mice (in utero studies) Natl Toxicol Program Tech Rep Ser. 2006;522:1–184. [PubMed] [Google Scholar]
- Olivero OA, Anderson LM, Diwan BA, Haines DC, Harbaugh SW, Moskal TJ, Jones AB, Rice JM, Riggs CW, Logsdon D, Yuspa SH, Poirier MC. Transplacental effects of 3′-azido-2′,3′-dideoxythymidine (AZT): Tumorigenicity in mice and genotoxicity in mice and monkeys. J Natl Cancer Inst. 1997;89:1602–1608. doi: 10.1093/jnci/89.21.1602. [DOI] [PubMed] [Google Scholar]
- Olivero OA, Shearer GM, Chougnet CA, Kovacs AA, Landay AL, Baker R, Stek AM, Khoury MM, Proia LA, Kessler HA, Sha BE, Tarone RE, Poirier MC. Incorporation of zidovudine into leukocyte DNA from HIV-1-positive adults and pregnant women, and cord blood from infants exposed in utero. AIDS. 1999;13:919–925. doi: 10.1097/00002030-199905280-00007. [DOI] [PubMed] [Google Scholar]
- Pamujula S, Graves RA, Freeman T, Srinivasan V, Bostanian LA, Kishore V, Mandal TK. Oral delivery of spray dried PLGA/amifostine nanoparticles. J Pharm Pharmacol. 2004;56:1119–1125. doi: 10.1211/0022357044210. [DOI] [PubMed] [Google Scholar]
- Perno CF, Yarchoan R, Cooney DA, Hartman NR, Gartner S, Popovic M, Hao Z, Gerrard TL, Wilson YA, Johns DG, Broder S. Inhibition of human immunodeficiency virus (HIV-1/HTLV-IIIBa-L) replication in fresh and cultured human peripheral blood monocytes/macrophages by azidothymidine and related 2′,3′-dideoxynucleosides. J Exp Med. 1988;168:1111–1125. doi: 10.1084/jem.168.3.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips MD, Nascimbeni B, Tice RR, Shelby MD. Induction of micronuclei in mouse bone marrow cells: An evaluation of nucleoside analogues used in the treatment of AIDS. Environ Mol Mutagen. 1991;18:168–183. doi: 10.1002/em.2850180305. [DOI] [PubMed] [Google Scholar]
- Poirier MC, Olivero OA, Walker DM, Walker VE. Perinatal genotoxicity and carcinogenicity of anti-retroviral nucleoside analog drugs. Toxicol Appl Pharmacol. 2004;199:151–161. doi: 10.1016/j.taap.2003.11.034. [DOI] [PubMed] [Google Scholar]
- Sauerbrei A, Eichhor U, Scheibenzuber M, Wutzler P. Hexon denaturation of human adenoviruses by different groups of biocides. J Hosp Infect. 2007;65:264–270. doi: 10.1016/j.jhin.2006.10.017. [DOI] [PubMed] [Google Scholar]
- Srinivasan V, Pendergrass JA, Jr, Kumar KS, Landauer MR, Seed TM. Radioprotection, pharmacokinetic and behavioural studies in mouse implanted with biodegradable drug (amifostine) pellets. Int J Radiat Biol. 2002;78:535–543. doi: 10.1080/095530002317577358. [DOI] [PubMed] [Google Scholar]
- Sussman HE, Olivero OA, Meng Q, Pietras SM, Poirier MC, O’Neill JP, Finette BA, Bauer MJ, Walker VE. Genotoxicity of 3′-azido-3′-deoxythymidine in the human lymphoblastoid cell line, TK6: Relationships between DNA incorporation, mutant frequency, and spectrum of deletion mutations in HPRT. Mutat Res. 1999;429:249–259. doi: 10.1016/s0027-5107(99)00111-6. [DOI] [PubMed] [Google Scholar]
- The Big Picture Book of Viruses. Available at: http://www.virology.net/Big_Virology/BVFamilyGenome.html.
- Thorne C, Newell ML. Safety of agents used to prevent mother-to-child transmission of HIV: Is there any cause for concern? Drug Saf. 2007;30:203–213. doi: 10.2165/00002018-200730030-00004. [DOI] [PubMed] [Google Scholar]
- Torres SM, Walker DM, Carter MM, Cook DL, Jr, McCash CL, Cordova EM, Olivero OA, Poirier MC, Walker VE. Mutagenicity of zidovudine, lamivudine, and abacavir following in vitro exposure of human lymphoblastoid cells or in utero exposure of CD-1 mice to single agents or drug combinations. Environ Mol Mutagen. 2007;48:224–238. doi: 10.1002/em.20264. [DOI] [PubMed] [Google Scholar]
- Walker DM, Hardisty J, Rucker R, Funk K, Wolfe C, Seilkop SK, Walker VE. Transplacental carcinogenicity of 3-azido-3-deoxythymidine in B6C3F1 mice and F344 rats. Environ Mol Mutagen. 2007;48:283–298. doi: 10.1002/em.20297. [DOI] [PubMed] [Google Scholar]
- Walker VE, Poirier MC. Special issue on health risks of perinatal exposure to nucleoside reverse transcriptase inhibitors. Environ Mol Mutagen. 2007;48:159–165. doi: 10.1002/em.20296. [DOI] [PubMed] [Google Scholar]
- Watts DH. Treating HIV during pregnancy: An update on safety issues. Drug Saf. 2006;29:467–490. doi: 10.2165/00002018-200629060-00002. [DOI] [PubMed] [Google Scholar]
- Witt KL, Cunningham CK, Patterson KB, Kissling GE, Dertinger SD, Livingston E, Bishop JB. Elevated frequencies of micro-nucleated erythrocytes in infants exposed to zidovudine in utero and postpartum to prevent mother-to-child transmission of HIV. Environ Mol Mutagen. 2007;48:322–329. doi: 10.1002/em.20266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wutzler P, Thust R. Genetic risks of antiviral nucleoside analogues—A survey. Antiviral Res. 2001;49:55–74. doi: 10.1016/s0166-3542(00)00139-x. [DOI] [PubMed] [Google Scholar]