

Abstract
Going beyond single gene function to cut deeper into gene regulatory networks requires multiple mutations combined in a single animal. Such analysis of two or more genes needs to be complemented with in situ hybridization of other genes, or immunohistochemistry of their proteins, both in whole mounted developing organs or sections for detailed resolution of the cellular and tissue expression alterations. Combining multiple gene alterations requires the use of cre or flipase to conditionally delete genes and avoid embryonic lethality. Required breeding schemes dramatically enhance effort and cost proportional to the number of genes mutated, with an outcome of very few animals with the full repertoire of genetic modifications desired. Amortizing the vast amount of effort and time to obtain these few precious specimens that are carrying multiple mutations necessitates tissue optimization. Moreover, investigating a single animal with multiple techniques makes it easier to correlate gene deletion defects with expression profiles. We have developed a technique to obtain a more thorough analysis of a given animal; with the ability to analyze several different histologically recognizable structures as well as gene and protein expression all from the same specimen in both whole mounted organs and sections. Although mice have been utilized to demonstrate the effectiveness of this technique it can be applied to a wide array of animals. To do this we combine lipophilic dye tracing, whole mount in situ hybridization, immunohistochemistry, and histology to extract the maximal possible amount of data.
Keywords: Neuroscience, Issue 49, lipophilic dye, in situ hybridization, immunohistochemistry, histology, neuronal tracing
Protocol
1. Lipophilic Dye Injection
Tissue Preparation:
All tissue dissections and manipulations are carried out in animals fixed in 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (pH 7.4) by transcardial perfusion using peristaltic pumps with appropriately sized needles. Tissue can be stored in 4% PFA in the refrigerator for up to 6 months. All preparations and manipulations are conducted in 0.4 % or higher PFA until mounting with glycerol for imaging. This amount of PFA effectively eliminates RNases and preserves the RNA for in situ hybridization.
Dye Storage:
All dyes should be stored in a dark closed compartment to minimize air circulation and exposure to bright daylight, as they are photosensitive. To avoid cross contamination, a separate set of instruments should be used to handle each dye.
First, an application site for the dye must be chosen and extraneous tissue extracted in order to visually confirm the chosen site, and access for placement of the dye. Choosing the application site is the most important step in this process. Choosing a good site to label the neuronal population under consideration and not extraneous structures can be challenging. The accuracy of the placement into a given tract under visual control in the dissecting scope requires a certain level of understanding of the neuroanatomy in question. Dye can either be placed centrally or peripherally to label peripheral or central projections respectively as needed for a given project (i.e. brain, peripheral nerve, ear, eye). Lipophic dyes will diffuse in all processes belonging to a given neuron, both retrogradely and anterogradely, filling a given neuron or all neurons projecting into a given track.
NeuroVue dye from MTTI comes pre-loaded onto filter strips for easy application. Dye can also be dissolved in 100% DMSO (work under the hood) and thin hair can be soaked in this solution and subsequently dried for smaller applications. The preloaded filter paper dye is cut into appropriately sized triangular pieces with microscissors. The size of the dye will depend on the size of the specimen, size of the structure being labeled, and number of colors being used simultaneously. Be sure to cut as small of a piece as possible to avoid labeling other structures. After using microsissors for cutting and forceps for handling the dye agitate the instruments in alcohol to remove any dye residue, then air dry completely, or dab with paper. This is to avoid contaminating the specimen with residual dye on instruments that may label unwanted structures.
For insertion into soft tissue such as brain the filter can be directly inserted using a point of the filter triangle to pierce the tissue. For more rigid tissues make an incision to allow easier insertion of dye. Do not use a dissecting needle to push filter into tissue. This can cause inaccurate placement and disruption of the tissue. Insert alternate wavelengths of NeuroVue dye as needed for labeling of additional neuronal populations.
After inserting the dye, and verifying its position, specimens are placed in a securely closed vial with 4% PFA and incubated at 36°C in the dark for 2-14 days depending on age and diffusion distance to be covered (approximately 2 mm per day at 36°C).
A dissecting scope with epiflouresence can be used to assess if the dye has diffused to the desired location, and the specimen can be placed back in the incubator if the diffusion is deemed to be insufficient. Using a normal dissecting scope without epifluorescene to detect diffusion will underestimate the true length the dye has diffused. Also, if dye concentration is high enough to asses visually it may cause absorption quenching when using emitted fluorescence.
The desired tissue of interest is dissected out and whole mounted onto a slide with glycerol and coverslipped for imaging with a confocal microscope. Confocal settings are as described in 1, 2. If tissue size inhibits whole mounting on a slide, serial sectioning is needed (see below). More details on preparation of tissue for imaging and dye spectral properties are available at: http://www.mtarget.com/mtti/documents/NeuroVuebrochuremar2010.pdf
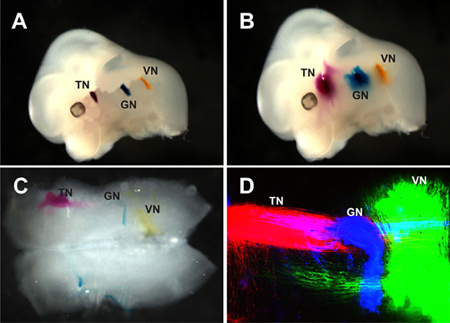 Figure 1. Schematic overview of experiment. The ear is exposed to allow for visualization of all structures. Lipophilic dye is then injected and allowed to diffuse. After diffusion distinct neuronal populations can be seen labeled by the different dyes. Next, in situ hybridization (Sox2) is performed on the ear and labels mRNA expression. Immunohistochemistry is then carried out (Myo7, Tubulin) to visualize protein expression. Followed by histological sections in which both in situ hybridization and immunohistochemistry can be visualized.
Figure 1. Schematic overview of experiment. The ear is exposed to allow for visualization of all structures. Lipophilic dye is then injected and allowed to diffuse. After diffusion distinct neuronal populations can be seen labeled by the different dyes. Next, in situ hybridization (Sox2) is performed on the ear and labels mRNA expression. Immunohistochemistry is then carried out (Myo7, Tubulin) to visualize protein expression. Followed by histological sections in which both in situ hybridization and immunohistochemistry can be visualized.
2. in Situ Hybridization
In case you want to start with in situ hybridization (after which tracing with lipophilic dyes will be impossible), refer to tissue preparation in Part 1. Each wash, unless indicated, is carried out with 2 mL of solution at room temperature on an inverter/mixer. Be sure to work in a clean, RNase free environment.
- Dehydrate and then rehydrate samples through a graded methanol series.
- 100% 1 hour to overnight @ 4°C (optional: store samples at -20°C in 100% methanol)
- 75% x 5 min. @ 4°C
- 50% x 5 min. @ 4°C
- 25% x 15 min. @ 4°C (or until tissue sinks)
- Transfer samples to a 2 mL eppendorf tube (RNase free).
- Wash three times in PBS (1x PBS) and turn hybridization oven on for future use (60°C).
- PBS x 5 min.
- PBS x 5 min.
- PBS x 5 min.
- Digest tissue with 2 μL of 20mg/ mL stock Proteinase K (Ambion Cat# AM2546) in 2.0 mL fresh PBS. Actual PK digestion time depends upon the age of the embryo. The following is a rough estimate of times. Digestion should be closely monitored as deproteination is a critical step. Under-digestion will result in poor probe penetration while over-digestion will result in a loss of structural integrity. A good indicator of digestion is a change in the tissue from opaque to almost clear.
Table 1. Proteinase K digestion time on mouse tissue. AGE (embryonic day) TIME (minutes) E8.5 6 E9.5 10 E10.5 13 E11.5 15 E12.5 17 E14.5 18 E16.5+ 20+ Stop digestion by incubating samples in 4% PFA for 5 min.
- Wash in PBS.
- PBS x 1 min.
- PBS x 5 min.
- PBS x 5 min.
- PBS x 5 min.
Discard PBS paying special attention to eliminate as much as possible.
- Prehybridize samples: Incubate at 60°C on inverter in 1.8 mL hybridization mix for at least 1 hour. Set IsoTemp heat block to 85°C for future use.
- As one hour nears, denature salmon sperm DNA (Invitrogen Cat No. 15632-011) by incubation at 85°C for 10 min. Set on ice until use.
After at least 1 hour of prehybridization, add 200 μL denatured ssDNA and approximately 100ng of DIG-labeled riboprobe to each sample. Hybridize overnight at 60°C in hybridization oven.
- Replace hybridization mix with 2 mL 2X SSC. Set IsoTemp heat blocks to 37°C and 70°C for future use.
- Wash with 2X SSC x 10 min. @ 60°C in hybridization oven.
- Wash with 2X SSC x 10 min. @ 60°C in hybridization oven.
- Wash with 2X SSC x 10 min. @ 60°C in hybridization oven.
- Wash with 2X SSC x 60 min. @ 70°C in IsoTemp heat block.
Wash with PBS x 5 min. Thaw RNase A enzyme on ice for future use.
Replace PBS and add 1.0 μL of RNase A Enzyme (Fermentas EN0531 10mg/ mL) x 60 min. @ 37° C in IsoTemp heat block (a 90 min. incubation period is preferred if the probe is highly concentrated).
- Discard PBS/RNase A and wash 4 times.
- 1X wash solution x 10 min.
- 1X wash solution x 10 min.
- 1X wash solution x 10 min.
- 1X wash solution x 60 min. @ 70°C in IsoTemp heat block.
- Incubate in 1X Block Buffer for 1 hour.
- At this time, prepare 2.0 mL of block buffer and 1 μL (1:2000) Anti-Digoxigenin antibody per sample. Invert briefly and let sit at 4°C until use.
After 1 hour discard block buffer and add 2.0 mL pre-absorbed block buffer + antibody to samples.
Incubate overnight.
Discard block solution.
- Wash with 1X wash buffer.
- 1X wash buffer x 5 min.
- 1X wash buffer x 5 min.
- 1X wash buffer for 1 hour x 5-6 changes.
Wash with 1X wash buffer overnight.
Rinse with 1X detection buffer for 10 min.
Transfer samples in detection buffer to well plate.
- Remove buffer and detect with BM Purple (Roche 11442074001) until desired signal strength is obtained (with longer probes this may mean overnight). BM purple is light sensitive cover with foil or a box.
- Mix BM Purple well before use.
- Make sure that samples are free floating and not stuck to wells.
- Check signal after 1 hour.
Rinse samples with 1X detection buffer in wells x 5 min.
Image or store samples in 4% PFA at 4°C.
*Samples can be imaged at this stage and/or after the immunohistochemistry step is added (Part 3 below).
Solutions:
Nuclease free water: Mix 1 mL DEPC (Sigma) with 1L sterile ultrapure water. Autoclave.
1X PBS: Mix 1 PBS packet (Sigma) in 1L nuclease free water and filter sterilize.
1X Wash solution: Mix 450 mL nuclease free water + 50 mL 10x wash solution and filter sterilize.
Hybridization Mix: 50% Formamide by volume (25 mL); 50% 2X SSC by volume (25 mL); 6% dextran sulfate by mass (3g).
1X Detection Buffer: Mix 50 mL 10X detection buffer with 450 mL nuclease free water and filter sterilize.
1X Block Buffer: 5 mL 10X Maleic Acid Buffer (Roche buffer set); 40 mL nuclease free water; 5 mL 10X Block Buffer.
2X SSC: Mix 50 mL 20X SSC with 450 mL nuclease free water and filter sterilize.
Graded methanol: Dilute 100% methanol with nuclease free water to 75%, 50%, and 25% concentrations.
3. Immunohistochemistry
Steps 1 and 2 can be omitted if Part 2 was completed. In that case, make sure that all glycerol or other mounting medium is washed off. Note that not all antibodies will still be able to detect their epitope after proteinase K digestion. We have a short list of antibodies that can be used in combination with in situ hybridization in mammalian tissue as they retain their specificity (see Table 2).
Graded ethanol series to rid tissue of lipophilic dyes (if not completed to effect in Part 2): 50% ethanol 5min., 70% ethanol 5 min., 95-100% ethanol overnight or as needed until desired effect, 70% ethanol 5 min., 50% ethanol 5min.
Rehydrate by incrementally adding PBS in 5-10 minutes.
Block tissue for 1 hr in 2.5% normal goat serum (NGS) and 0.5% Triton X-100 @ RT on shaker. (1:1 5% NGS:1.0% TritonX-100 in 1x PBS)
Incubate with primary antibody(s) diluted in block solution 48-72 hrs @ 4°C on shaker.
Wash with PBS for 1hr x 3 changes.
Block as in step 3.
Incubate with secondary antibody(s) diluted in block solution for 12-24hrs @ 4°C on shaker (Alexa Fluor® dyes from Invitrogen were used).
Wash as in step 5. (Option: counter stain with Hoechst nuclear stain as first wash dilute 10mg/ mL stock 1:2000 in PBS. Follow with 1 hr. washes in PBS x 2-3 changes @ RT on shaker)
Imaging: Specimens can be imaged at this step with both transmission and epifluorescent microscopy, preferentially using a confocal imaging system for added resolution. To do this we routinely whole mount ears in glycerol using coverslips as spacers to avoid over compression of the tissue. In addition or instead of this whole mount imaging, specimens can be embedded in epoxy resin and serially sectioned for added histological details. To benefit from the combined whole mount/section analysis, avoid bleaching the fluorescent signal in the confocal imaging step.
Solutions
Ethanol solutions: Dilute 100% ethanol in distilled water to final concentrations of 50% and 70%.
Block Solution: 1:1 dilution 5% NGS:1.0% TritonX-100 in 1x PBS (final working concentrations: 2.5% NGS; 0.5% TritonX-100).
1X PBS: Mix 1 PBS packet (Sigma) in 1L distilled water.
5% NGS: Thaw 2.5 mL NGS and mix with 47.5 mL 1x PBS. Store at 4°C.
1.0% TritonX-100: Mix 49.5 mL 1x PBS with 0.5 mL TritonX-100 (leave on shaker @ RT to mix for 1 hour). Store at 4°C.
Hoechst stain stock solution: Dissolve 10mg Hoechst dye per milliliter of 1x PBS.
| Table 2. Antibodies that work with proteinase K digested tissue. | ||
| Cat # | Item | Vender |
| 25-6790 | Anti-Myosin-VIIA Polyclonal | Proteus Biosciences |
| 25-6791 | Anti-Myosin-VI Polyclonal | Proteus Biosciences |
| 9661 | Cleaved Caspase-3 Polyclonal | Cell Signaling |
| T7451 | Anti-Acetylated Tubulin Monoclonal | Sigma |
| PRB-238C | Prox1 polyclonal | Covance |
4. Histology
Histology can be introduced after Part 1 to increase resolution of the lipophilic dye tracing in thick whole mounts such as juvenile brains or after completion of Parts 2 or 3. For serial brain sections we recommend the Compresstome VF-700 microtome (Precisionary Instruments Inc., Greenville, North Carolina) to obtain uniform section thickness. Frozen sections are less optimal due to greater dye leakage during the sectioning process 3. Preparation of serial sections and mounting in glycerol 4 is the preferred method of tissue preparation when whole mounts or surface mounts do not allow adequate visualization of tissue regions of interest. Tissue can also subsequently be embedded in epoxy resin and cut at 1-2 μm thickness using glass or diamond knifes for TEM. When using this technique most immunofluorescence will remain and is even more stable after plastic embedding, however, all lipophilic tracing will be entirely lost at this stage as they dissolve in alcohol and epoxy resin. Take precaution as samples will be light sensitive until they are embedded.
For Compresstome VF-700 microtome sectioning, follow the recommendations of Precisionary Instruments Inc.
Epoxy Embedding/Sectioning:
Fixation: 2.5% gluteraldehyde/1% PFA in 0.1M phosphate buffer (pH 7.4) 1hr to overnight.
In a glass sample vial dehydrate tissue: 30% ethanol (5min.), 50% ethanol (5min.), 70% ethanol (5min. to overnight), 95% ethanol (5x 10min. each), and absolute ethanol (5x 10min. each).
Transitional Solvent: 1:1 absolute ethanol:propylene oxide (PO) for 5 min.
Solvent: PO only 5x 10min. each.
- Infiltration with resin:
- 1:1 PO:resin overnight on shaker.
- Pour solution with sample(s) into flat embedding mold and leave on counter 4-6 hrs to evaporate PO. Alternatively, remove cap of vial and leave on shaker for 4 hrs (works well with larger tissue).
- Transfer samples to 100% resin for 4 hrs.
Embed in resin in final mold with label. Polymerize by incubating at 60°C for 24-48 hours.
Cut block with a razor blade as needed to minimize extraneous resin. Cut 1-2 um serial sections on an Ultramicrotome (an Ultracut E Reichert-Jung was used). Mount and image using epifluorescent microscopy. Optional: Stain a portion of the sections with histological stains (i.e. Stevenel's blue) if desired (realize immunofluorescence will not be sustained in these sections).
Solutions
Ethanol solutions: Dilute 100% ethanol in distilled water to 30%, 50%, 70%, and 95% concentrations.
Fixation solution: Mix 2.5 mL 50% Gluteraldehyde, 5 mL 10% Paraformaldehyde, and 42.5 mL 0.1M phosphate buffer pH 7.4 (final concentrations: 2.5% gluteraldehyde; 4% PFA). Store at 4°C.
Epoxy Resin: Make according to instructions supplied with Poly/Bed 812 Embedding Media/DMP-30 Kit (Polysciences, Inc. #08792-1). Store at -20°C.
5. Representative Results
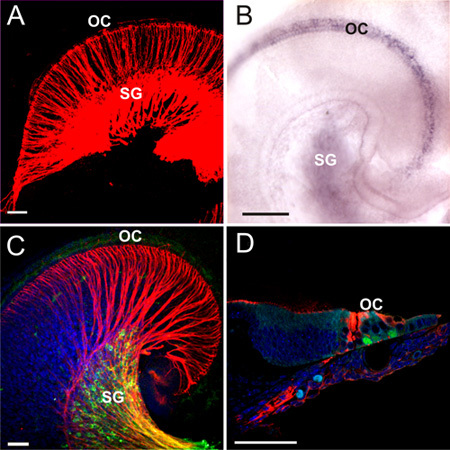 Figure 2. Lipophilic dye placement and imaging in a mouse embryo. Three different wavelength lipophilic dyes were injected and incubated to allow the visualization of the trigeminal nerve (TN), glossopharyngeal nerve (GN), and vagus nerve (VN) central projections. A) Lipophilic dye placement into the peripheral portions of three cranial nerves to allow diffusion to the brainstem for visualization of their central processes. The TN is labled with NeuroVue Red, GN: NeurVue Maroon, and VN: NeuroVue Jade. B) Same mouse as in (A) after incubation. Some diffusion can be seen with brightfield microscopy. C) Same mouse as in (A and B). The hindbrain has been dissected and flat mounted. Some labeling of the central processes of the three nerves labeled can be seen with brightfield microscopy. D) Confocal image of (C). With use of the confocal specific neurons can be seen, and use of three different dyes allows for the assessment of the distribution of each population in relation to the others. Dyes were imaged sequentially. NeuroVue Jade was imaged at an excitation of 488 nm and emission 500-550 nm. NeuroVue Red was imaged at an excitation of 535 nm and emission 550-600 nm. NeuroVue Maroon was imaged at an excitation of 643 nm and emission at 650-700 nm.
Figure 2. Lipophilic dye placement and imaging in a mouse embryo. Three different wavelength lipophilic dyes were injected and incubated to allow the visualization of the trigeminal nerve (TN), glossopharyngeal nerve (GN), and vagus nerve (VN) central projections. A) Lipophilic dye placement into the peripheral portions of three cranial nerves to allow diffusion to the brainstem for visualization of their central processes. The TN is labled with NeuroVue Red, GN: NeurVue Maroon, and VN: NeuroVue Jade. B) Same mouse as in (A) after incubation. Some diffusion can be seen with brightfield microscopy. C) Same mouse as in (A and B). The hindbrain has been dissected and flat mounted. Some labeling of the central processes of the three nerves labeled can be seen with brightfield microscopy. D) Confocal image of (C). With use of the confocal specific neurons can be seen, and use of three different dyes allows for the assessment of the distribution of each population in relation to the others. Dyes were imaged sequentially. NeuroVue Jade was imaged at an excitation of 488 nm and emission 500-550 nm. NeuroVue Red was imaged at an excitation of 535 nm and emission 550-600 nm. NeuroVue Maroon was imaged at an excitation of 643 nm and emission at 650-700 nm.
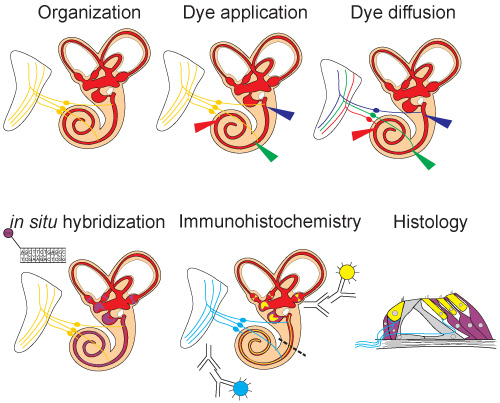 Figure 3. Schematic overview of experiment. The ear is exposed to allow for visualization of all structures. Lipophilic dye is then injected and allowed to diffuse. After diffusion distinct neuronal populations can be seen labled by the different dyes. Next, in situ hybridization (sox2) is performed on the ear and labels mRNA expression. Immunohistochemistry is then carried out (Myo7, Tubulin) to visualize protein expression. Followed by histological sections in which both in situ hybridization and immunohistochemistry can be visualized.
Figure 3. Schematic overview of experiment. The ear is exposed to allow for visualization of all structures. Lipophilic dye is then injected and allowed to diffuse. After diffusion distinct neuronal populations can be seen labled by the different dyes. Next, in situ hybridization (sox2) is performed on the ear and labels mRNA expression. Immunohistochemistry is then carried out (Myo7, Tubulin) to visualize protein expression. Followed by histological sections in which both in situ hybridization and immunohistochemistry can be visualized.
Discussion
In this video, we demonstrate a method to combine four techniques in order to maximize data collection and cohesiveness by correlating data within a given animal (figure 3). This approach will reduce the amount of breeding and thus time needed to obtain publishable data while improving information about co-localization and correlative effects of mutants. Although embryonic mice were utilized in this video, adult mouse as well as other animals such as chicken, xenopus, and zebrafish can be analyzed using these techniques as well. When using other species proteinase k digestion may need to be altered. Here we use different fluorescing NeuroVue dyes to specifically label up to three neuronal populations of interest. The benefit of these dyes is that they can be used in mutant mice, which may be problematic when relying solely on immunohistochemistry that may or may not be affected by the mutations to be analyzed, and they label neurons retrogradely and anterogradely 5. A recent study has also increased the number of neuron populations that can be labeled simultaneously to six 2. After, the lipophilic dye tracing the same tissue of interest can then be analyzed using in situ hybridization, for analysis of gene transcription and can be subsequently analyzed using immunohistochemistry for protein distribution. The latter analysis can be supplemented by detailed histology which can add to the phenotype characterization 6. This multifactorial analysis has the potential to gain new insights through correlative analysis within the same animal that may otherwise be improbable or at least in need of more extensive protocols and mouse breeding.
Disclosures
Mouse tissue was collected in accordance with the guidelines and regulations set forth by the University of Iowa Institutional Animal Care and Use Committee (ACURF 0804066).
Acknowledgments
Confocal images were obtained at the University of Iowa Carver Center for Imaging. Funding was provided by an SBIR grant (MH079805) to B.F. Additional support for breeding the mice for this project was provided by NIDCD (5R01DC00559007) to B.F.
References
- Jensen-Smith H, Gray B, Muirhead K, Ohlsson-Wilhelm B, Fritzsch B. Long-distance three-color neuronal tracing in fixed tissue using NeuroVue dyes. Immunol Invest. 2007;36:763–789. doi: 10.1080/08820130701706711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonniges J. Photo- and bio-physical characterization of novel violet and near-infrared lipophilic fluorophores for neuronal tracing. Journal of Microscopy. 2010. [DOI] [PubMed]
- Bartheld CSvon, Cunningham DE, Rubel EW. Neuronal tracing with DiI: decalcification, cryosectioning, and photoconversion for light and electron microscopic analysis. J Histochem Cytochem. 1990;38:725–733. doi: 10.1177/38.5.2185313. [DOI] [PubMed] [Google Scholar]
- Gurung B, Fritzsch B. Time course of embryonic midbrain thalamic and thalamic auditory connection development in mice as revealed by carbocyannine dye injection. J Comp Neurol. 2004;479:309–327. doi: 10.1002/cne.20328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzsch B, Nichols DH. DiI reveals a prenatal arrival of efferents at the differentiating otocyst of mice. Hear Res. 1993;65:51–60. doi: 10.1016/0378-5955(93)90200-k. [DOI] [PubMed] [Google Scholar]
- Jahan I, Kersigo J, Pan N, Fritzsch B. Neurod1 suppresses hair cell differentiation in ear ganglia and regulates hair cell subtype development in the cochlea. PloS ONE. 2010;5:e11661–e11661. doi: 10.1371/journal.pone.0011661. [DOI] [PMC free article] [PubMed] [Google Scholar]