

Abstract
The developing Drosophila melanogaster embryo undergoes a number of cell shape changes that are highly amenable to live confocal imaging. Cell shape changes in the fly are analogous to those in higher organisms, and they drive tissue morphogenesis. So, in many cases, their study has direct implications for understanding human disease (Table 1)1-5. On the sub-cellular scale, these cell shape changes are the product of activities ranging from gene expression to signal transduction, cell polarity, cytoskeletal remodeling and membrane trafficking. Thus, the Drosophila embryo provides not only the context to evaluate cell shape changes as they relate to tissue morphogenesis, but also offers a completely physiological environment to study the sub-cellular activities that shape cells.
The protocol described here is designed to image a specific cell shape change called cellularization. Cellularization is a process of dramatic plasma membrane growth, and it ultimately converts the syncytial embryo into the cellular blastoderm. That is, at interphase of mitotic cycle 14, the plasma membrane simultaneously invaginates around each of ~6000 cortically anchored nuclei to generate a sheet of primary epithelial cells. Counter to previous suggestions, cellularization is not driven by Myosin-2 contractility6, but is instead fueled largely by exocytosis of membrane from internal stores7. Thus, cellularization is an excellent system for studying membrane trafficking during cell shape changes that require plasma membrane invagination or expansion, such as cytokinesis or transverse-tubule (T-tubule) morphogenesis in muscle.
Note that this protocol is easily applied to the imaging of other cell shape changes in the fly embryo, and only requires slight adaptations such as changing the stage of embryo collection, or using "embryo glue" to mount the embryo in a specific orientation (Table 1)8-19. In all cases, the workflow is basically the same (Figure 1). Standard methods for cloning and Drosophila transgenesis are used to prepare stable fly stocks that express a protein of interest, fused to Green Fluorescent Protein (GFP) or its variants, and these flies provide a renewable source of embryos. Alternatively, fluorescent proteins/probes are directly introduced into fly embryos via straightforward micro-injection techniques9-10. Then, depending on the developmental event and cell shape change to be imaged, embryos are collected and staged by morphology on a dissecting microscope, and finally positioned and mounted for time-lapse imaging on a confocal microscope.
Protocol
1. Assemble Embryo Collection Cups
Cut the bottom off of a 100 mL Tri-corn beaker with a razor, making the edge as smooth as possible. The cups are easier to handle if you also trim the three corners off of the top, though this is not absolutely necessary.
Cut a square of wire mesh (6 cm x 6 cm). On a pre-warmed hot plate, inside of a fume hood, layer the square of wire mesh on top of a piece of heavy-duty aluminum foil. Push the cut bottom edge of the cup firmly onto the hot mesh. Wait for a few seconds, and lift the cup with the mesh now attached. If the foil also sticks, just peel it off.
Cool the cup overnight. Remove excess mesh with scissors, and sand off any sharp edges with fine-grain sandpaper.
2. Make Apple Juice Agar Plates
In a 6L flask, combine 100 g BD Bacto Agar and 3L distilled water. Autoclave the agar for 30 minutes on the slow exhaust setting.
In a 2L flask with a stir bar, combine 100 g sucrose, 1L apple juice, and 6 g p-Hydroxybenzoic acid. Heat the solution to boiling while stirring on a hot plate. Do not boil for more than 2 minutes. Allow the apple juice mixture to cool, and then add with stir bar to the agar. Mix completely.
Allow the combined solution to cool in a 60°C water bath before pouring into 60x15 mm petri dishes. Alternatively, a peristaltic pump can be used to dispense the agar. Allow the plates to cool to room temperature for at least 4 hours. Stack in Rubbermaid containers with a layer of wet paper towels and store at 4°C.
Notes:
In the fall or winter, it may be necessary to add an additional 100 mL of water to the agar.
The brand of petri dishes is important! Only the BD Falcon dishes fit the Tri-corn beakers. Specific ordering information can be found in the Materials table below.
3. Add GFP Flies to the Embryo Collection Cups
Expand GFP stocks according to your collection needs by setting up bottles of flies approximately two weeks prior to imaging. One bottle of flies is usually more than sufficient to fill one embryo collection cup with a minimum of 50 females and 30 males. For best laying, flies should be newly eclosed (i.e. less than 5 days post-hatching).
Make a yeast paste by filling a small cup with roughly equal parts Red Star active dry yeast and distilled water, and stir until the yeast is dissolved. The paste should approximate the consistency of wet peanut butter. More yeast or water can be added to alter the consistency. Yeast paste can be used for several days, and should be stored, covered, at 4°C.
Once the yeast paste is ready, remove apple juice plates from 4°C storage. Streak the apple juice plates with the yeast paste, and allow them to warm to 22-25°C, which will encourage egg laying.
Fold a circular filter paper in half, then in half again. Trim to 8-9 cm diameter, and make accordion folds in the quarter circle. Unfold the circle, invert it, and insert it in a collection cup until it touches the mesh. Make sure that the paper is secure in the cup so that it does not fall and crush the flies. The filter paper is optional, but does provide a welcoming environment for mating, and maintains humidity in the cup.
Label a cup with stock name and date. Transfer the flies from the bottle to the collection cup by gently shaking the inverted bottle over the cup, and immediately cover the cup with a prepared apple juice plate. Secure the plate to the cup with a rubber band. Set cups mesh side up in an area with direct light. Make sure that no shadows fall on the cup, as flies will not lay well in the dark.
Change the apple juice plate as needed. Two-hour collections, at room temperature, work well for imaging cellularization.
4. Prepare a Mounting Chamber
To prepare a mounting chamber for the embryos, cut a piece of double-sided tape 2-3 cm long and place it on a slide, aligning the long axis of the tape and slide. Cut a second 2-3 cm long piece of double-sided tape and layer it on top of the other piece of tape, making sure their edges are aligned.
Using a razor blade, make two cuts approximately 3 mm apart in the center of the tape and perpendicular to the long axis of the slide. Remove the tape between the cuts to make a channel. Pipet a drop of Halocarbon 27 Oil into the channel. This channel is where the embryos will be mounted.
Notes:
Only the ½ inch Scotch double-sided tape is the right thickness to accommodate the embryos.
Halocarbon 27 Oil is oxygen permeable, and so allows oxygen exchange, while preventing embryo dehydration. Due to its refractive index, Halocarbon 27 Oil is also ideal for staging and imaging embryos.
5. Dechorionate Embryos
To collect and dechorionate embryos, pour enough 50% bleach on to the apple juice agar plate to fully submerge the entire surface. Using a dissecting microscope, such as the Zeiss Discovery V8 with transmitted light, watch for the embryos to release from their chorion. As soon as the chorion loosens and releases from a few embryos (30-60 seconds), pour bleach and embryos into a cell strainer.
Wash embryos in the strainer immediately and vigorously with distilled water from a squirt bottle. Dab strainer on paper towels. If any pink is seen on the towel after dabbing, continue washing the embryos with water.
Use a moist paintbrush to transfer 10-50 embryos to a clean apple juice plate (with no yeast paste). Wick away water with the torn edge of a paper towel, and immediately cover the embryos with a small amount of Halocarbon 27 Oil.
Notes:
Do not use Clorox bleach. Instead, use a brand such as Austin's A-1 Commercial Bleach, which is <6% sodium hypochlorite.
Over-bleaching or insufficient rinsing will result in mushy embryos with irregular cellularization.
6. Stage and Mount Embryos
Using the dissecting microscope with transmitted light, follow the morphological guidelines of Bownes20 to stage embryos on the plate. Using forceps, transfer 5 mitotic cycle 11 or 12 embryos, corresponding to Bownes stage 4, to the channel of the mounting chamber. Arrange embryos in a line, with dorsal and ventral sides visible and lateral side down. Embryos are easily arranged in this orientation in oil alone. Do not crowd embryos together as they will deprive their neighbors of oxygen once the cover slip is applied below. (Leave approximately half of an embryo length between them).
Lay one edge of a 25x25 mm cover slip on the double-sided tape at one side of the channel. Drop the cover slip to cover the channel. If air is caught underneath, apply a small amount of Halocarbon 27 Oil at the edge of the cover slip. Capillary action will pull the oil into the channel and push out the air.
Notes:
Another excellent resource for staging embryos is the following: Campos-Ortega, J. A. and Hartenstein, V. (1985). The Embryonic development of Drosophila melanogaster. Springer-Verlag, Berlin. While this book is no longer published, used copies are regularly available on Amazon.com.
Always use the Halocarbon 27 Oil sparingly. This will make the mounting easier. It will also prevent accidental oozing of the oil onto the microscope objective in the following imaging steps.
7. Image Embryos
How you proceed here will of course be dictated by the cell shape change that you are imaging, and the question that you are addressing. For any shape change, place your mounting chamber on either an upright or inverted microscope, find your embryos by transmitted light, and then switch to confocal imaging to focus on a region of interest. Adhere to best practices for confocal microscopy, such as those discussed at Nikon's MicroscopyU (http://www.microscopyu.com/articles/confocal). Be particularly rigorous in exposing your living embryos to as little laser power as possible, to prevent photobleaching and phototoxicity.
For cellularization, we image on a Zeiss 710 laser scanning confocal microscope, using a C-Apochromat 40x/1.2W Corr objective. This microscope is housed in a temperature-controlled room, with temperatures ranging from 18-20°C. While temperature control is not absolutely necessary, it is better for the microscope and makes the imaging more consistent. We regularly image in a single plane, near the middle of the embryo, to follow the plasma membrane invaginations in cross-section (Figure 2). For simply following invagination dynamics, 30-60 second intervals are sufficient for time-lapse imaging, and embryos should withstand imaging for >90 minutes with no obvious signs of oxygen deprivation.
After acquiring the images, they can be analyzed using any number of image analysis packages, including freeware from the National Institutes of Health, called ImageJ (http://rsbweb.nih.gov/ij). The data can then be presented as movies (Movie 1), sequences (Figure 2), or kymographs21.
Note:
The water immersion C-Apochromat 40x/1.2W Corr objective is well-suited for imaging near the embryo mid-section due to its high aperture, which limits aberrations for deep tissue imaging in aqueous samples, and gives high resolution and high fluorescent signal. In addition, this objective has a very long working distance (220 μm). However, other water or glycerine immersion objectives will perform well, particularly if the cell shape change of interest can be imaged near the embryo surface.
8. Alternative Method: Mount Embryos with "embryo-glue"
To image a cell shape change other than cellularization, it may be necessary to mount embryos in a specific orientation that is not easily maintained in oil alone. To do so, use an alternate mounting method to the one described previously. Start by making "embryo-glue" by combining 20 cm of double-sided tape with 250 μL of heptane in a scintillation vial. Place the scintillation vial on a nutator or rotating platform, and mix overnight.
Dip a yellow pipette tip into the embryo glue, and then trace the tip along a slide, leaving a trail of glue. While the heptane is evaporating, align your staged embryos in the required orientation on a block of agar. (Remember that the embryo surface to be imaged should be facing the agar. For example, to image cell shape change during ventral furrow formation, mount embryos with their ventral side facing the agar.)
Invert the slide with glue, and gently press the trail of glue against the embryos on the agar block. Now invert the slide again. The embryos will be stuck in the appropriate orientation. Add two layers of double-sided tape on either side of the embryos, cover them with Halocarbon 27 Oil, and apply a coverslip. Proceed with the imaging as described above.
9. Representative Results:
If the embryos are healthy and the imaging is optimal, then cellularization should take 50-60 minutes, and the plasma membrane invaginations should ingress almost 40 microns. However, if the embryos are over-bleached, oxygen deprived or damaged by phototoxicity, then invagination will either slow or stop, particularly in the imaged area. Such deterioration of embryo health often results in altered development, and a failure to hatch as larvae. Thus, for a rigorous test to assay embryo health after imaging, keep your slides in a humidified chamber and watch for hatching the next day.
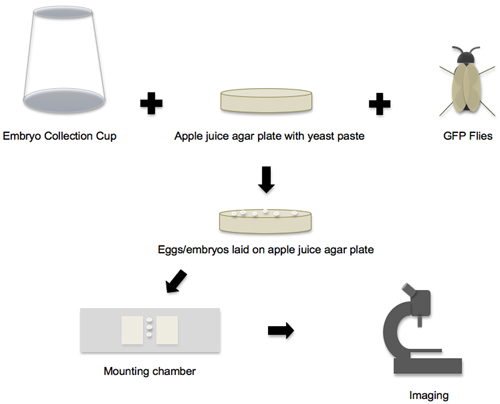 Figure 1. Workflow from embryo collection to imaging. The workflow of the protocol can be broken down into four main phases. In the first phase, all the individual supplies and components are prepared, and then the embryo collection cup, apple juice agar plate and GFP flies are put together to create the embryo-laying environment. In the second phase, the eggs and embryos that are laid on the apple juice agar plates are collected. In the third phase, the embryos are removed from the plate, staged and transferred to the mounting chamber. In the fourth phase, the mounted embryos are imaged on a confocal microscope.
Figure 1. Workflow from embryo collection to imaging. The workflow of the protocol can be broken down into four main phases. In the first phase, all the individual supplies and components are prepared, and then the embryo collection cup, apple juice agar plate and GFP flies are put together to create the embryo-laying environment. In the second phase, the eggs and embryos that are laid on the apple juice agar plates are collected. In the third phase, the embryos are removed from the plate, staged and transferred to the mounting chamber. In the fourth phase, the mounted embryos are imaged on a confocal microscope.
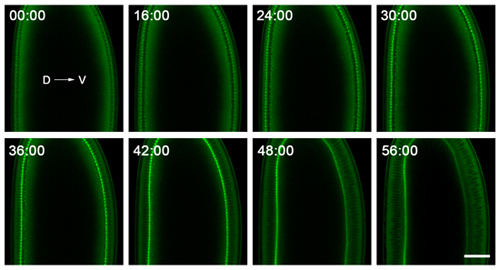 Figure 2. Representative data from time-lapse imaging of cellularization. Embryos are mounted with dorsal (D) and ventral (V) sides clearly visible, and are imaged near their middle to follow the plasma membrane invaginations in cross-section. The embryo shown here expresses a GFP-Myosin-2 probe6, which concentrates at the tips of the plasma membrane invaginations. Thus, tracking the ingression of this front over time gives the rate at which the plasma membrane invaginates. The 0:00 minute time point corresponds to cellularization onset. Shortly after the 56:00 minute time point, gastrulation starts on the ventral side of the embryo. Bar is 40 microns.
Figure 2. Representative data from time-lapse imaging of cellularization. Embryos are mounted with dorsal (D) and ventral (V) sides clearly visible, and are imaged near their middle to follow the plasma membrane invaginations in cross-section. The embryo shown here expresses a GFP-Myosin-2 probe6, which concentrates at the tips of the plasma membrane invaginations. Thus, tracking the ingression of this front over time gives the rate at which the plasma membrane invaginates. The 0:00 minute time point corresponds to cellularization onset. Shortly after the 56:00 minute time point, gastrulation starts on the ventral side of the embryo. Bar is 40 microns.
Movie 1. Representative movie from time-lapse imaging of cellularization. This movie corresponds to Figure 2. To record the entire process of cellularization, imaging started in the prior mitotic cycle 13, capturing pseudocleavage furrow regression, and continued until gastrulation movements were seen on the ventral side of the embryo. Images were collected at one minute intervals. The intensities were increased post-acquisition to make it easier to see the gastrulation movements. Click here for movie
| Developmental event and timing* | Cell shape changes related to | A link to disease or human health | Recent references with live imaging |
| Pseudo-cleavage furrow formation (4; 90 minutes pf) | Cytokinesis | Polyploidy and cancer progression1 | Mavrakis et al., 20098 Cao et al., 20109 |
| Cellularization (5; 130 minutes pf) | Cytokinesis | Polyploidy and cancer progression1 | Cao et al., 200810 Sokac & Wieschaus, 200811 |
| Ventral furrow formation; Mesoderm invagination (6; 180 minutes pf) | Apical constriction; Epithelial-mesenchymal transition | Cancer metastasis2 | Fox & Peifer, 200712 Martin et al., 200913 |
| Germband extension (7; 195 minutes pf) | Convergent extension | Neural tube defects3 | Bertet et al., 200414 Blankenship et al., 200615 |
| Tracheogenesis (11; 320 minutes pf) | Epithelial tube formation and branching | Angiogenesis4 | Caussinus et al., 200816 Gervais & Casanova, 201017 |
| Dorsal closure (14; 620 minutes pf) | Apical constriction | Wound healing5 | Gorfinkiel et al., 200918 Solon et al., 200919 |
Table 1. Examples of cell shape changes imaged in living fly embryos *The Bownes stage number and time post-fertilization (pf), when each event starts, are listed according to Campos-Ortega,1985.
| Fly stock | Labels | Original reference |
| Spider-GFP (95-1) | Plasma membrane | Morin et al., 200122 |
| Resille-GFP (117-2) | Plasma membrane | Morin et al., 200122 |
| GAP43-Venus | Plasma membrane | Mavrakis et al., 20098 |
| Spaghetti Squash-GFP (Sqh-GFP) | Myosin-2 | Royou et al., 20026 |
| E-cadherin-GFP (Ecad-GFP) | Cell-cell junctions | Oda et al., 200123 |
| GFP-Moesin | F-actin | Kiehart et al., 200024 |
| Utrophin-Venus (Utro-Venus) | F-actin | Sokac et al., unpublished results |
Table 2. Useful stocks for imaging cell shape change in fly embryos
Discussion
The protocol described herein will permit the live, confocal imaging of a number of cell shape changes in the developing fly embryo. GFP stocks for imaging can be prepared by an individual lab (Table 2), but many such stocks are also publicly available from centers such as Bloomington Drosophila Stock Center at Indiana University (http://flystocks.bio.indiana.edu) and FlyTrap Stock Center at Yale University (http://flytrap.med.yale.edu). Once acquired these live imaging data can then be paired with quantitative analysis to thoroughly define protein functions, sub-cellular activities, and biophysical parameters that drive cell shape change and tissue morphogenesis.
Disclosures
No conflicts of interest declared.
Acknowledgments
We gratefully acknowledge Eric Wieschaus, who provided the foundation on which this protocol was developed. Our work is supported by a Verna & Marrs McLean Department of Biochemistry and Molecular Biology Start-up Award, Baylor College of Medicine.
References
- Sagona AP, Stenmark H. Cytokinesis and cancer. FEBS Lett. 2010;584:2652–2661. doi: 10.1016/j.febslet.2010.03.044. [DOI] [PubMed] [Google Scholar]
- Baum B, Settleman J, Quinlan MP. Transitions between epithelial and mesenchymal states in development and disease. Semin Cell Dev Biol. 2008;19:294–308. doi: 10.1016/j.semcdb.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Kibar Z, Capra V, Gros P. Toward understanding the genetic basis of neural tube defects. Clin Genet. 2007;71:295–310. doi: 10.1111/j.1399-0004.2007.00793.x. [DOI] [PubMed] [Google Scholar]
- Jazwinska A, Ribeiro C, Affolter M. Epithelial tube morphogenesis during Drosophila tracheal development requires Piopio, a luminal ZP protein. Nat Cell Biol. 2003;5:895–901. doi: 10.1038/ncb1049. [DOI] [PubMed] [Google Scholar]
- Martin P, Parkhurst SM. Parallels between tissue repair and embryo morphogenesis. Development. 2004;131:3021–3034. doi: 10.1242/dev.01253. [DOI] [PubMed] [Google Scholar]
- Royou A, Field C, Sisson JC, Sullivan W, Karess R. Reassessing the role and dynamics of nonmuscle myosin II during furrow formation in early Drosophila embryos. Mol Biol Cell. 2004;15:838–850. doi: 10.1091/mbc.E03-06-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecuit T, Wieschaus E. Polarized insertion of new membrane from a cytoplasmic reservoir during cleavage of the Drosophila embryo. J Cell Biol. 2000;150:849–860. doi: 10.1083/jcb.150.4.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrakis M, Rikhy R, Lippincott-Schwartz J. Plasma membrane polarity and compartmentalization are established before cellularization in the fly embryo. Dev Cell. 2009;16:93–104. doi: 10.1016/j.devcel.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Crest J, Fasulo B, Sullivan W. Cortical Actin Dynamics Facilitate Early-Stage Centrosome Separation. Curr Biol. 2010 doi: 10.1016/j.cub.2010.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Albertson R, Riggs B, Field CM, Sullivan WNuf. a Rab11 effector, maintains cytokinetic furrow integrity by promoting local actin polymerization. J Cell Biol. 2008;182:301–313. doi: 10.1083/jcb.200712036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokac AM, Wieschaus E. Local actin-dependent endocytosis is zygotically controlled to initiate Drosophila cellularization. Dev Cell. 2008;14:775–786. doi: 10.1016/j.devcel.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox DT, Peifer M. Abelson kinase (Abl) and RhoGEF2 regulate actin organization during cell constriction in Drosophila. Development. 2007;134:567–578. doi: 10.1242/dev.02748. [DOI] [PubMed] [Google Scholar]
- Martin AC, Kaschube M, Wieschaus EF. Pulsed contractions of an actin-myosin network drive apical constriction. Nature. 2009;457:495–499. doi: 10.1038/nature07522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertet C, Sulak L, Lecuit T. Myosin-dependent junction remodelling controls planar cell intercalation and axis elongation. Nature. 2004;429:667–671. doi: 10.1038/nature02590. [DOI] [PubMed] [Google Scholar]
- Blankenship JT, Backovic ST, Sanny JS, Weitz O, Zallen JA. Multicellular rosette formation links planar cell polarity to tissue morphogenesis. Dev Cell. 2006;11:459–470. doi: 10.1016/j.devcel.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Caussinus E, Colombelli J, Affolter M. Tip-cell migration controls stalk-cell intercalation during Drosophila tracheal tube elongation. Curr Biol. 2008;18:1727–1734. doi: 10.1016/j.cub.2008.10.062. [DOI] [PubMed] [Google Scholar]
- Gervais L, Casanova J. In vivo coupling of cell elongation and lumen formation in a single cell. Curr Biol. 2010;20:359–366. doi: 10.1016/j.cub.2009.12.043. [DOI] [PubMed] [Google Scholar]
- Gorfinkiel N, Blanchard GB, Adams RJ, Arias Martinez, A Mechanical control of global cell behaviour during dorsal closure in Drosophila. Development. 2009;136:1889–1898. doi: 10.1242/dev.030866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solon J, Kaya-Copur A, Colombelli J, Brunner D. Pulsed forces timed by a ratchet-like mechanism drive directed tissue movement during dorsal closure. Cell. 2009;137:1331–1342. doi: 10.1016/j.cell.2009.03.050. [DOI] [PubMed] [Google Scholar]
- Bownes M. A photographic study of development in the living embryo of Drosophila melanogaster. J Embryol Exp Morphol. 1975;33:789–801. [PubMed] [Google Scholar]
- Sokac AM, Wieschaus E. Zygotically controlled F-actin establishes cortical compartments to stabilize furrows during Drosophila cellularization. J Cell Sci. 2008;121:1815–1824. doi: 10.1242/jcs.025171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin X, Daneman R, Zavortink M, Chia W. A protein trap strategy to detect GFP-tagged proteins expressed from their endogenous loci in Drosophila. Proc Natl Acad Sci U S A. 2001;98:15050–15055. doi: 10.1073/pnas.261408198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda H, Tsukita S. Real-time imaging of cell-cell adherens junctions reveals that Drosophila mesoderm invagination begins with two phases of apical constriction of cells. J Cell Sci. 2001;114:493–501. doi: 10.1242/jcs.114.3.493. [DOI] [PubMed] [Google Scholar]
- Kiehart DP, Galbraith CG, Edwards KA, Rickoll WL, Montague RA. Multiple forces contribute to cell sheet morphogenesis for dorsal closure in Drosophila. J Cell Biol. 2000;149:471–490. doi: 10.1083/jcb.149.2.471. [DOI] [PMC free article] [PubMed] [Google Scholar]