

Abstract
BMPs are responsible for a wide range of developmental and biological effects. BMP receptors activate (phosphorylate) the Smad1/5/8 effectors, which then, form a complex with Smad4 and translocate to the nucleus where they function as transcription factors to initiate BMP specific downstream effects 1. Traditional immuno-fluorescence techniques with antibodies against phospho-Smad peptides exhibit low sensitivity, high background and offer gross quantification as they rely on intensity of the antibody signal particularly if this is photosensitive fluorescent. In addition, phospho-Smads may not all be in complex with Smad4 and engaged in active transcription.
In situ PLA is a technology capable of detecting protein interactions with high specificity and sensitivity 2-4. This new technology couples antibody recognition with the amplification of DNA surrogate of the protein. It generates a localized, discrete signal in a form of spots revealing the exact position of the recognition event. The number of signals can be counted and compared providing a measurement. We applied in situ PLA, using the Duolink kit, with a combination of antibodies that allows the detection of the BMP signaling effectors phospho-Smad1/5/8 and Smad4 only when these are in proximity i.e. in a complex, which occurs only with signaling activation. This allowed for the first time, the visualization and measurement of endogenous BMP signaling with high specificity and sensitivity in a time course experiment under BMP4 stimulation.
Protocol
1. Plating of Cells and Treatment with BMP
Culture Neuro2a cells in GMEM supplemented with 10% FBS, 1x Non-Essential amino acids (NEAM), 1x sodium pyruvate and 1x L-glutamine. Split the cells using trypsin (0.05%-EDTA) and plate 15.000-20.000 cells per well in a 16-well chamber slide. Culture HEK293T or Cos7 cells in DMEM supplemented with 10% FBS, 1x L-glutamine and 1x Penicillin/Streptomycin. Sterilize polysine slides by immersing them in 70% ethanol and using Immedge pen draw 1 cm2 wells. Split the cells using trypsin (0.05%-EDTA) and plate 20.000-25.000 cells per well in 50 μl medium.
Incubate the cells at 37°C in a humidified, 5% CO2 incubator.
After 24 h replace the medium with serum free GMEM (supplemented with 0.1% albumin, 1x NEAM, 1x sodium pyruvate and 1x L-glutamine) for Neuro2a or with serum free DMEM (supplemented with L-Glutamine and 1x Penicillin/Streptomycin) for HEK293T/Cos7 and incubate the cells for 2-3 h. Keep three wells with cells under normal culturing conditions (i.e. 10% FBS) to use as controls.
Treat the cells with dorsomorphin (inhibitor of the BMP pathway, 2 μM) or BMP4 (25 ng/ml) for the desired time (e.g. for 10 min-1 h) 5.
2. Fixation and Permeabilization
Prepare the fixative, 4% PFA. Weigh 4 g of PFA to prepare a 4% solution in 100 ml PBS. Warm the solution at 50°C until it is clear. Filter the solution through a 0.22 μm filter and store at 4°C until use. Use fresh fixative (not stored for more than 2 days).
Aspirate the medium from the wells and wash with 1xPBS. Do not pipette the solutions directly onto the cells as this may result in detachment of the cells. Before going to the next step, if chamber slides are used remove the chambers, but leave the silicon around the wells.
Add 50 μl 4% PFA and incubate for 10min, at RT, without agitation.
Wash the cells with PBS for 3 x 5 min in a Coplin jar with agitation at RT.
Treat the cells with 0,5% Triton X-100 in PBS for 10 min without agitation at RT.
Wash the cells with 0,05% Tween 20 in TBS (TBS-T) for 3 x 5 min in a Coplin jar with agitation at RT.
3. Blocking
Tap off the TBS-T. Add one drop of Duolink II Blocking Solution (1x) per well.
Incubate the slides in a pre-heated humidity chamber for 1 h at 37°C.
4. Primary Antibodies
Mix and dilute a-P-Smad1/5/8 (rabbit polyclonal) at 1:100 and a-Smad4 (mouse monoclonal) at 1:100 in the Duolink II Antibody Diluent (1x). Prepare also a-P-Smad1/5/8 and a-Smad4 alone at the same concentrations to be used for controls.
Tap off the Blocking Solution from the slides. Remove as much blocking solution as possible, but take extra care not to let the cells dry before adding the antibody. Add 40 μl of the antibody solutions to the wells. In one well add only Antibody Diluent as an additional negative control.
5. PLA probes
Dilute the two PLA probes (Duolink II anti-Mouse MINUS and Duolink II anti-Rabbit PLUS) 1:5 in Antibody Diluent.
Tap off the primary antibody solution from the slides. Wash the slides 2 times 5 min each with 1x Duolink II Wash Buffer A in a Coplin jar with agitation at RT.
Tap off the Wash Buffer A from the slides and add the PLA probe solution (40 μl/well).Incubate the slides in a pre-heated humidity chamber for 1 h at 37°C.
6. Ligation
Vortex the Duolink II Ligation stock (5x) and dilute 1:5 in high purity water and mix. When calculating the volume of the water, take into account the volume of the Ligase that will be added at a final dilution of 1:40 just before the addition of the mix to the wells.
Tap off the PLA probe solution from the slides. Wash the slides in 1x Wash Buffer A for 2 times 5 min each in a Coplin jar with agitation at RT.
Take out the Ligase from the freezer using a freezing block (-20°C). Add the Ligase to the Ligation solution (prepared in 6.1) at a 1:40 dilution and vortex.
Tap off the Wash Buffer A from the slides and add the Ligation-Ligase solution to each well (40 μl/well). Incubate the slides in a pre-heated humidity chamber for 30 min at 37°C.
7. Amplification
Note: Light sensitive reagents. Keep the slides protected from light.
Dilute the Duolink II Amplification stock (5x) 1:5 in high purity water and mix. When calculating the volume of the water take into account the volume of the Polymerase that will be added at a final dilution of 1:80 just before the addition of the mix to the wells.
Tap off the Ligation-Ligase solution from the slides. Wash the slides in 1x Wash Buffer A with for 2 times 2 min each in a Coplin jar with agitation at RT.
Take out the Polymerase from the freezer using a freezing block (-20°C). Add the Polymerase to the Amplification solution (prepared in 7.1) at a 1:80 dilution and vortex.
Tap off the Wash Buffer A from the slides and add the Amplification-Polymerase solution to each well (40 μl/well). Incubate the slides in a pre-heated humidity chamber for 100 min at 37°C.
8. Preparation for Imaging
Note: Light sensitive reagents. Keep the slides protected from light.
Tap off the Amplification-Polymerase solution from the slides and wash for 2 times 10min each in 1x Duolink II Wash Buffer B in a Coplin jar with agitation at RT.
Dip the slides in 0.1 x Wash Buffer B.
Remove the silicone from the 16-well slide completely. Add ~ 40μl of the Duolink II Mounting Medium with DAPI on the coverslip and gently place it over the samples pressing it slightly so that there are no air bubbles under the cover slip. Fix and seal the coverslip on the slide using nail polish. Wait for at least 15min before proceeding to imaging.
Analyze the samples with a confocal or fluorescence microscope. Obtain digital images.
9. Quantification
Note: Light sensitive reagents. Keep the slides protected from light.
Use Duolink ImageTool to count the signals on the images to obtain a measurement of the signaling level.
Compare the measurements and make a graph.
10. Representative Results
The result from an in situ PLA experiment shows as discrete fluorescent spots. The location of the signal depends on the specific proteins studied.
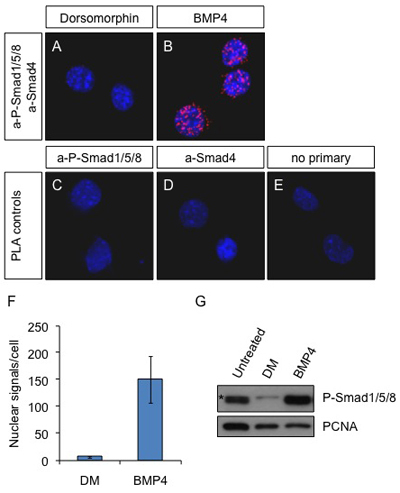 Figure 1.
In situ PLA on Neuro2a cells after BMP4 stimulation. Cells were treated with 2 μM dorsomorphin (inhibitor of the BMP pathway) (A) or 25 ng/ml BMP4 (B) for 60 min or left untreated (C-E). Antibodies against P-Smad1/5/8 and Smad4 were used to detect the active complexes in A and B. The primary antibodies a-P-Smad1/5/8 alone (C) and a-Smad4 alone (D) or omission of the primary antibodies (E) were used as controls. Blue: DAPI; Red: PLA signal. Pictures were obtained with a Leica SP5 confocal microscope.
Figure 1.
In situ PLA on Neuro2a cells after BMP4 stimulation. Cells were treated with 2 μM dorsomorphin (inhibitor of the BMP pathway) (A) or 25 ng/ml BMP4 (B) for 60 min or left untreated (C-E). Antibodies against P-Smad1/5/8 and Smad4 were used to detect the active complexes in A and B. The primary antibodies a-P-Smad1/5/8 alone (C) and a-Smad4 alone (D) or omission of the primary antibodies (E) were used as controls. Blue: DAPI; Red: PLA signal. Pictures were obtained with a Leica SP5 confocal microscope.
(F) The PLA signals were counted with the Duolink ImageTool software and the average number of spots in the nucleus per cell is presented in the graph. (G) Neuro2a cells were left untreated or treated with Dorsomorphin (2 μM) or BMP4 (25 ng/ml) for 60 min. The cells were lysed and the proteins were subjected to SDS-PAGE and analysed by immunoblotting with a-P-Smad1/5/8 and a-PCNA as loading control. (*) non-specific band. Note that the a-P-Smad1/5/8 antibody cannot distinguish between the 3 different proteins present in complex with Smad4.
Discussion
The visualization of protein complexes in situ is in great demand, particularly for studies in signaling where protein interaction and protein modification are the means that cells use for sending a signal from their surface to the nucleus. It has not been possible to visualize and quantify complexes between two endogenous proteins in situ with immunofluorescence before. Co-localization of antibodies exhibits low resolution and cannot be used to visualize true interaction. PLA is a new technique that researchers have started using in different systems with great success 6, 7. Here, we demonstrate how to use PLA not only to visualize but also to quantify endogenous complexes between Smad effectors activated downstream of BMP stimulation over time. We used different tissue culture cells including Neuro2a and relied on commercial antibodies raised in different species (mouse a-Smad4 and rabbit a-P-Smad1/5/8) to achieve this (fig 1). This method allowed us, for the first time, to see the activation of BMP effector-complexes over time and not just the presence of the phosphorylated Smad1/5/8, which may not all be engaged in active complexes with Smad4. We counted the signals (fig 1F) and compared the measurement with the quantification obtained from immunoblot of the same cells 8 (figure 1G). We concluded that the number of spots provide an accurate comparative measurement of signaling levels over time. The technique was also applied on other cell lines (HEK293T and Cos7) with similar results (data not shown).
The principle of the technology is based on two unique probes provided with the Duolink kit. Each PLA probe consists of a secondary antibody attached to a unique synthetic oligonucleotide, which acts as a reporter. The proximity of the probes allows DNA ligation at the exact location where these probes are attached in proximity. The distance of the oligonucleotides, which allows DNA hybridization and ligation is small (<40nm) and therefore, only proteins that interact can allow ligation. The ligated DNA can then be amplified and detected with hybridization of labeled oligonucleotides of the amplified sequence. The amplification is specific as it depends on DNA-DNA hybridization principles and also provides high sensitivity as the ligated DNA is amplified several fold resulting in enhancement of the initial ligation event. The amplified DNA is detected by hybridization with labeled oligonucleotides and produces a visible and rather large spot. As the spots can be counted, the PLA signal provides not only the exact spatial information (the location of the interaction events) but also an objective way of quantifying these events 2-4.
Other techniques that are employed for the detection of protein interactions include co-immunoprecipitation, immunofluoresence and Fluorescent Resonance Energy Transfer (FRET). Co-immunoprecipitation assays are widely used enabling the isolation of protein complexes. The method is accompanied by immunoblotting which means that the proteins must be expressed at relatively high levels in order to be detected. Problems of high background or non-specific binding of some proteins to the beads are sometimes difficult to overcome in co-immunoprecipitation. In addition, co-immunoprecipitation assays allow the detection of proteins that can be part of a protein complex but cannot distinguish those that are in very close proximity interacting physically with each other. Furthermore, co-immunoprecipitations cannot provide quantitation in single cell resolution or information on the localization of the protein complexes in the cell. Detection of co-existence of protein complexes in a cell or a cell compartment by immunofluoresence is based on the overlap of diffused signals and therefore, has low spatial resolution and cannot provide accurate information on protein interactions. Quantification of the immunofluoresence signal can be done by visual estimation only if the difference is several fold, and cannot be accurate or measurable as the PLA spots. FRET allows the visualization of protein complexes in live cells and overcomes the need for antibodies and the exposure of the epitope. However, FRET usually depends on the overexpression of artificial fused proteins, which may not reflect the endogenous protein complexes as in the PLA.
Blocking conditions in the PLA protocol are critical as antibody aggregation may occur and increase the background. Alternative blocking conditions can be found in antibody product sheets. Appropriate controls with primary and secondary antibody omission provide information on what may have gone wrong and they need to be always included. It is important to screen different antibodies that will give the least background when they are used alone and the best signal when they are together. Fixation of cells is critical as over-fixing may cause protein complexes to fully denature and degrade, whereas insufficient fixation may lead to loss of protein complexes during the steps of the procedure. This can be controlled by lowering the concentration of the fixative to 3% or by limiting the fixation time. However, the fixation depends also on whether the primary antibodies recognize denatured proteins or not. As the PLA technique is very sensitive, special care is needed to keep the incubation times and micro conditions equal among different samples.
The samples should not be left to dry at any case before the final step. Drying of the samples can lead to increased background. The complete removal of the blocking solution is also critical in order to avoid background. The wash buffers should always be brought to room temperature before use, as low temperature slows the enzymatic reactions. For further troubleshooting refer to the manual of the Kit.
It is useful to use stains to ascertain the sub-cellular localization of the PLA signal (e.g. DAPI, actin etc). Here we have used DAPI (in the mounting medium) to stain the nuclei. PLA is compatible with the use of immunofluorescence with antibodies to determine cell type or cellular compartment (not shown here). Accurate quantification can be done by counting the signals and the use of specific software (Duolink Image Tool, Olink Biosience).
In conclusion we have demonstrated how to use in situ PLA, using the Duolink kit to detect BMP signaling in fixed cells and presented its advantages: easy to use, highly sensitive, no background, and a unique ability to quantify active BMP signaling in situ.
The limitation of applying this technique to study different protein interactions depends on the availability and quality of the primary antibodies that recognize the proteins under investigation.
Disclosures
The production of this video-article was sponsored by OLINK who makes a reagent used in the proximity ligation assay procedure.
Acknowledgments
This research is funded by the MRC and the BBSRC grants. We thank Agata Zieba and Katerina Pardalis in Ulf Landegren's Lab, Uppsala University for helping with setting up the technique and useful advice. Olink Biosience for technical help and the presentation.
References
- Miyazono K, Kamiya Y, Morikawa MBone. morphogenetic protein receptors and signal transduction. J Biochem. 2010;147:35–51. doi: 10.1093/jb/mvp148. [DOI] [PubMed] [Google Scholar]
- Gustafsdottir SM. Proximity ligation assays for sensitive and specific protein analyses. Anal Biochem. 2005;345:2–9. doi: 10.1016/j.ab.2005.01.018. [DOI] [PubMed] [Google Scholar]
- Soderberg O. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- Soderberg O. Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods. 2008;45:227–232. doi: 10.1016/j.ymeth.2008.06.014. [DOI] [PubMed] [Google Scholar]
- Yu PB. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat Chem Biol. 2008;4:33–41. doi: 10.1038/nchembio.2007.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baan B. In situ proximity ligation detection of c-Jun/AP-1 dimers reveals increased levels of c-Jun/Fra1 complexes in aggressive breast cancer cell lines in vitro and in vivo. Mol Cell Proteomics. 9:1982–1990. doi: 10.1074/mcp.M110.000943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massinen S. Functional interaction of DYX1C1 with estrogen receptors suggests involvement of hormonal pathways in dyslexia. Hum Mol Genet. 2009;18:2802–2812. doi: 10.1093/hmg/ddp215. [DOI] [PubMed] [Google Scholar]
- Thymiakou E, Zannis VI, Kardassis D. Physical and functional interactions between liver X receptor/retinoid X receptor and Sp1 modulate the transcriptional induction of the human ATP binding cassette transporter A1 gene by oxysterols and retinoids. Biochemistry. 2007;46:11473–11483. doi: 10.1021/bi700994m. [DOI] [PubMed] [Google Scholar]