

Abstract
Here we describe a method for preparing and culturing primary cells dissociated from Drosophila gastrula embryos. In brief, a large amount of staged embryos from young and healthy flies are collected, sterilized, and then physically dissociated into a single cell suspension using a glass homogenizer. After being plated on culture plates or chamber slides at an appropriate density in culture medium, these cells can further differentiate into several morphologically-distinct cell types, which can be identified by their specific cell markers. Furthermore, we present conditions for treating these cells with double stranded (ds) RNAs to elicit gene knockdown. Efficient RNAi in Drosophila primary cells is accomplished by simply bathing the cells in dsRNA-containing culture medium. The ability to carry out effective RNAi perturbation, together with other molecular, biochemical, cell imaging analyses, will allow a variety of questions to be answered in Drosophila primary cells, especially those related to differentiated muscle and neuronal cells.
Protocol
1. Preparation of fly cages
Grow Drosophila (wild-type strains such as Oregon-R or any genotype) in convenient containers such as 32-oz insect cups.
Transfer newly eclosed flies to large embryo-collection cages or fly population cages.
Move the cages into a room with a temperature of ~25 °C and humidity ~65% in a light 12-h and a dark 12-h cycle to facilitate the collection of synchronous embryos.
Maintain the flies in the cages by feeding them killed yeast paste streaked on molasses plates. Change the molasses plates (with killed yeast paste) once a day for 2-3 d.
2. Collection of synchronous embryos
On the day of primary-cell preparation, feed the flies wth fresh plates coated with killed yeast paste for a 1-2 h pre-lay period.
Just before the 12-h dark cycle, replace the pre lay plates with fresh molasses plates streaked with a minimal amount of yeast paste. Discard the pre lay collection, which contains asynchronous older embryos.
Collect embryos for a period of 1-2 h.
Remove the plates containing the relatively synchronous population of embryos and store at 25°C for 5-6 h. Embryos will be at the advanced gastrula stage at this time.
3. Collection and washing of embryos
Loosen the embryos from the plates with a wet paintbrush, and rinse thoroughly with tepid tap water through a stacked set of sieves, so that the embryos collect in the finest mesh sieve at the bottom. Rinse for a few minutes to remove as much yeast and fly debris as possible.
Tip the sieves so that the embryos accumulate at one side, and gently wash them off from the sieve into a dechorionation basket.
Move to the tissue culture hood area without letting the embryos dry out.
4. Homogenization of embryos and plating of cells
Sterilize and dechorionate the embryos by immersing them in 50% (vol/vol) bleach for 5-10 min. Rinse the embryos off the wall of the basket with 70% (vol/vol) ethanol. Alternatively, wash the bleach off embryos with distilled water before ethanol treatment. From this point onward embryos should be handled in sterile conditions and in the tissue culture hood.
Rinse the embryos thoroughly to remove the bleach and/or ethanol with autoclaved distilled water.
During the dechorionation step, fill a Dounce homogenizer with the appropriate volume of room temperature M3 medium based on the amount of embryos. The ratio between the volume of embryos and that of media should not be over 0.5-ml embryos: 40-ml media (or ~100-200 embryos/ml).
Place the rinsed embryos in a sterilized beaker with the amount of M3 media enough to submerge the embryos. Let the embryos soak for 2-5 min.
Disassemble the dechorionation basket and blot the Nitex mesh dry with sterilized paper towels.
Transfer the embryos into the homogenizer with a sterilized paintbrush.
Homogenize gently with short strokes using a loose pestle without reaching the bottom of the tube until all the embryos are suspended. Then gently, but firmly, homogenize with eight full strokes. Do not twist the pestle as it moves up and down.
Transfer the homogenate into a sterile 50-ml conical centrifuge tube either by pouring or pipetting, and cap the tube.
Centrifuge for 10 min at 40g, room temperature to pellet the tissue debris, large cell clumps and vitelline membranes. Transfer the supernatant containing the cells and yolk to a clean tube and repeat the centrifugation step for 5 min.
Carefully transfer the supernatant by pouring or pipetting into a clean tube and spin for 10 min at 360g, room temperature to pellet the cells. Discard the supernatant. Resuspend the cell pellet in 10 ml of culture medium (serum-free medium for RNAi experiments).
Count viable cells using a hemocytometer to estimate the cell density. Meanwhile, repeat step 4.10 to pellet the cells.
Resuspend the cells to a desired concentration. To start, try a range of 1~5 x106 cells /ml and plate out at 1.7 to 2.5 x105 cells /cm2.
5. Primary-cell RNAi for cells grown on a 384-well plate
Dispense 5 μl of dsRNAs into each well at a final concentration of ~250 ng per well in a 384-well plate.
Use a multi-channel pipette, dispense 10 ml (1~4x106 cells/ml) of primary cells per well in a serum-free M3 medium in a 384-well plate containing a different dsRNA in each well. Alternatively, cells can be cultured on 8-well glass chamber slides for confocal imaging.
Centrifuge the plates for 1 min at 300g, room temperature. Culture the cells in serum-free medium at 25°C for 22 h or at 18°C for 1-2 d.
Add 30 ml of serum-containing culture medium to each well.
Centrifuge the plates for 1 min at 300g, room temperature.
Place the plates in a humid chamber and culture the primary cells for an additional 5~7 d at 25°C.
Image the cells either live or after immunofluorescence staining.
6. Representative Results:
In optimal conditions, cells in the culture will look healthy with a round morphology. The culture will have little cell debris, and be 50~80% confluent after initial plating. These cells will undergo morphological changes after being cultured for an extended period of time. It usually takes about 5-8 days for cells in the culture to be fully differentiated at 25°C. Good quality primary cultures are usually judged by how well the cells-of-interest are differentiated in culture. For example, primary muscle cells often have a branch-like morphology and become multinucleated myotubes with stripe-like myofibrils consisting of a series of sarcomeres. Fully differentiated myotubes will usually spontaneously move in the culture. In contrast, differentiated primary neurons form clusters with their cell bodies, and connect with other clusters with their axon cables.
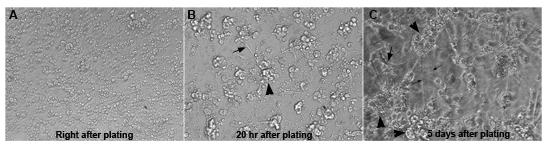 Figure 1. Primary cells undergo morphologic changes after being plated in the culture
(A) A bright field image of primary cells right after being plated in a well of a 384-well plate. The majority of cells are round and intact and well separated. (B) A bright field image of primary cell culture 20 hours after plating. Primary cells already form clusters (big arrowhead) and muscles with a branch-like morphology (long arrow) can be recognized at this stage. (C) A phase-contrast image of primary culture 5 days after plating. The culture looks much more advanced, with neuronal extensions (small arrows), neuronal clusters (big arrowheads) and muscles (medium-sized arrow).
Figure 1. Primary cells undergo morphologic changes after being plated in the culture
(A) A bright field image of primary cells right after being plated in a well of a 384-well plate. The majority of cells are round and intact and well separated. (B) A bright field image of primary cell culture 20 hours after plating. Primary cells already form clusters (big arrowhead) and muscles with a branch-like morphology (long arrow) can be recognized at this stage. (C) A phase-contrast image of primary culture 5 days after plating. The culture looks much more advanced, with neuronal extensions (small arrows), neuronal clusters (big arrowheads) and muscles (medium-sized arrow).
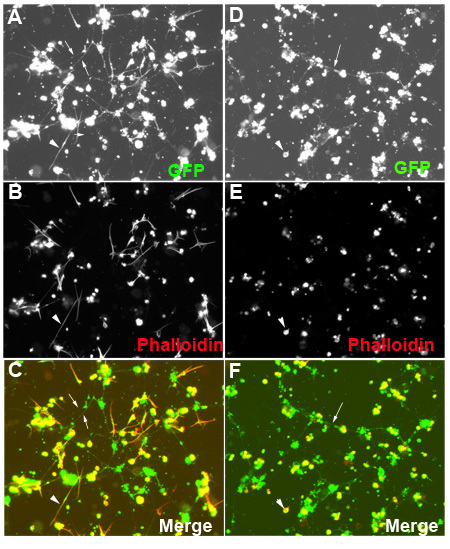 Figure 2. Examples of primary cells treated with dsRNAs cultured in a 384-well plate
Primary cells were isolated from the embryos carrying Dmef2-Gal4, D42-Gal4, UAS-mito-GFP transgenes, in which Dmef2-Gal4 and D42-Gal4 drive expression of mito-GFP in muscles and motor neurons, respectively. The cells were treated with dsRNAs targeting either lacZ (A-C) or inflated(if) (D-F). Both primary muscles and neurons can be seen by GFP (A,C,D,F). The white arrows point to the neuronal extensions. Phalloidin staining of actin (arrowheads) reveals a branch-like muscle structure in the culture treated with lacZ dsRNAs (B and C), but round-up muscle morphology in the culture treated with if dsRNAs (E and F). In the merged images (C, F), muscles are stained yellow, but red negative shows neurons and their extensions (white arrows).
Figure 2. Examples of primary cells treated with dsRNAs cultured in a 384-well plate
Primary cells were isolated from the embryos carrying Dmef2-Gal4, D42-Gal4, UAS-mito-GFP transgenes, in which Dmef2-Gal4 and D42-Gal4 drive expression of mito-GFP in muscles and motor neurons, respectively. The cells were treated with dsRNAs targeting either lacZ (A-C) or inflated(if) (D-F). Both primary muscles and neurons can be seen by GFP (A,C,D,F). The white arrows point to the neuronal extensions. Phalloidin staining of actin (arrowheads) reveals a branch-like muscle structure in the culture treated with lacZ dsRNAs (B and C), but round-up muscle morphology in the culture treated with if dsRNAs (E and F). In the merged images (C, F), muscles are stained yellow, but red negative shows neurons and their extensions (white arrows).
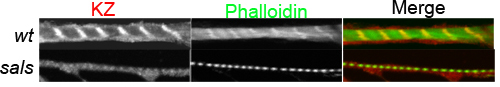 Figure 3. Confocal micrographs of primary muscles cultured on human vitronectin-coated cover glass slides
Primary muscles were immunofluorescently stained with anti-Drosophila Titin (KZ) (red in the merge) and with phalloidin for actin (green in the merge). The top panels are the wild-type control muscle and bottom ones are the sals(CG31374) RNAi muscle with shortened
sarcomeres.
Figure 3. Confocal micrographs of primary muscles cultured on human vitronectin-coated cover glass slides
Primary muscles were immunofluorescently stained with anti-Drosophila Titin (KZ) (red in the merge) and with phalloidin for actin (green in the merge). The top panels are the wild-type control muscle and bottom ones are the sals(CG31374) RNAi muscle with shortened
sarcomeres.
Discussion
The developmental pattern of primary cultures of Drosophila cells can vary greatly, possibly due to the several variables during the primary cell preparation process. First of all, flies used for egg laying should be young (within a week old) and healthy (free of viral or bacterial infection). Unfertilized or infected embryos must be avoided, as they are useless and cells derived from those embryos cannot differentiate and will only survive for 1-2 days. Second, during the homogenization process, it is important not to overload the homogenizer with too many embryos. Overloading the homogenizer with too many embryos will cause an increase in the amount of debris and the number of dead cells, which will eventually compromise cell differentiation. Finally, cells should be plated at an appropriate density to ensure good differentiation of primary cells. We found that the differentiation of both neurons and muscle cells is dramatically reduced when primary cells are seeded initially at a too high density.
For the phenotypic analysis, 384-well plates are usually used to grow primary cells for screening or imaging analysis at a lower magnification. Images with a much higher magnification and better resolution can be achieved by growing cells on a cover-glass chamber slide, followed by imaging either live or stained cells using a confocal microscope. As most primary cells are adherent, the area for growing cells in the well of chamber slides should be used as a guideline for estimation of both the number of cells and the amount of medium required for each well. In addition, cover-glass chamber slides can be coated with extracellular matrix (ECM) proteins such as human vitronectin, laminin or collagen IV to allow differentiation of cells-of-interest. For example, primary muscles differentiate poorly on the non-coated cover-glass, but well on the ECM-coated cover-glass. Finally, in order to reduce autofluorescence emitted from culture medium, the regular culture medium such as Shield and Sang M3, should be replaced with a fly physiological saline buffer, such as HL6, right before imaging.
In principle, this protocol can be used for preparation of primary cells from flies with any genotypes, such as from homozygous viable and fertile mutant stocks, and from those whose embryos can be selected manually or by a sorter based on expression of GFP, or express tissue-specific Gal4, UAS-gene X genotypes. In combination with other experimentation methods, this protocol will allow a variety of questions to be addressed, especially those that are related to differentiated cells such as neurons and muscles, and are difficult to be tackled in cultured cell lines.
Disclosures
No conflicts of interest declared.
Acknowledgments
J.B. was supported by the Damon Runyon Cancer Research Foundation Fellowship DRG-1716-02. J.Z. was supported by NIH/NIGMS Fellowship F32 GM082174-02. N.P. is an investigator of the Howard Hughes Medical Institute.
References
- Ashburner M, Roote J. Sullivan W, Ashburner M, Hawley RS. Laboratory culture of Drosophila. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 1999. pp. 588–599. [Google Scholar]
- Bai J, Hartwig JH, Perrimon NSALS. a WH2-domain-containing protein, promotes sarcomeric actin filament elongation from pointed ends during Drosophila muscle growth. Dev. Cell. 2007;13:828–842. doi: 10.1016/j.devcel.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Bai J. RNA interference screening in Drosophila primary cells for genes involved in muscle assembly and maintenance . Development. 2008;135:1439–1449. doi: 10.1242/dev.012849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepp KJ. Identification of neural outgrowth genes using genome-wide RNAi. PloS. Genet. 2008;4:e1000111–e1000111. doi: 10.1371/journal.pgen.1000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donady JJ, Fyrberg E. Mass culturing of Drosophila embryonic cells in vitro. TCA Manual. 1977;3:685–687. [Google Scholar]
- Seecof RL, Alleaume N, Teplitz RL, Gerson I. Differentiation of neurons and myocytes in cell cultures made from Drosophila gastrulae. Exp. Cell Res. 1971;69:161–173. doi: 10.1016/0014-4827(71)90321-1. [DOI] [PubMed] [Google Scholar]
- Bai J, Sepp KJ, Perrimon N. Culture of Drosophila primary cells dissociated from gastrula embryos and their use in RNAi screening. Nature Protocols. 2009;4:1502–1512. doi: 10.1038/nprot.2009.147. [DOI] [PubMed] [Google Scholar]