

Abstract
Electrospinning is a technique for producing micro- to nano-scale fibers. Fibers can be electrospun with varying degrees of alignment, from highly aligned to completely random. In addition, fibers can be spun from a variety of materials, including biodegradable polymers such as poly-L-lactic acid (PLLA). These characteristics make electrospun fibers suitable for a variety of scaffolding applications in tissue engineering. Our focus is on the use of aligned electrospun fibers for nerve regeneration. We have previously shown that aligned electrospun PLLA fibers direct the outgrowth of both primary sensory and motor neurons in vitro. We maintain that the use of a primary cell culture system is essential when evaluating biomaterials to model real neurons found in vivo as closely as possible. Here, we describe techniques used in our laboratory to electrospin fibrous scaffolds and culture dorsal root ganglia explants, as well as dissociated sensory and motor neurons, on electrospun scaffolds. However, the electrospinning and/or culture techniques presented here are easily adapted for use in other applications.
Keywords: Neuroscience, Issue 48, Electrospinning, motor neurons, nanofibers, scaffold, primary cell culture, DRG
Protocol
1. Poly-L-lactic Acid (PLLA) Spinning Solution
Dissolve 0.4 g PLLA in 9 mL chloroform by stirring over low heat.
Add 1 mL dimethylformamide to the solution, bringing the final concentration of the solution to 4% PLLA (w/v) in chloroform:DMF 9:1 (v/v).
Place the solution in a polypropylene or glass syringe with a blunt 23ga metal tip.
2. Spinning Substrate Preparation1
Make an 8% (w/v) solution of 85:15 PLGA (poly-lactic-co-glycolic acid) in chloroform by stirring over low heat.
Coat clean glass cover slips in PLGA by covering the surface of each cover slip with the PLGA solution. Allow the PLGA to dry to a thin film (approx. 30 min).
3. Electrospinning2
Secure PLGA coated glass cover slips to collector with conductive carbon tape. For aligned fibers, the collector is a motor-driven wheel. For random fibers, the collector is a stationary plate.
Place syringe in pump with tip 20 cm from collector wheel. Set pump to approximately 2 mL/hr and if using a wheel, set the motor to 300-400 RPM. If possible, apply a -2 kV DC bias to the collector and +15 kV to metal tip. In the absence of access to a bipolar power supply, ground the collector.
Fibers will jet from the syringe tip and collect on the rotating wheel. Continue spinning until the desired density of fibers is obtained. Swipe the metal tip with a paper towel affixed to a non-conductive rod periodically to prevent clogging at the tip.
4. Moating1
Following spinning, 'moat' the electrospun samples by pipetting a line of PLGA around the perimeter of the cover slip. Allow the PLGA to dry. This will maintain the alignment of the fibers during culture.
5. Protein Coating
Electrospun fibers and glass controls need to be coated in protein prior to cell culture. Cover the substrates in a protein solution for at least one hour and then aspirate excess solution and continue aspirating until the substrates are dry. Possible protein solutions include: PLL (poly-L-lysine) (100 µg/ mL), laminin (25 µg/ mL) or fibronectin (50 µg/ mL). If PLL is used, substrates should be rinsed in sterile water and dried twice following the initial coating.
6. Obtain E15 Rat Embryos3
*All experiments were done in accordance with the NIH Guide for Care and Use of Laboratory Animals as approved by the University of Michigan Committee on Use and Care of Animals.
Preparation: Check if the animal is pregnant. Thaw 3 mL trypsin, 3 mL FBS and warm a bottle of L15. Fire polish a Pasteur pipette. Disinfect all surgical instruments in 70% ethanol for 30 min.
Prepare and warm plating media (see Table 1).
Euthanasia: Perform an IP injection of ketamine/xylazine. Wait 10-15 minutes and check for lack of corneal and pedal withdrawal reflexes. Once reflexes are absent, perform an IC injection of pentobarbital (FatalPlus).
Tent the skin of the abdomen. Cut through the skin and muscle layers to expose the abdominal cavity. Locate the uterus and detach it at the cervix and ovaries.
Remove the embryos from the uterus with scissors and place them immediately into warm L15. (All the following procedures are completed under a dissecting microscope and embryos and dissected cords should be submerged in warm L15 at all times.) Decapitate the embryos by closing forceps around the chin and below the occipital bone of the skull.
7. Dissection3
Place the embryo on one side. Protect the spinal cord with one set of forceps while using the other set to strip away limbs and abdominal organs.
Turn the embryo and hold it steady with one set of forceps. Snip along the length of the embryo, from the neck to the tail, until the entire cord is exposed.
Grasp the end of the cord carefully and pull it over itself towards the tail to remove. The cord will pull free with membrane and dorsal root ganglia (DRG) attached.
- If DRG explant or dissociated sensory neuron culture is to be performed, snip the DRG off the cord and transfer them to a fresh dish of warm L15.
- For DRG explant culture, skip to step 10.2
- For dissociated sensory neuron culture, skip to step 9
- For motor neuron culture, the DRG will be removed with the membrane in step 7.5
Remove the membrane from the spinal cord by grasping it at the top end of the cord and peeling it down, similar to removing a long sock. Repeat for all embryos and then chop the cords into small (2-3 mm) pieces.
8. Motor Neuron (MN) Processing5,6,7
Pour the chopped cords and L15 into a conical and spin at 1000 RPM for 2-3 min to pellet the pieces.
Pour off the supernatant and add 3 mL of trypsin to the pelleted cords. Incubate in a 37°C water bath for 20 min.
Add 3 mL FBS to conical and spin at 1000 RPM for 2-3 min to re-pellet.
Pour off the supernatant and coat a fire polished Pasteur pipette with FBS.
Add one Pasteur pipette-full of L15 to the conical and then triturate gently until the solution is homogenous with no visible clumps. Avoid bubbles.
Prepare a 9% solution of Optiprep in L15. Prepare one tube with 3 mL of the Optiprep solution for every two embryos (if there is an odd number of embryos, round up).
Drop the homogenized solution onto the Optiprep solution, dividing it evenly among all the tubes. Spin the tubes at 2000 RPM for 15 min.
Carefully collect the top 2 mL from each tube and pool in a 50 mL conical. Fill the conical with L15 and spin at 1000 RPM for 5 min to rinse the cells of Optiprep.
Pour off the supernatant and resuspend the cells in a small amount of plating media (250-500 µl). Count the cells using trypan blue exclusion to identify live cells. Skip to step 10.1
9. Sensory Neuron (SN) Processing7
Pour the DRG removed from the spinal cords and L15 into a conical tube and spin at 1000 RPM for 2-3 min to pellet the pieces.
Pour off the supernatant and add 3 mL of trypsin to the pelleted DRG. Incubate in a 37°C water bath for 10 min.
Add 3 mL FBS to conical and spin at 1000 RPM for 2-3 min to re-pellet.
Pour off the supernatant and coat a fire polished Pasteur pipette with FBS.
Add one Pasteur pipette-full of L15 to the conical and then triturate gently until the solution is homogenous with no visible clumps. Avoid bubbles.
Spin the homogenate at 1000 RPM for 5 min. Pour off the supernatant and resuspend the cells in a small amount of SN plating media (250-500 µl). Count the cells using trypan blue exclusion to identify live cells. Continue to step 10.1
10. Plating and Culture
- For dissociated SN or MN culture:
- Cover the substrates with a few drops of media then add an appropriate volume of cell suspension. Dissociated cells should be plated at a low density (25-50 cells/mm2). Allow the cells to adhere for at least one hour before flooding the wells with media.
- For DRG explant culture:
- Cover the substrates with a few drops of media then add DRG to the media. Arrange the DRG so they are evenly distributed across the substrate (approximately 4 DRG will fit on a 22x22 mm cover slip). Allow the DRG to adhere for at least 4 hrs before flooding the wells with media.
Cultures can be maintained for up to 4 days without media change. For longer cultures, half media changes should be done with feed media. Feed media is the same composition as plating media minus the L-glutamine.
11. Immunocytochemistry1,5,8
Fixing cells: Aspirate media and replace with warm 4% PFA (paraformaldehyde) for 30 min. Then perform 3X 5 min 1x PBS washes.
Blocking/permeabilization: Prepare block/perm solution (1.25% bovine serum albumin in 1X PBS, 0.05% Triton X-100 and 2.0% normal goat serum). Aspirate PBS and apply block/perm solution to samples for 30 minutes. Then perform 1X 5 min 1x PBS washes.
Primary antibody: Prepare primary antibody working solution (10% normal goat serum, 1.25% bovine serum albumin, 0.1% Na azide, 0.05% Triton X-100 and to 10 mL with 1x PBS). Add the appropriate amount of primary antibody (see Table 2). Line several dishes with parafilm. Place a 200µl bead of primary antibody solution on the parafilm for each substrate. Invert the substrates, floating them cell side down on the primary antibody working solution overnight (if the room is air conditioned or particularly dry, a piece of water saturated paper can be added to a corner of the dish to prevent evaporation of the working solution).
Secondary antibody: Remove the substrates from the primary (the working solution can be collected, stored at 4°C and reused) and perform 2X 5 min 1x PBS washes. Apply 200 µl secondary antibody diluted 1:200 in PBS (also inverted on parafilm lined dish) for 4 hr.
Remove from secondary then perform 2X 5 min 1x PBS washes. Mount substrates on slides with Prolong Gold containing DAPI or another suitable nuclear counter stain.
12. Representative Results:
The typical morphology of aligned and random electrospun fibers are shown in Figure 1. We typically obtain MN purity of >90% neurons7,8 with this protocol and we have maintained DRG cultures for as long as one week without any significant fibroblast growth. Figure 2 shows typical MN appearance on glass and fibers after 24 hrs of culture and immunostaining. Immunostained DRG cultured for three days on glass and fibers are depicted in Figure 3.
 Figure 1. Fiber alignment is manipulated by choice of collector. A.) Aligned fibers spun onto a rotating wheel collector and B.) random fibers spun onto a stationary plate collector. Scale bars = 20 μm.
Figure 1. Fiber alignment is manipulated by choice of collector. A.) Aligned fibers spun onto a rotating wheel collector and B.) random fibers spun onto a stationary plate collector. Scale bars = 20 μm.
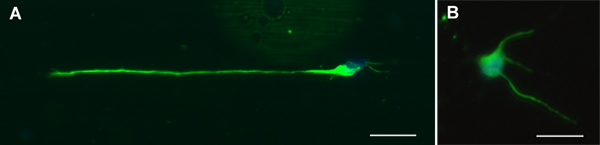 Figure 2. Representative motor neurons stained for TuJ1 (green) and DAPI (blue) after 24 hrs of culture on PLL coated A.) fibers and B.) glass. Scale bars = 20 μm.
Figure 2. Representative motor neurons stained for TuJ1 (green) and DAPI (blue) after 24 hrs of culture on PLL coated A.) fibers and B.) glass. Scale bars = 20 μm.
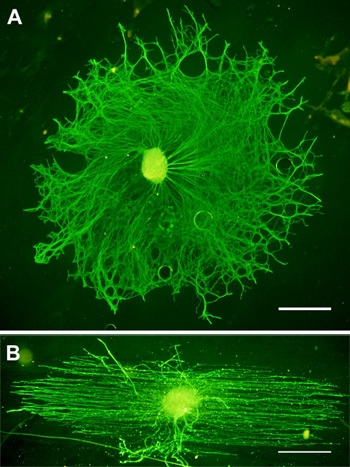 Figure 3. Representative DRG stained for neurofilament (green) after 3 days of culture on PLL coated A.) glass and B.) fibers. Scale bars = 200 μm.
Figure 3. Representative DRG stained for neurofilament (green) after 3 days of culture on PLL coated A.) glass and B.) fibers. Scale bars = 200 μm.
| Stock solution: | MN media: | DRG/SN media: |
| 10 mg mL-1 albumin | 12.5 μl | -- |
| 10 mg mL-1 apo-transferrin | 50 μl | 40 μl |
| 50 μg mL-1 β-estradiol | 2.7 μl | 2.2 μl |
| 0.1 mg mL-1 biotin | 50 μl | -- |
| 16 mg mL-1 catalase | 8 μl | -- |
| 15 mg mL-1 D- galactose | 50 μl | -- |
| 50 μg mL-1 hydrocortisone | 3.7 μl | 2.9 μl |
| 0.63 mg mL-1 progesterone | 0.5 μl | -- |
| 16 mg mL-1 putrescine | 50 μl | -- |
| 50 μg mL-1 selenium | 3.4 μl | 4.1 μl |
| 2.5 mg mL-1 superoxide dismutase | 50 μl | -- |
| * 500 ng mL-1 nerve growth factor | -- | 1 mL |
| * 100X PSN | 0.5 mL | 0.5 mL |
| * B27 | 1 mL | 1 mL |
| * 2 mM L-glutamine | 35 μl | 35 μl |
| * Neurobasal | to 50 mL | to 50 mL |
Table 1. Add starred (*) components to media just prior to use. The remaining components can be prepared as a stock solution and kept at -20°C until needed6,7.
| Primary (concentration) | Neurons: | Glia: | Fibroblasts: |
| anti-β-tubulin (TuJ1) (1:1000) | ✓ | X | X |
| anti-neurofilament (1:1000) | ✓ | X | X |
| anti-S-100 (1:250) | X | ✓ | ✓ |
| anti-NGFR p75 (1:500) | ✓ | ✓ | X |
Table 2. The selection of primary antibody is dependent on the goals of the investigator. The above are several antibodies and the concentrations we have used with success in our laboratory. S-100 is particularly useful to identify Schwann cells and/or check for contaminating fibroblasts, but note that it must be used in combination with NGFR p75 to distinguish between glia and fibroblasts4. We have also noticed that NGFR p75 stains neurites very lightly while neurofilament or TuJ1 are excellent choices if the goal is to visualize individual neurites.
Discussion
This protocol has several critical steps. The first involves the proper production of the electrospun fiber substrates. In liquid media, the PLLA fibers, PLGA film and PLGA moat will separate from the glass cover slip as a single unit. If the PLGA is omitted, the PLLA fibers will not remain a flat sheet - they will curl up into an unusable tangle. Thus, the PLGA film is included as a suitable substrate to maintain fiber alignment during culture. The moat is added after spinning both to ensure that the fibers are securely fixed to the PLGA film and to add structural rigidity to the film. The moat also serves as a useful handle when the substrates are manipulated during fixing, staining and mounting. Trituration is the second critical step. Every effort should be made to avoid the formation of bubbles. Additionally, we have obtained much higher yields when a fire-polished pipette is used for this step. To fire polish, light a Bunsen burner and attach a bulb to the pipette. Pass the tip of the pipette quickly through the flame 2-3 times while continuously squeezing and releasing the bulb - air flow through the pipette will help prevent the tip from closing completely. Examine the tip of the pipette to ensure the hole is still open and that the edges appear slightly rounded before use.
Electrospinning is a very versatile process; the electrospinning parameters can be modified to produce fibers with a variety of morphologies. Tan et al. (2005) details a systematic study of parameters affecting the diameter of PLLA electrospun fibers. Wang et al. (2009) presents a detailed study of the parameters influencing the alignment of electrospun PLLA fibers.
Disclosures
No conflicts of interest declared.
Acknowledgments
NIH K08 EB003996
References
- Corey JM, Gertz CC, Wang BS, Birrell LK, Johnson SL, Martin DC, Feldman EL. The design of electrospun PLLA nanofiber scaffolds compatible with serum-free growth of primary motor and sensory neurons. Acta. Biomater. 2008;4:863–875. doi: 10.1016/j.actbio.2008.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DY, Johnson MA, Vohden RA, Chen D, Martin DC. Tailored nanofiber morphologies using modulated electrospinning for biomedical applications. Mat. Res. Soc. Symp. Proc. 2003;736:D3.8.1–D3.8.6. [Google Scholar]
- Banker G, Goslin K. Culturing Nerve Cells. MIT Press; 1998. Chapter 19. [Google Scholar]
- Kameda Y. Expression of glial progenitor markers p75NTR and S100 protein in the developing mouse parathyroid gland. Cell Tissue Res. 2007;327:15–23. doi: 10.1007/s00441-006-0315-0. [DOI] [PubMed] [Google Scholar]
- Corey JM, Lin DY, Mycek KB, Chen Q, Samuel S, Feldman EL, Martin DC. Aligned electrospun nanofibers specify the direction of dorsal root ganglia neurite growth. J. Biomed. Mater. Res. A. 2007;83:636–645. doi: 10.1002/jbm.a.31285. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Mobley BC, Hiller A, Feldman EL. IGF-I prevents glutamate-induced motor neuron programmed cell death. Neurobiol. Dis. 2004;16:407–416. doi: 10.1016/j.nbd.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Russell JW, Windebank AJ, Schenone A, Feldman EL. Insulin-like growth factor-I prevents apoptosis in neurons after nerve growth factor withdrawal. J. Neurobiol. 1998;36:455–467. doi: 10.1002/(sici)1097-4695(19980915)36:4<455::aid-neu1>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Gertz CC, Leach MK, Birrel LK, Martin DC, Feldman EL, Corey JM. Accelerated neuritogenesis and maturation of primary spinal motor neurons in response to nanofibers. Dev. Neurobiol. 2010;70:589–603. doi: 10.1002/dneu.20792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S-H, Kotaki M, Ramakrishna S. Systematic parameter study for ultra-fine fiber fabrication via electrospinning process. Polymer. 2005;46:6128–6134. [Google Scholar]
- Wang HB, Mullins ME, Cregg JM, Hurtado A, Oudega M, Trombley MT, Gilbert RJ. Creation of highly aligned electrospun poly-L-lactic acid fibers for nerve regeneration applications. J. Neural Eng. 2009;6 doi: 10.1088/1741-2560/6/1/016001. [DOI] [PubMed] [Google Scholar]