

Abstract
Pre-implantation genetic diagnosis (PGD) is an established alternative to pre-natal diagnosis, and involves selecting pre-implantation embryos from a cohort generated by assisted reproduction technology (ART). This selection may be required because of familial monogenic disease (e.g. cystic fibrosis), or because one partner carries a chromosome rearrangement (e.g. a two-way reciprocal translocation). PGD is available for couples who have had previous affected children, and/or in the case of chromosome rearrangements, recurrent miscarriages, or infertility. Oocytes aspirated following ovarian stimulation are fertilized by in vitro immersion in semen (IVF) or by intracytoplasmic injection of an individual spermatozoon (ICSI). Pre-implantation cleavage-stage embryos are biopsied, usually by the removal of a single cell on day 3 post-fertilization, and the biopsied cell is tested to establish the genetic status of the embryo. Fluorescence in situ hybridization (FISH) on the fixed nuclei of biopsied cells with target-specific DNA probes is the technique of choice to detect chromosome imbalance associated with chromosome rearrangements, and to select female embryos in families with X-linked disease for which there is no mutation-specific test. FISH has also been used to screen embryos for spontaneous chromosome aneuploidy (also known as PGS or PGD-AS) in order to try and improve the efficiency of assisted reproduction; however, the predictive value of this test using the spreading and FISH technique described here is likely to be unacceptably low in most people's hands and it is not recommended for routine clinical use. We describe the selection of suitable probes for single-cell FISH, spreading techniques for blastomere nuclei, and in situ hybridization and signal scoring, applied to PGD in a clinical setting.
Protocol
1. Lysing and Spreading a Nucleus from a Biopsied Blastomere
Cell lysis buffer for spreading cells (0.2% Tween20 in 0.01 M HCl, pH 2.0) should be prepared 24 hours in advance and stored at -20°C. Prepare 100 mL and filter 20 mL into a 30 mL sterile universal container using a sterile 20 mL syringe and syringe filter. Dispense 1 mL aliquots into 10-15 1.7 mL sterile microcentrifuge tubes, close and label the tubes prior to freezing. It is recommended to have two different batches is use, the new batch and the previous batch, which has been tested and can be used if the new batch is unsatisfactory.
Defrost the lysis buffer at room temperature 30 minutes before the biopsy procedure. For practical purposes the working temperature will be between room temperature and hand temperature.
Score a small circle (approximately 5 mm diameter) on the underside of an amine-coated slide (e.g. Genetix) using a diamond pen and pre-label the slide with the case number, unique slide number, and biopsy date. Use a separate slide for each blastomere in numerical order, and label with the embryo number. Slides should be labeled with a hard pencil such as 4H, and "blotted" with a latex glove to remove any graphite dust.
Place a small volume of lysis buffer within the circle.
Transfer the biopsied blastomere into the lysis buffer. If necessary add lysis buffer within the circle until the cell begins to lyse; the cell should lyse completely and the cytoplasm disperse before the buffer dries.
Observe the nucleus to ensure that it remains within the circle and is not lost; if the cell does not have a nucleus or has multiple nuclei, biopsy another cell.
Leave the slide to air-dry at room temperature.
2. In Situ Hybridization of a Single Blastomere Nucleus
Prepare an ethanol series (70, 90 and 100%) made up in sterile distilled water.
Turn on and set the hot block (e.g. Hybaid Omnislide or Vysis Hybrite) to 75°C.
Defrost probes, vortex, and centrifuge. The reagents should be witnessed and verified to be correct before making up the probe mixture. Pipette volumes as specified by the manufacturer to make up the probe mixture in a 0.65 mL sterile microcentrifuge tube and vortex and centrifuge before use. The total volume of probe mixture should be sufficient to allow 2 μL per nucleus to be tested and rounded up to the nearest 10 μL to allow a safety margin.
Pre-wash the slides in coplin jars using phosphate-buffered saline (PBS) (pH 7.0: 0.14 M NaCl, 3 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4) for 5 min at room temperature.
Rinse the slides twice in sterile distilled water.
Dehydrate the slides with the ethanol series (70, 90, and 100%) for 2 min each at room temperature and air-dry. Ensure slides are fully immersed and if any graphite dust floats to the surface, soak up with a clean tissue.
Record the position of the nucleus within the circle by visualizing with a phase contrast microscope.
Dehydrate with 100% ethanol for 2 min at room temperature and air-dry.
Apply 2 μL of probe mixture, and cover with a 9 x 9 mm coverslip (one quarter of an 18 x 18 mm no.1 coverslip).
Seal the edges of the coverslip with rubber cement (e.g. Cow Gum; Cow Proofing).
Codenature the slides on a hot block at 75°C for 5 min, and then hybridize the slides overnight (16-20 h) in a humidified chamber at 37°C. Probe mixes that consist entirely of centromere probes (i.e. for sex-linked cases) will give a satisfactory result after 60 min of hybridization.
Prepare a water bath with sufficient coplin jars and heat to 71°C.
Prepare a 0.4x standard saline citrate (SSC) stringent wash solution (pH 7.0 at 71°C, 0.06 M NaCl, 6 mM C6H5Na3O7.2H2O) and heat in the water bath.
Dispense 50 mL of stringent wash per coplin jar required and check that the temperature is 71°C immediately prior to use using a clean thermometer.
Carefully remove the rubber cement from each slide and rinse off the coverslip using 4x SSC/0.05% Tween20 (pH 7.0) at room temperature.
Wash the slides in the 0.4x SSC stringent wash at 71°C for 5 min. Wash no more than 6 slides per coplin jar.
Wash the slides in 4x SSC/0.05% Tween20 at room temperature for 2 min.
If the probe mix contains indirectly labelled probe(s), drain the slides of excess liquid and apply 20 μL of fluorescently conjugated antibody under a 20 x 20 mm square of Parafilm.
Incubate in a humidified chamber at 37°C for 15 min.
Remove the Parafilm and wash once in 4x SSC/0.05% Tween20 at room temperature for 2 min.
Wash twice for 2 min in PBS at room temperature and drain the slides.
Apply 6 μL of DAPI/Vectashield (160 ng of 4',6-diamidino-2-phenylindole dihydrochloride in 1 mL of Vectashield mounting medium, Vector Laboratories) to a 22 x 22 mm no. 0 coverslip and invert the slide over the coverslip.
Blot and seal the edges of the coverslip with clear nail varnish.
3. Analysis using a Fluorescence Microscope Suitably Equipped with Appropriate Filters for the Probes Used.
Score signals by direct visualization using a fluorescence microscope and single band-pass filters for each fluorochrome in the assay. Each nucleus should be scored by two analysts. A general guideline should lead to scoring of a single signal where two closely spaced signals are less than one domain (signal-width) apart; however, judgment based on experience needs to be exercised to interpret signals of varying size, intensity, and separation.
Use imaging software (e.g. Isis, MetaSystems, Altlussheim, Germany; CytoVision, Genetix) to capture an image of the nucleus for confirmation of the visual diagnosis, and for image archiving as part of a laboratory quality assurance plan.
4. Representative Results:
Metaphase and interphase nuclei from cultured peripheral blood lymphocytes should be examined to confirm that the selected probes are specific for the translocation chromosomes, informative for the breakpoints (the subtelomere probes should hybridize only to the translocated segments and the centromere probe(s) to the centric segment(s)) and the signals in interphase nuclei should be bright and discrete. Scoring the number of signals for each probe in 100 interphase nuclei from each partner is recommended to assess the efficiency of the assay. In this case 2 signals were scored in 95%-99% of nuclei for each probe. Figure 1 shows a metaphase and interphase nucleus from a preparation for a reciprocal translocation between the short arms of chromosomes 5 and 9.
Signals in interphase nuclei from embryo blastomeres should be bright and discrete and scored using separate band pass filters for the colors used. Figure 2 shows a blastomere nucleus with a normal signal pattern (2 copies for all the loci tested) consistent with a normal or balanced chromosome complement for chromosome 5 and 9, and a nucleus with an abnormal signal pattern consistent with an unbalanced product of the translocation that has monosomy (one copy) for the translocated segment of chromosome 5 and trisomy (3 copies) for the translocated segment of chromosome 9.
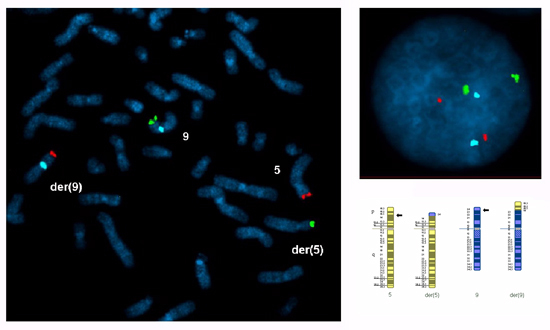 Figure 1. A metaphase spread and an interphase nucleus prepared from cultured peripheral blood lymphocytes from a carrier of a reciprocal translocation between the short arms of chromosomes 5 and 9 with breakpoints at 5p14.3 and 9p24.1: 46,XY,t(5;9)(p14.3;p24.1).ish t(5;9)(5ptel48-,9ptel30+;9ptel30-,5ptel48+,9cen+). FISH probes were selected for both translocated segments (5p14.3→5pter, red Cytocell subtelomere5ptel48 TexasRed ; 9p24.1→9pter, green Cytocell subtelomere 9ptel30 FITC) and the centric segment of chromosome 9 (9p24.1→9qter, blue Abbott CEP 9 alpha satellite SpectrumAqua).
Figure 1. A metaphase spread and an interphase nucleus prepared from cultured peripheral blood lymphocytes from a carrier of a reciprocal translocation between the short arms of chromosomes 5 and 9 with breakpoints at 5p14.3 and 9p24.1: 46,XY,t(5;9)(p14.3;p24.1).ish t(5;9)(5ptel48-,9ptel30+;9ptel30-,5ptel48+,9cen+). FISH probes were selected for both translocated segments (5p14.3→5pter, red Cytocell subtelomere5ptel48 TexasRed ; 9p24.1→9pter, green Cytocell subtelomere 9ptel30 FITC) and the centric segment of chromosome 9 (9p24.1→9qter, blue Abbott CEP 9 alpha satellite SpectrumAqua).
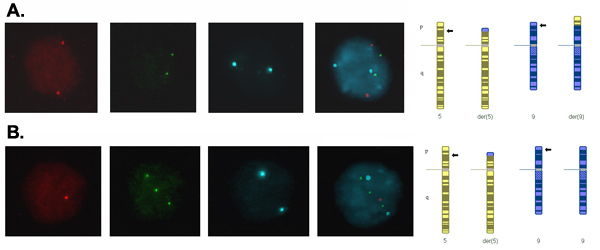 Figure 2. Signals in interphase blastomere nuclei from day-3 embryos captured using a different filter for each fluorochrome and merged to form a composite image. (A) Two signals for each probe indicating two copies of each locus, which is consistent with a normal or balanced complement for the translocation chromosomes. (B) One red signal, three green signals and two blue signals indicating one copy of the translocated segment of chromosome 5, three copies of the translocated segment of chromosome 9 and two copies of the centric segment of chromosome 9, which is consistent with adjacent-1 segregation of the translocation resulting in an unbalanced product with monosomy for 5p14.3→5pter and trisomy for 9p24.1→9pter.
Figure 2. Signals in interphase blastomere nuclei from day-3 embryos captured using a different filter for each fluorochrome and merged to form a composite image. (A) Two signals for each probe indicating two copies of each locus, which is consistent with a normal or balanced complement for the translocation chromosomes. (B) One red signal, three green signals and two blue signals indicating one copy of the translocated segment of chromosome 5, three copies of the translocated segment of chromosome 9 and two copies of the centric segment of chromosome 9, which is consistent with adjacent-1 segregation of the translocation resulting in an unbalanced product with monosomy for 5p14.3→5pter and trisomy for 9p24.1→9pter.
Discussion
The application of fluorescence in situ hybridization (FISH) to a single embryo cell (blastomere) presents special challenges both in practicalities and in interpretation of the signal pattern. The biopsied cell needs to be spread within a pre-defined area on the slide in order to facilitate its localization following FISH; extreme care needs to be taken in ensuring that the cell is lysed, that the cytoplasm has been dispersed, and that the nucleus is visible and intact; and, as the diagnosis depends on the results from this single cell, stringent scoring and interpretation guidelines should be applied. However, in experienced hands, FISH is a robust technique for pre-implantation genetic diagnosis (PGD) in clinical practice. The principle of PGD by FISH is that target-specific DNA probes labelled with different fluorochromes or haptens can be used to detect the copy number of specific loci, and thereby to detect chromosome imbalance associated with meiotic segregation of chromosome rearrangements (1), including Robertsonian translocations, reciprocal translocations, inversions, and complex rearrangements (2). FISH can also be used to select female embryos in families with X-linked disease, for which there is no mutation-specific test (3, 4). More controversially, FISH has also been used to screen for sporadic chromosome aneuploidy in order to try and improve the efficiency of assisted reproduction (5, 6); however, the predictive value of this test using FSIH is likely to be unacceptably low in most people's hands and it is not recommended for routine clinical use (7). This article concentrates on the technical aspects and the limitations of FISH applied to clinical single-cell diagnosis.
Methods of spreading and fixing single blastomeres include methanol/acetic acid (5, 8), Tween/HCl (9), and a combination of Tween/HCl and methanol/acetic acid (10). Variations include hypotonic treatment of cells prior to spreading and/or pepsin and paraformaldehyde treatment after fixation. The method should be appropriately validated for the laboratory (14). The Tween/HCl method is described in detail in this article. The Tween/HCl method is technically simple and highly reproducible in different laboratories. This method can be used to prepare single nuclei for the FISH diagnosis of sex determination and chromosome rearrangements with acceptable diagnostic accuracy (7).
Probe mixes can combine directly labelled and indirectly labelled probes, and probes from different manufacturers. Probes for known polymorphic chromosome regions (11, 12), or those known to cross-hybridize significantly with other chromosomes (13) should be avoided, although can be used if shown to be specific and suitable for PGD by prior testing on both reproductive partners. Available fluorochromes/haptens and strategies for discriminating probes include the following: Texas Red (TR), fluorescein isothiocyanate (FITC), SpectrumGreen (Vysis), SpectrumOrange (Vysis), SpectrumAqua, biotinylated probes detected with TR-avidin, FITC-avidin, or Cy-5 streptavidin (visualized using a FarRed filter), a mix of red and green probes to produce a yellow signal, a second round of hybridization and a third color created by sequential capturing of SpectrumOrange using a TexasRed and a SpectrumGold filter).
A probe set containing three probes, specific for the centromere regions of the X and Y chromosomes and one autosome, is recommended for sex determination (14); the autosomal probe is used to establish ploidy and thereby to differentiate between trisomy X (2n, 47,XXX) and triploidy (3n, 69,XXX), and between tetrasomy X (48,XXXX) and tetraploidy (4n, 92,XXXX). A typical probe set applied in this setting is the Abbott AneuVysion mix containing alpha-satellite X, Y, and 18; this probe set has been demonstrated to have a very low polymorphism rate, and therefore pre-treatment work-up of the reproductive partners is not required (14).
The probe mix for any specific rearrangement should: Ideally contain probes at least sufficient to detect all the expected products of the rearrangement with chromosome imbalance. If this is not possible, probe mixes where the undetected unbalanced products have been assessed to be non-viable in a recognizable pregnancy and are likely to have very low prevalence may be acceptable (14). Be tested on cultured lymphocyte metaphases from both reproductive partners. At least ten metaphase spreads should be examined for probe specificity, polymorphisms and cross-hybridization, and, for the chromosome rearrangement carrier, to ensure that the probes hybridize as expected to the different segments of the rearrangement. In addition, at least 100 interphase nuclei from these preparations should be scored to assess signal specificity, brightness, and discreteness (14).
Commercial PGS probe sets are available (e.g. Abbott MultiVysion PB or PGT), targeting the chromosomes most frequently found to be aneuploid in products of conception, and comprising a single probe per chromosome targeted. Typically the nucleus may have a second hybridization with probes for additional chromosomes providing an assay for chromosomes 13, 15, 16, 18, 21, 22, and XY (14). The predictive value of this test using the spreading and FISH technique described here is likely to be unacceptably low in most people's hands and it is not recommended for routine clinical use (7, 15).
Techniques such as comparative genomic hybridization (CGH) applied to single cells are able to test for the copy number of every chromosome (Wilton et al. 2001), and the use of single nucleotide polymorphisms (SNP) arrays and quantitative analysis (Wells et al. 2008) or "karyomapping" genotyping (Handyside et al. 2009) are promising alternative techniques to detect chromosome aneuploidy. However, the resolution limit and accuracy for segment imbalance remain uncertain and it is likely that FISH will continue to be the technique of choice for chromosome rearrangements involving small segments.
Disclosures
No conflicts of interest declared.
References
- Scriven PN, Handyside AH, Ogilvie CM. Chromosome translocations: segregation modes and strategies for preimplantation genetic diagnosis. Prenat. Diagn. 1998;18:1437–1449. [PubMed] [Google Scholar]
- Mackie Ogilvie C, Scriven PN. Harper JC. Preimplantation Genetic Diagnosis. 2nd Ed. Cambridge University Press; 2009. Preimplantation genetic diagnosis for chromosome rearrangements; pp. 194–201. [Google Scholar]
- Harper JC, Coonen E, Ramaekers FCS, Delhanty JDA, Handyside AH, Winston RML, Hopman AHN. Identification of the sex of human preimplantation embryos in two hours using an improved spreading method and fluorescent in situ hybridisation using directly labelled probes. Hum. Reprod. 1994;9:721–724. doi: 10.1093/oxfordjournals.humrep.a138577. [DOI] [PubMed] [Google Scholar]
- Mackie Ogilvie C, Scriven PN. Harper JC. Preimplantation Genetic Diagnosis. 2nd Ed. Cambridge University Press; 2009. Preimplantation genetic diagnosis for sex-linked diseases and sex-selection for non-medical reasons; pp. 230–235. [Google Scholar]
- Munné S, Lee A, Rosenwaks Z, Grifo J, Cohen J. Diagnosis of major chromosome aneuploidies in human preimplantation embryos. Hum. Reprod. 1993;8:2185–2192. doi: 10.1093/oxfordjournals.humrep.a138001. [DOI] [PubMed] [Google Scholar]
- Mackie Ogilvie C, Scriven PN. Harper JC. Preimplantation Genetic Diagnosis. 2nd Ed. Cambridge University Press; 2009. Preimplantation genetic diagnosis for infertility (preimplantation genetic screening) pp. 203–229. [Google Scholar]
- Scriven PN, Bossuyt PM. Diagnostic accuracy: theoretical models for preimplantation genetic testing of a single nucleus using the fluorescence in situ hybridization technique. Hum Reprod. Cytogenetics. 2010;5:394–400. doi: 10.1093/humrep/deq196. [DOI] [PubMed] [Google Scholar]
- Coonen E, Dumoulin JC, Ramaekers FC, Hopman AH. Optimal preparation of preimplantation embryo interphase nuclei for analysis by fluorescence in-situ hybridization. Hum. Reprod. 1994;9:533–537. doi: 10.1093/oxfordjournals.humrep.a138540. [DOI] [PubMed] [Google Scholar]
- Dozortsev DI, McGinnis KT. An improved fixation technique for fluorescence in situ hybridization for preimplantation genetic diagnosis. Fertil. Steril. 2001;76:186–188. doi: 10.1016/s0015-0282(01)01836-2. [DOI] [PubMed] [Google Scholar]
- Hsu LY, Benn PA, Tannenbaum HL, Perlis T, Carlson AD. Chromosomal polymorphisms of 1, 9, 16, and Y in 4 major ethnic groups: a large prenatal study. Am. J. Med. Genet. 1987;26:95–101. doi: 10.1002/ajmg.1320260116. [DOI] [PubMed] [Google Scholar]
- Shim SH, Pan A, Huang XL, Tonk VS, Varma SK, Milunsky JM, Wyandt HE. FISH variants with D15Z1. J. Assoc. Genet. Technol. 2003;29:146–151. [PubMed] [Google Scholar]
- Knight SJ, Flint J. Perfect endings: a review of subtelomeric probes and their use in clinical diagnosis. J. Med. Genet. 2000;37:401–409. doi: 10.1136/jmg.37.6.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornhill AR, de Die-Smulders CE, Geraedts JP, Harper JC, Harton GL, Lavery SA, Moutou C, Robinson MD, Schmutzler AG, Scriven PN, Sermon KD, Wilton L. ESHRE PGD Consortium (PGS) ESHRE PGD Consortium 'Best practice guidelines for clinical preimplantation genetic diagnosis (PGD) and preimplantation genetic screening (PGS)'. Hum. Reprod. 2005;20:35–48. doi: 10.1093/humrep/deh579. [DOI] [PubMed] [Google Scholar]
- Munné S, Wells D, Cohen J. Technology requirements for preimplantation genetic diagnosis to improve assisted reproduction outcomes. Fertil. Steril. 2010;94:408–430. doi: 10.1016/j.fertnstert.2009.02.091. [DOI] [PubMed] [Google Scholar]
- Wilton L, Williamson R, McBain J, Edgar D, Voullaire L. Birth of a healthy infant after preimplantation confirmation of euploidy by comparative genomic hybridization. N. Engl. J. Med. 2001;345:1537–1541. doi: 10.1056/NEJMoa011052. [DOI] [PubMed] [Google Scholar]
- Wells D, Alfarawati S, Fragouli E. Use of comprehensive chromosomal screening for embryo assessment: microarrays and CGH. Mol. Hum. Reprod. 2008;14:703–710. doi: 10.1093/molehr/gan062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handyside AH, Harton GL, Mariani B, Thornhill AR, Affara N, Shaw MA, Griffin DK. Karyomapping, a universal method for genome wide analysis of genetic disease based on mapping crossovers between parental haplotypes. J. Med. Genet. 2010;47:651–658. doi: 10.1136/jmg.2009.069971. [DOI] [PubMed] [Google Scholar]