

Abstract
The budding yeast, Saccharomyces cerevisiae, is a powerful model system for defining fundamental mechanisms of many important cellular processes, including those with direct relevance to human disease. Because of its short generation time and well-characterized genome, a major experimental advantage of the yeast model system is the ability to perform genetic screens to identify genes and pathways that are involved in a given process. Over the last thirty years such genetic screens have been used to elucidate the cell cycle, secretory pathway, and many more highly conserved aspects of eukaryotic cell biology 1-5. In the last few years, several genomewide libraries of yeast strains and plasmids have been generated 6-10. These collections now allow for the systematic interrogation of gene function using gain- and loss-of-function approaches 11-16. Here we provide a detailed protocol for the use of a high-throughput yeast transformation protocol with a liquid handling robot to perform a plasmid overexpression screen, using an arrayed library of 5,500 yeast plasmids. We have been using these screens to identify genetic modifiers of toxicity associated with the accumulation of aggregation-prone human neurodegenerative disease proteins. The methods presented here are readily adaptable to the study of other cellular phenotypes of interest.
Protocol
1. Preparations for yeast transformation
This protocol is designed for ten 96-well plates but can be scaled up or down accordingly. We have found that this protocol does not work well for more than twenty 96-well plates per round of transformation. The entire transformation procedure (from step I.3) will take approximately eight hours.
Aliquot 5-10μL of plasmid DNA (100 ng/μl) from the Yeast FLEXGene ORF library into each well of a round-bottom 96-well plate with Biorobot RapidPlate liquid handler. Place uncovered 96-well plates in clean bench fume hood overnight to dry. We have found similar transformation efficiencies with CEN and 2μ yeast plasmids.
Inoculate 200mL of yeast peptone dextrose (YPD) media (10g/L yeast extract, 20g/L peptone, 20g/L glucose) with query strain in a 500mL baffled Erlenmeyer flask. Incubate overnight at 30°C with shaking (200 rpm). If required, selective media can also be used for these starter cultures.
The following morning, dilute the query strain to OD600=0.1 in 2L of YPD. Shake for 4-6 hours at 30°C (200 rpm) until the culture reaches OD600=0.8-1.0. For optimal growth, grow up two 1L cultures in separate 2.8 L baffled flasks. If required, selective media can also be used instead of YPD.
2. Yeast transformation
Harvest cells by centrifugation. Fill four disposable 225mL conical tubes with yeast culture and spin at 3000rpm (˜1800 x g) for 5 minutes in tabletop centrifuge (Eppendorf 5810R). Pour off supernatant and repeat until all cells are harvested.
Wash cells in 200mL of autoclaved distilled H2O. Resuspend cell pellet by shaking or vortexing. Combine cells in one 225mL conical tube. Spin at 3000rpm for 5 minutes. Remove supernatant.
Prepare 0.1MLiOAc/1XTE solution (0.1M LiOAc, 10mM Tris, 1mM EDTA, pH 8 in autoclaved distilled water). Wash cells in 100mL 0.1MLiOAc/1XTE. Spin at 3000rpm for 5 minutes. Remove supernatant.
Resuspend cell pellet in 70mL 0.1M LiOAc/1xTE. Shake and/or vortex to ensure pellet is completely resuspended.
Incubate with shaking at 30°C for 30 minutes (200 rpm).
Add β-mercaptoethanol to 0.1M. Incubate with shaking at 30°C for 30 minutes (200 rpm). Note: This step can be omitted if using yeast strains that are sensitive to β-mercaptoethanol. We have found that this step is not essential for successful transformation, however it does improve transformation efficiency.
Boil 2mL of sonicated salmon sperm DNA (10mg/mL) for 15 minutes, and then chill on ice.
Add 2mL of boiled salmon sperm DNA to cell mixture. Caution: A precipitate may form upon addition of the salmon sperm DNA to the cell mixture. Using a pipetman and 200 μl tip, carefully remove any precipitate that forms because this may interfere with downstream pipetting.
Aliquot 50μL of cell mixture to each well of the 96-well plate using the BioRobot Rapidplate (can also use multi-channel pipette). Do not mix.
Incubate at room temperature for 30 minutes without shaking.
Prepare 200mL of 40%PEG-3350/10%DMSO/0.1M LiOAc (160mL of 50% PEG-3350, 20mL DMSO, 20mL 1M LiOAc). Prepare solution immediately before use.
Add 125μL of PEG/LioAc/DMSO solution to each well. Mix by aspirating and dispensing cell suspension eight times with RapidPlate.
Incubate at room temperature for 30 minutes without shaking.
Heat shock the cells at 42°C for 15 minutes in a dry incubator. Do not stack plates. Note: We have found that this heat shock step is not essential for successful transformation. For certain yeast strains (for example, temperature sensitive mutants), heat shock is deleterious. We have previously reduced the heat shock time to one minute and have been able to successfully transform a yeast strain harboring a ypt1 temperature sensitive mutation using this protocol.
Spin plates at 2500rpm (˜1100 x g) for 5 minutes, using centrifuge adapters to accommodate 96-well plates. Remove PEG solution by inverting plates over waste bucket. Blot inverted plate on paper towel to remove residual liquid (cells will remain on bottom of wells).
Rinse cells by adding 200μL of minimal media (e.g. SD/-Ura) to each well with RapidPlate.
Spin plates at 2500rpm for 5 minutes and remove supernatant by inverting over waste bucket and blotting on paper towels.
Add 200μL of minimal media (e.g. SD/-Ura) to each well with RapidPlate.
Incubate at 30°C for 2 days without shaking. After 2 days, small colonies of cells should begin to form at the bottom of each successfully transformed well.
3. Spotting assay
Dispense 200μL of raffinose based minimal media (e.g. SRaf/-Ura) into each well of a new flat-bottom 96-well plate.
Mix each well of transformation plate by aspirating and dispensing cell suspension eight times with Rapidplate.
Inoculate SRaf plates with 5μL of cell mixture from transformation plate.
Incubate at 30°C for one day. Small colonies should be present at the bottom of each well after one day.
Spot colonies on SD/-Ura and SGal/-Ura plates in hood. Sterilize 96-bolt replicator (frogger) by flaming. Mix colonies with frogger before spotting on selective media plates. After spotting, allow plates to dry in hood for 5-10 minutes.
Incubate at 30°C for 2-3 days.
Photograph plates with digital camera, and visually compare growth of colonies on SGal/-Ura plates to find colonies in which query strain toxicity is enhanced (slower growth/less dense colonies) or suppressed (faster growth/more dense colonies).
4. Representative results:
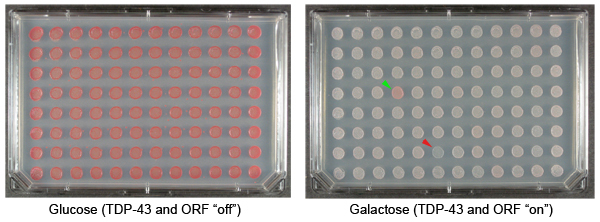 Figure 1. Yeast plasmid overexpression screen to identify suppressors and enhancers of TDP-43 toxicity. TDP-43 is a human protein that has been implicated in the pathogenesis of amyotrophic lateral sclerosis (Lou Gehrig’s disease). Cytoplasmic aggregates of TDP-43 accumulate in the brain and spinal cord neurons of ALS patients 17. Expressing TDP-43 in yeast cells results in aggregation and cytotoxicity 18. We have used this model system to define mechanisms of TDP-43 toxicity 19,20. Shown are repesentative examples of plates from our yeast TDP-43 toxicity modifier screen. These plates display colonies with an integrated galactose-inducible TDP-43 plasmid and also transformed with plasmids from the FLEXGene ORF expression library. The plate on the left contains glucose, which represses expression of TDP-43 or the FLEXGene plasmids. The plate on the right contains galactose, which induces the expression of TDP-43 and the ORFs in the FLEXGene plasmids. The green arrowhead indicates a colony transformed with a plasmid that suppresses the toxicity of TDP-43. The red arrowhead indicates a colony transformed with a plasmid that enhances the toxicity of TDP-43.
Figure 1. Yeast plasmid overexpression screen to identify suppressors and enhancers of TDP-43 toxicity. TDP-43 is a human protein that has been implicated in the pathogenesis of amyotrophic lateral sclerosis (Lou Gehrig’s disease). Cytoplasmic aggregates of TDP-43 accumulate in the brain and spinal cord neurons of ALS patients 17. Expressing TDP-43 in yeast cells results in aggregation and cytotoxicity 18. We have used this model system to define mechanisms of TDP-43 toxicity 19,20. Shown are repesentative examples of plates from our yeast TDP-43 toxicity modifier screen. These plates display colonies with an integrated galactose-inducible TDP-43 plasmid and also transformed with plasmids from the FLEXGene ORF expression library. The plate on the left contains glucose, which represses expression of TDP-43 or the FLEXGene plasmids. The plate on the right contains galactose, which induces the expression of TDP-43 and the ORFs in the FLEXGene plasmids. The green arrowhead indicates a colony transformed with a plasmid that suppresses the toxicity of TDP-43. The red arrowhead indicates a colony transformed with a plasmid that enhances the toxicity of TDP-43.
Discussion
Here we present a protocol to perform a high-throughput plasmid overexpression screen in yeast. This approach allows for the rapid and unbiased screening for genetic modifiers of many different cellular phenotypes. Using this approach, a researcher can screen a significant portion of the yeast genome in a matter of weeks. This unbiased approach also allows for the identification of modifiers, which may not have been predicted based on previous findings. We have used this approach to identify modifiers of toxicity associated with the aggregation of human neurodegenerative disease proteins 19,21-23. However, owing to the adaptability of this protocol to study other cellular processes, this protocol will be useful to researchers addressing a wide range of important biological questions. For example, we have also found this screening approach useful for identifying genes and pathways involved in adaptation to environmental stressors, including heavy metals and oxidative stress. Here we have focused on one plasmid library, the Yeast FLEXGene library 9. However, there are several other yeast plasmid libraries available, which could also be used for these screens 6,8.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work was supported by a grant from the Packard Center for ALS Research at Johns Hopkins (A.D.G.), an NIH Director’s New Innovator Award 1DP2OD004417-01 (A.D.G), NIH R01 NS065317 (A.D.G.), the Rita Allen Foundation Scholar Award. A.D.G. is a Pew Scholar in the Biomedical Sciences, supported by The Pew Charitable Trusts.
References
- Nurse P. The Nobel Prize and beyond: an interview with Sir Paul Nurse. Interview by Susan R. Owens. EMBO Rep. 2002;3:204–206. doi: 10.1093/embo-reports/kvf060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH. Nobel Lecture. Yeast and cancer. Biosci Rep. 2002;22:373–394. doi: 10.1023/a:1020918107706. [DOI] [PubMed] [Google Scholar]
- Stevens T, Esmon B, Schekman R. Early stages in the yeast secretory pathway are required for transport of carboxypeptidase Y to the vacuole. Cell. 1982;30:439–448. doi: 10.1016/0092-8674(82)90241-0. [DOI] [PubMed] [Google Scholar]
- Novick P, Ferro S, Schekman R. Order of events in the yeast secretory pathway. Cell. 1981;25:461–469. doi: 10.1016/0092-8674(81)90064-7. [DOI] [PubMed] [Google Scholar]
- Novick P, Field C, Schekman R. Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell. 1980;21:205–215. doi: 10.1016/0092-8674(80)90128-2. [DOI] [PubMed] [Google Scholar]
- Sopko R. Mapping pathways and phenotypes by systematic gene overexpression. Mol Cell. 2006;21:319–330. doi: 10.1016/j.molcel.2005.12.011. [DOI] [PubMed] [Google Scholar]
- Alberti S, Gitler AD, Lindquist S. A suite of Gateway((R)) cloning vectors for high-throughput genetic analysis in Saccharomyces cerevisiae. Yeast. 2007;24:913–919. doi: 10.1002/yea.1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelperin DM. Biochemical and genetic analysis of the yeast proteome with a movable ORF collection. Genes Dev. 2005;19:2816–2826. doi: 10.1101/gad.1362105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y. Approaching a complete repository of sequence-verified protein-encoding clones for Saccharomyces cerevisiae. Genome Res. 2007;17:536–543. doi: 10.1101/gr.6037607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- Boone C, Bussey H, Andrews BJ. Exploring genetic interactions and networks with yeast. Nat Rev Genet. 2007;8:437–449. doi: 10.1038/nrg2085. [DOI] [PubMed] [Google Scholar]
- Mnaimneh S. Exploration of essential gene functions via titratable promoter alleles. Cell. 2004;118:31–44. doi: 10.1016/j.cell.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Parsons AB. Integration of chemical-genetic and genetic interaction data links bioactive compounds to cellular target pathways. Nat Biotechnol. 2004;22:62–69. doi: 10.1038/nbt919. [DOI] [PubMed] [Google Scholar]
- Schuldiner M. Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell. 2005;123:507–519. doi: 10.1016/j.cell.2005.08.031. [DOI] [PubMed] [Google Scholar]
- Tong AH. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294:2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- Tong AH. Global mapping of the yeast genetic interaction network. Science. 2004;303:808–813. doi: 10.1126/science.1091317. [DOI] [PubMed] [Google Scholar]
- Neumann M. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Johnson BS, McCaffery JM, Lindquist S, Gitler AD. A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc Natl Acad Sci U S A. 2008;105:6439–6444. doi: 10.1073/pnas.0802082105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elden AC. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069–1075. doi: 10.1038/nature09320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BS. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem. 2009;284:20329–20339. doi: 10.1074/jbc.M109.010264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AA. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science. 2006;313:324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler AD. Beer and Bread to Brains and Beyond: Can Yeast Cells Teach Us about Neurodegenerative Disease? Neurosignals. 2008;16:52–62. doi: 10.1159/000109759. [DOI] [PubMed] [Google Scholar]
- Gitler AD. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet. 2009;41:308–315. doi: 10.1038/ng.300. [DOI] [PMC free article] [PubMed] [Google Scholar]