

Abstract
Dynamic live cell imaging allows direct visualization of real-time interactions between cells of the immune system1, 2; however, the lack of spatial and temporal control between the phagocytic cell and microbe has rendered focused observations into the initial interactions of host response to pathogens difficult. Historically, intercellular contact events such as phagocytosis3 have been imaged by mixing two cell types, and then continuously scanning the field-of-view to find serendipitous intercellular contacts at the appropriate stage of interaction. The stochastic nature of these events renders this process tedious, and it is difficult to observe early or fleeting events in cell-cell contact by this approach. This method requires finding cell pairs that are on the verge of contact, and observing them until they consummate their contact, or do not. To address these limitations, we use optical trapping as a non-invasive, non-destructive, but fast and effective method to position cells in culture.
Optical traps, or optical tweezers, are increasingly utilized in biological research to capture and physically manipulate cells and other micron-sized particles in three dimensions4. Radiation pressure was first observed and applied to optical tweezer systems in 19705, 6, and was first used to control biological specimens in 19877. Since then, optical tweezers have matured into a technology to probe a variety of biological phenomena8-13.
We describe a method14 that advances live cell imaging by integrating an optical trap with spinning disk confocal microscopy with temperature and humidity control to provide exquisite spatial and temporal control of pathogenic organisms in a physiological environment to facilitate interactions with host cells, as determined by the operator. Live, pathogenic organisms like Candida albicans and Aspergillus fumigatus, which can cause potentially lethal, invasive infections in immunocompromised individuals15, 16 (e.g. AIDS, chemotherapy, and organ transplantation patients), were optically trapped using non-destructive laser intensities and moved adjacent to macrophages, which can phagocytose the pathogen. High resolution, transmitted light and fluorescence-based movies established the ability to observe early events of phagocytosis in living cells. To demonstrate the broad applicability in immunology, primary T-cells were also trapped and manipulated to form synapses with anti-CD3 coated microspheres in vivo, and time-lapse imaging of synapse formation was also obtained. By providing a method to exert fine spatial control of live pathogens with respect to immune cells, cellular interactions can be captured by fluorescence microscopy with minimal perturbation to cells and can yield powerful insight into early responses of innate and adaptive immunity.
Keywords: Immunology, Issue 53, Optical trapping, optical tweezers, T-cell, pathogen, live cell imaging, spinning disk confocal microscopy, Aspergillus fumigatus, Candida albicans, fungi
Protocol
1. Culture conditions of pathogens for optical trapping
Grow A. fumigatus (B-5233/RGD12-8) on a semi-solid agar media containing SBD (Sabouraud dextrose) at 30°C for 3 days.
Grow C. albicans (SC5314) in YPD (Yeast-Peptone Dextrose) liquid culture containing 100 μg/mL ampicillin overnight in a shaker incubator at 30°C.
2. Preparation of pathogens for fluorescent labeling
Harvest desired amount of pathogens and transfer to a 1.5 mL reaction tube.
Add 300 μL of phosphate buffered saline (PBS) to reaction tube.
Sonicate mixture for 30 seconds.
Centrifuge at 4000 rpm for 1 minute.
Aspirate supernatant, leaving pellet undisturbed.
Repeat (2.4) and (2.5) two more times.
Resuspend in 500μL of PBS.
3. Labeling of pathogens with fluorescent dye
Dissolve 1 mg of dye of interest (e.g. Alexa Fluor 488, Alexa Fluor 647) in 100 μL dimethylformamide (DMF) (concentration of 10 mg/mL).
Add 3 μL of dye mixture to reaction tubes containing washed pathogens.
Rotate or shake sample at 37°C for 1 hour.
Wash sample with PBS 3X by centrifuging at 4000 rpm for 1 minute.
Resuspend in 300 μL of PBS.
4. Harvesting T-cells from whole blood
Obtain whole blood (fresh).
Warm blood, PBS + 2% fetal bovine serum (FBS), and histopaque to room temperature.
Add RosetteSep Human CD4+ T Cell Enrichment Cocktail at 50 μL/mL of whole blood.
Rotate sample and incubate for 20 minutes at room temperature.
Dilute sample with an equal volume of PBS + 2% FBS and mix gently.
Layer diluted sample on top of histopaque, minimizing mixing
Centrifuge for 20 minutes at 1200 x g at room temperature with the brake off.
Remove the enriched cells.
Wash the enriched cells 2x with PBS + 2% FBS solution.
Lyse red blood cells for 2 minutes with red blood cell lysing buffer
Add 10 mL of PBS + 2% FBS and centrifuge lysed red blood cells at 1500 rpm for 5 minutes
Aspirate supernatant, careful not to disturb the pellet
Resuspend in IMDM media containing 10% fetal bovine serum
5. Preparation of RAW 264.7 macrophages into chamber slides
Prepare DMEM (Dulbecco's modified Eagle's medium) to contain 10% FBS, 1% penicillin/streptomycin, and 1% L-glutamine.
Warm media, trypsin, and PBS in warm bath to 37°C.
Wash plate 2x with PBS
Aspirate PBS between each wash.
Add 5 mL trypsin to plate to cover surface (for a 10 cm tissue culture plate).
Incubate for 5 min at 37°C.
Gently knock side of plate to detach cells from plate surface. Be careful not to splash trypsin outside of the plate.
Add 5 mL or equivalent amount of media to trypsin.
Aspirate mixture into a reaction tube.
Centrifuge at 1000 x g for 3 minutes.
Aspirate media, careful not to disturb the pellet.
Resuspend in 10 mL of media.
Add 400 μL of media to each chamber of the chamber slide.
Add 5 μL of cell suspension to each chamber.
Grow overnight in incubator at 37°C with 5% CO2.
6. Addition of pathogens to sample
Pipet 5-10 μL of labeled pathogens of interest (from steps 1-3) into chamber.
Mix thoroughly by pipeting up and down, careful not to touch the bottom of the chamber to disturb the adhered macrophages.
7. Loading sample onto spinning disk confocal microscope (in video, scan through components)
Turn on all components to spinning disk confocal microscope.
Align microscope for DIC imaging.
Remove chamber slide from incubator.
Insert chamber slide into specialized stage.
Remove top of chamber slide (necessary for DIC imaging).
8. Preparation for optical trapping (in video, scan through components and how it's integrated into microscope)
Turn on shutter for optical trap.
Turn on IR laser.
Open shutter (on the laser) to optical trap.
Confirm shutter in front of IR laser is closed by checking with IR card.
9. Selection and manipulation of pathogen with optical trap
Focus on macrophages on adhered slide.
Find pathogens freely fluctuating in solution adjacent to macrophages.
Move stage such that the pathogen is in the vicinity of the trap.
Open shutter and engage the trap.
Move sample to bring macrophages into contact with the stationary trapped pathogen.
Image with spinning disk confocal microscope, either in DIC, fluorescence, or combination of both. Typically, trapping is done in DIC, and real-time imaging is done with fluorescence.
10. Representative Results:
To exert full spatial and temporal control of pathogens and beads in an in vivo and in vitro environment, we designed a custom-built trapping apparatus integrated onto a spinning disk confocal microscope (schematic shown in Fig. 1). At full strength, the laser provided 350 mW of power and after coupling the light with the various optical components in the optical trap apparatus, ˜80 mW of power at the TIRF objective, as measured with a power meter, was used to form the trap in the chamber.
In order to position an object in the chamber relative to the trapped object, the stage was moved while holding the trapped object stationary with the trapping laser. The stage was moved at slow enough velocities, so that the drag force on the trapped particle did not exceed the maximum trapping force.
Separate populations of C. albicans (typical size - ˜5 μm) were labeled with each of three colors (AF488, AF568, and AF647, corresponding to green, blue, and red in the figure, respectively) to illustrate imaging with multiple fluorescence channels while simultaneously optically trapping the pathogen. A single C. albicans was trapped and moved in a square pattern through a cluster of other yeast, as indicated by gray arrows, demonstrating the ability to capture and manipulate the specific location of a single pathogen chosen by the operator even in a crowded environment (Fig. 2A).
To illustrate further the flexibility of this system to trap the different shape morphologies exhibited by pathogenic organisms, the optical tweezer was also able to hold and situate a C. albicans particle with a pseudohyphae. The C. albicans labeled with AF647 (red) was moved along a trajectory as outlined by the white arrow and placed next to fluorescent GFP-LC3-RAW cells (Fig. 2B). The yeast portion of the C. albicans was trapped as the pseudohyphae trailed along.
We also positioned Aspergillus fumigatus next to a RAW mouse macrophage cell in order to analyze the absolute time frame of phagocytosis with this particular cell line and pathogen (Fig. 3A). Once the pathogen makes contact with the cell, the trap is turned off, and time-lapse imaging is employed to observe the subsequent cellular events (Fig. 3B). The optical trap was also used to capture primary T-cells isolated from blood and directed adjacent to beads coated with anti-CD3 antibody in order for the T-cell to form an immunological synapse with the bead (Fig. 4), show the additional versatility to directly trap and manipulate immune cells.
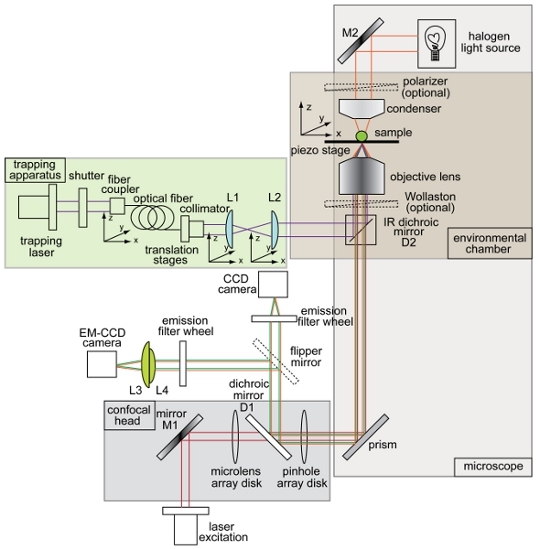 Figure 1. Overview and schematic of combined optical trap and spinning disk confocal microscope setup. Instrument layout showing the trapping beam (purple), illumination path for brightfield imaging (orange), fluorescence excitation beam (red), fluorescence emission (green), charge-coupled device (CCD) camera, electron-multiplying charge coupled device (EM-CCD) camera, dichroic mirrors (D1 and D2), mirrors (M1 and M2), and lenses (L1, L2, L3, L4). All other components of the trap-confocal microscope system are labeled in the figure.
Figure 1. Overview and schematic of combined optical trap and spinning disk confocal microscope setup. Instrument layout showing the trapping beam (purple), illumination path for brightfield imaging (orange), fluorescence excitation beam (red), fluorescence emission (green), charge-coupled device (CCD) camera, electron-multiplying charge coupled device (EM-CCD) camera, dichroic mirrors (D1 and D2), mirrors (M1 and M2), and lenses (L1, L2, L3, L4). All other components of the trap-confocal microscope system are labeled in the figure.
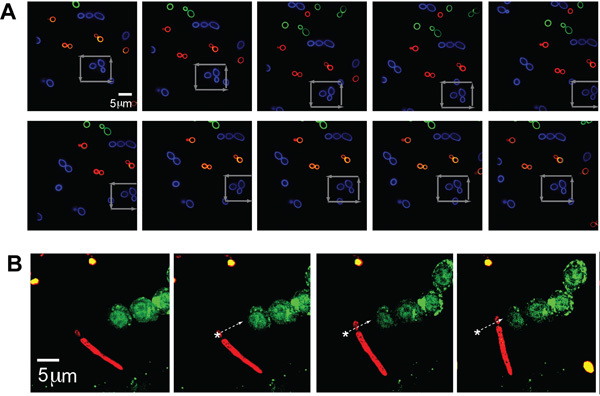 Figure 2. (A) Fluorescence images of trapped and manipulated C. albicans. A trapped fluorescently-labeled (Alexa Fluor 488 (AF488), blue) organism in a field of other fluorescently-labeled C. albicans (AF568, green and AF647, red). The stage is moved around the trapped, blue CA particle as indicated by the gray arrows. (B) Trapped pseudo-hyphal form of C. albicans next to GFP-LC3 RAW cell. Fluorescently labeled psuedohyphal form of CA (red, AF647) optically trapped and moved adjacent to GFP-LC3 expressed RAW macrophages. The CA organism is moved along the trajectory as indicated by the white arrow.
Figure 2. (A) Fluorescence images of trapped and manipulated C. albicans. A trapped fluorescently-labeled (Alexa Fluor 488 (AF488), blue) organism in a field of other fluorescently-labeled C. albicans (AF568, green and AF647, red). The stage is moved around the trapped, blue CA particle as indicated by the gray arrows. (B) Trapped pseudo-hyphal form of C. albicans next to GFP-LC3 RAW cell. Fluorescently labeled psuedohyphal form of CA (red, AF647) optically trapped and moved adjacent to GFP-LC3 expressed RAW macrophages. The CA organism is moved along the trajectory as indicated by the white arrow.
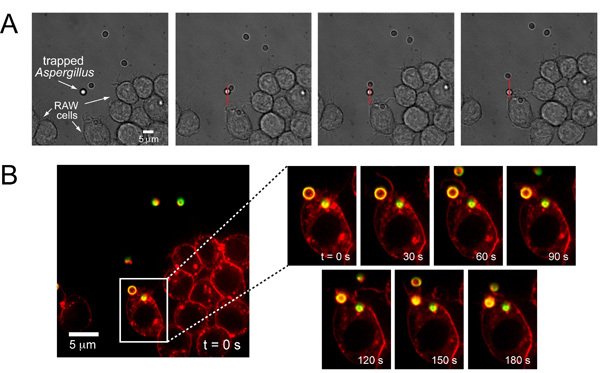 Figure 3. Trapping and positioning of A. fumigatus next to a phagocytic RAW cell. (A) Brightfield images of a trapped A. fumigatus, as indicated by the white arrow, moved and positioned along the path as indicated by the red arrow. The trapped pathogen is slightly out of focus due to the trap pushing the organism slightly above the focal plane. A. fumigatus is moved until it is placed adjacent to the desired RAW cell. (B) Fluorescence imaging of phagocytosis of A. fumigatus by RAW cell. After the trapped pathogen is placed next to a RAW cell, the phagocytosis process is activated. At 30 s, the membrane of the RAW cell starts to change and form a cup around the particle. At 60 s, the cup is fully formed. From 90s to 150s, the A. fumigatus is engulfed, and by 180 s, the particle is fully internalized
Figure 3. Trapping and positioning of A. fumigatus next to a phagocytic RAW cell. (A) Brightfield images of a trapped A. fumigatus, as indicated by the white arrow, moved and positioned along the path as indicated by the red arrow. The trapped pathogen is slightly out of focus due to the trap pushing the organism slightly above the focal plane. A. fumigatus is moved until it is placed adjacent to the desired RAW cell. (B) Fluorescence imaging of phagocytosis of A. fumigatus by RAW cell. After the trapped pathogen is placed next to a RAW cell, the phagocytosis process is activated. At 30 s, the membrane of the RAW cell starts to change and form a cup around the particle. At 60 s, the cup is fully formed. From 90s to 150s, the A. fumigatus is engulfed, and by 180 s, the particle is fully internalized
 Figure 4. Trapping primary T-cell to form synapse with anti-CD3 coated beads. DIC images of a primary T-cell moved by the optical trap to a position next to a bead coated with anti-CD3 antibody. The cell then forms an immunological synapse, which cannot be easily discerned in the images.
Figure 4. Trapping primary T-cell to form synapse with anti-CD3 coated beads. DIC images of a primary T-cell moved by the optical trap to a position next to a bead coated with anti-CD3 antibody. The cell then forms an immunological synapse, which cannot be easily discerned in the images.
Discussion
In this work we use an optical trap to capture pathogens with dimensions between 3 μm - 5 μm. Although pathogens of interest to our lab typically have these dimensions, the optical tweezer system described here is flexible to trap a large range of sizes. Indeed optical traps have been used to capture particles ranging from single atoms to cells approximately 10 μm in diameter. Additionally, this optical trapping system was able to capture particles of various shapes: spherical, elliptical, and extremely elongated particles, which is useful when working with biological pathogens.
With this method, phagocytosis and synapse formation, in real-time, were investigated, and structural changes on the cellular surface was analyzed. By positioning particles next to a cell, live pathogens were controlled and manipulated to activate the phagocytic machinery and study the immune response in living cells and monitor the absolute beginning of an event through the entire course of phagocytosis. As a result, the entire evolution of phagocytic events and machinery can be accurately measured. We followed phagocytosis of A. fumigatus as the pathogen is placed next to a macrophage, and watched the initial membrane changes as the particle was engulfed. We were able to accurately measure the entire time frame of this process by controlling when we wanted the process to start with the placement of the pathogen. This technique was also extended to observing synapse formation by primary T-cells. The optical tweezer moved a T-cell next to an anti-CD3 antibody coated bead, and synapse formation can be observed in real time.
To our knowledge, this method is the first to control and manipulate a pathogen next to a phagocytic cell, and in conjuction with confocal microscopy, was used to record and visualize the absolute beginning and end of phagocytosis in three dimensions with diffraction-limited spatio-temporal resolution. The technology was also utilized to control and manipulate primary T-cells to study immunological synapses, illustrating the versatility of this instrument for a variety of biological applications. Our system provides five excitation wavelengths, including UV excitation, enabling imaging of a variety of fluorophores. The trapping apparatus occupies a 4.5 ft2 footprint, able to be integrated on many conventional epifluorescent and confocal microscopes. Although this instrument was designed mainly for object manipulation, future modifications would include higher laser power, bead position detection capabilities, etc. would enable characterization of biomechanical forces in the cell-pathogen interaction.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work was supported by Massachusetts General Hospital Department of Medicine Internal Funds (J.M.T., M.K.M, M.L.C., J.M.V.), National Institute of Biomedical Imaging and Bioengineering grant T32EB006348 (C.E.C.), Massachusetts General Hospital's Center for Computational and Integrative Biology development fund and AI062773 (R.J.H.), grants AI062773, DK83756, and DK 043351 (R.J.X.), NSF 0643745 (M.J.L.), NIH R21CA133576 (M.J.L.), and National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (NIH) AI057999 (J.M.V.). We thank Nicholas C. Yoder for helpful discussions, and Charles Felts (RPI, Inc.) for technical assistance.
References
- Grakoui A. The immunological synapse: A molecular machine controlling T cell activation. Science. 1999;285:221–227. [PubMed] [Google Scholar]
- Monks CR. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. 1998;395:82–86. doi: 10.1038/25764. [DOI] [PubMed] [Google Scholar]
- Stuart LM, Ezekowitz RA. Phagocytosis and comparative innate immunity: Learning on the fly. Nat Rev Immunol. 2008;8:131–141. doi: 10.1038/nri2240. [DOI] [PubMed] [Google Scholar]
- Neuman KC, Block SM. Optical trapping. Rev Sci Instrum. 2004;75:2787–2809. doi: 10.1063/1.1785844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkin A. Optical trapping and manipulation of neutral particles using lasers. Proc Natl Acad Sci USA. 1997;94:4853–4860. doi: 10.1073/pnas.94.10.4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkin A. Acceleration and trapping of particles by radiation pressure. Phys Rev Lett. 1970;24:156–159. [Google Scholar]
- Ashkin A, Dziedzic J. Optical trapping and manipulation of viruses and bacteria Science. Nature. 1987;235:1517–1520. doi: 10.1126/science.3547653. [DOI] [PubMed] [Google Scholar]
- Khalil AS. Single M13 bacteriophage tethering and stretching. Proc Natl Acad Sci USA. 2007;104:4892–4897. doi: 10.1073/pnas.0605727104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil AS. Kinesin's cover-neck bundle folds forward to generate force. Proc Natl Acad Sci USA. 2008;105:19247–19252. doi: 10.1073/pnas.0805147105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z. Membrane tether formation from outer hair cells with optical tweezers. Biophys J. 2002. pp. 1386–1395. [DOI] [PMC free article] [PubMed]
- Kim S. The αβ T cell receptor is an anisotropic mechanosensor. J Biol Chem. 2009;284:31028–31028. doi: 10.1074/jbc.M109.052712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanty S, Mohanty K, Gupta P. Dynamics of interaction of RBC with optical tweezers. Opt. Express. 2005;13:4745–4751. doi: 10.1364/opex.13.004745. [DOI] [PubMed] [Google Scholar]
- Tam J. Control and manipulation of pathogens with an optical trap for live cell imaging of intercellular interactions. PLoS One. 2010;5:e15215–e15215. doi: 10.1371/journal.pone.0015215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SJ, Schranz J, Teutsch SM. Aspergillosis case-fatality rate: Systematic review of the literature. Clin Infect Dis. 2001;32:358–366. doi: 10.1086/318483. [DOI] [PubMed] [Google Scholar]
- Wey SB. Hospital-acquired candidemia - the attributable mortality and excess length of stay. Arch. Intern. Med. 1988;148:2642–2645. doi: 10.1001/archinte.148.12.2642. [DOI] [PubMed] [Google Scholar]