

Abstract
Using small molecule probes to understand gene function is an attractive approach that allows functional characterization of genes that are dispensable in standard laboratory conditions and provides insight into the mode of action of these compounds. Using chemogenomic assays we previously identified yeast Crg1, an uncharacterized SAM-dependent methyltransferase, as a novel interactor of the protein phosphatase inhibitor cantharidin. In this study we used a combinatorial approach that exploits contemporary high-throughput techniques available in Saccharomyces cerevisiae combined with rigorous biological follow-up to characterize the interaction of Crg1 with cantharidin. Biochemical analysis of this enzyme followed by a systematic analysis of the interactome and lipidome of CRG1 mutants revealed that Crg1, a stress-responsive SAM-dependent methyltransferase, methylates cantharidin in vitro. Chemogenomic assays uncovered that lipid-related processes are essential for cantharidin resistance in cells sensitized by deletion of the CRG1 gene. Lipidome-wide analysis of mutants further showed that cantharidin induces alterations in glycerophospholipid and sphingolipid abundance in a Crg1-dependent manner. We propose that Crg1 is a small molecule methyltransferase important for maintaining lipid homeostasis in response to drug perturbation. This approach demonstrates the value of combining chemical genomics with other systems-based methods for characterizing proteins and elucidating previously unknown mechanisms of action of small molecule inhibitors.
Author Summary
Chemical genetics uses small molecules to perturb biological systems to study gene function. By analogy with genetic lesions, chemical probes act as fast-acting, reversible, and “tunable” conditional alleles. Furthermore, small molecules can target multiple protein targets and target pathways simultaneously to uncover phenotypes that may be masked by genes encoding partially redundant proteins. Finally, potent chemical probes can be useful starting points for the development of human therapeutics. Here, we used cantharidin, a natural toxin, to uncover otherwise “hidden” phenotypes for a methyltransferase that has resisted characterization. This enzyme, Crg1, has no phenotype in standard conditions but is indispensible for survival in the presence of cantharidin. Using this chemical genetic relationship, we characterized novel functions of Crg1, and by combining diverse genomic assays with small molecule perturbation we characterized the mechanism of cantharidin cytotoxicity. These observations are relevant beyond yeast Crg1 because cantharidin and its analogues have potent anticancer activity, yet its therapeutic use has been limited to topical applications because of its cytotoxicity. Considering that methyltransferases are an extremely abundant and diverse class of cellular proteins, chemical probes such as cantharidin are critical for understanding their cellular roles and defining potential points of therapeutic intervention.
Introduction
Methyltransferases are a large class of enzymes comprising 0.6–1.6% of protein coding genes in most sequenced organisms [1]. S-adenosyl methionine (SAM)-dependent methyltransferases regulate a dynamic network of cellular signaling events and are required to maintain intracellular homeostasis in the face of external perturbations by catalyzing the methylation of a wide variety of substrates (proteins, nucleic acids, lipids and small molecules) [2]–[4]. The characterization and understanding of the roles of most methyltransferases remains challenging, however, due to their dispensability in standard growth conditions. Numerous studies from our lab and others have demonstrated that chemogenomic profiling of the Saccharomyces cerevisiae yeast deletion collection [5] is a powerful approach for the identification and subsequent characterization of genes required for growth in the presence of bioactive compounds [6]–[15]. Moreover, while most yeast genes (∼80%) are dispensable for growth in standard laboratory conditions, the presence of chemical and/or environmental perturbations of the cell, 97% of the yeast genome exhibits a fitness defect that would not otherwise have been revealed [15].
Well-established chemogenomic assays in yeast, such as drug-induced Haploinsufficiency Profiling (HIP), Homozygous Profiling (HOP) and Multicopy Suppression Profiling (MSP) are designed to identify small molecule-gene interactions. For example, HIP assay is used to detect compounds that target essential genes, and HOP and MSP are suitable for identification genetic modifiers of drug resistance [8]–[10], [13]. The combination of these chemogenomic assays allowed us to identify a novel gene, YHR209W, that we subsequently named CRG1 (Cantharidin Resistance Gene 1), due to its requirement for growth in the presence of the small molecule cantharidin [14]. Specifically, both CRG1 heterozygous and homozygous deletion strains exhibited sensitivity to the drug, and the overexpression of CRG1 conferred resistance to the drug. Nonetheless, Crg1 is uncharacterized, except for annotation derived from large-scale analyses [15]–[17].
Based on its primary sequence, Crg1 is predicted to encode a Class I S-adenosyl-methionine (SAM)-dependent methyltransferase [18]. Crg1 shares close sequence homology with trans-aconitate methyltransferase Tmt1 (BLAST-P expect value 2×10−31 and 3×10−34 for the full proteins and the methyltransferase domains, respectively). Tmt1 is known to modify and detoxify small molecules by methylation [19]–[21]. We have previously shown indirectly that Crg1 does not likely possess Tmt1 methyltransferase activity towards trans-aconitate, 3-isopropylmalate, and isopropylmaleate, indicating that these closely related proteins have divergent substrates [19], [21]. Bioinformatics analysis from our group has shown, however, that Crg1 clusters with a family of eight methyltransferases based on their methyl-accepting substrate specificity, including Tmt1, the lipid methyltransferases (Coq3, Coq5, and Erg6), and a tRNA methyltransferase, Trm9 [22]. All of these proteins methylate carboxylic acids present in small molecules to form methyl esters, suggesting that Crg1 might have a similar biochemical activity and catalyze the formation of a methyl ester.
Cantharidin, a natural product produced by Chinese blister beetles of the Meloidae family of Coleoptera, is used in Traditional Chinese Medicine for the treatment of a variety of cancers [23]. Cantharidin has potent anticancer activity characterized by cell cycle arrest in G2/M phase, apoptosis, and DNA damage, presumably as a result of the generation of reactive oxygen species [23]–[29], yet its use is limited due to renal and mucous membrane toxicity. Although the activity of cantharidin is usually attributed to its high affinity towards Type 1 and 2A serine/threonine protein phosphatases [30], [31], several studies suggest that cantharidin has additional cellular targets. Specifically, cantharidin has been reported to stimulate xanthine oxidase activity and to inhibit N-acyltransferase and cAMP phosphodiesterase in liver cells, suggesting a complex mode of action [32]–[34]. Using the HIP and HOP genome-wide assays, we discovered that a surprisingly large number of methyltransferase deletion mutants are sensitive to cantharidin, suggesting that, as a class, these enzymes may interact directly or indirectly with cantharidin and participate in the response to cantharidin stress [15]. Notably, among these methyltransferases only the overexpression of CRG1 is able to confer resistance to cantharidin.
To further explore the function of Crg1 and the mechanism of cantharidin cytotoxicity, we employed chemical genomics tools combined with conventional biological techniques. We demonstrated that Crg1 methylates cantharidin in vitro, and identified cantharidin-specific CRG1 genetic interactors. To extend our chemogenomic results we analyzed the lipid profile of mutants grown in the presence of cantharidin, and demonstrated that cantharidin resistance involves Crg1-dependent maintenance of lipid homeostasis.
Results
CRG1 Is a Functional Methyltransferase Required for Protein Phosphatase Inhibitor Resistance
To confirm our published cantharidin-specific response of CRG1 [14], we measured the growth of three strains 1) wild-type diploid strain BY4743, 2) a crg1 Δ/Δ homozygous deletion strain, and 3) a crg1Δ/Δ homozygous deletion strain overexpressing CRG1 (2 µ plasmid) as a function of cantharidin concentration. We observed that the gene dosage of the putative SAM-dependent methyltransferase CRG1 correlated with the sensitivity/resistance of these strains to cantharidin (Figure 1A). In agreement with this gene-dose dependent effect, crg1Δ/CRG1 heterozygous mutants grew worse than the wild-type strain but better than a crg1Δ/Δ homozygous mutant in the presence of cantharidin (500 µM) (Figure S1A). We found that cantharidin is more potent against cells grown in synthetically defined (SD) medium than in YPD medium (5 µM and 250 µM, IC20 for wild-type in SD and YPD, respectively; Figure S1B). The observed differential drug sensitivity in defined media and rich YPD media is a common phenomenon in our drug screens (unpublished data). We also tested structural analogues of cantharidin, including cantharidic acid and norcantharidin, and found that these compounds produced a similar gene-dose dependent response in crg1 mutants (Figure S1C).
Figure 1. Functional SAM-dependent methyltransferase Crg1 is required for cantharidin response.

(A) CRG1 gene dose is important for cantharidin tolerance. Wt, crg1Δ/Δ and CRG1-overexpressing crg1Δ/Δ mutants were assessed in the presence of cantharidin in YPD. Dose-response curves were obtained by plotting OD600 at saturation point vs. tested drug concentrations. (B) Chemical (left panel) and genetic inhibition (right panel) of protein phosphatases result in the induction of CRG1. Wt, glc7Δ/GLC7 heterozygous, sit4Δ/Δ and ptc1Δ/Δ homozygous deletion mutants grown to mid-exponential phase were incubated with or without cantharidin (250 µM). For each time point, total RNA was extracted, cDNA synthesized and the relative abundance of CRG1 transcript was analyzed by qRT-PCR. Data are the mean of at least three independent experimental replicates, and error bars represent the standard deviation. (C) Point mutations in conserved residues of the Crg1 methyltransferase domain reduce cantharidin tolerance. Site-specific mutations in the conserved motifs of methyltransferase domain are shown with the arrows. Mutated CRG1 ORFs were cloned under GAL1 promoter and transformed into crg1Δ/Δ cells. The transformants were grown in SD-URA with raffinose (2%) to mid-exponential phase and induced with galactose (2%). The fitness of point mutants based on saturation at final OD was assessed in the presence of cantharidin (6 µM). (D) A plot comparing the transcriptome profiles of crg1Δ/Δ and CRG1-overexpressing crg1Δ/Δ mutants. Exponentially grown cells were treated with cantharidin (250 µM) for 1 hour or DMSO, total RNA was extracted and synthesized cDNA was hybridized to Affymetrix Tiling arrays. Significantly different GO Biological processes are listed in Table S1. (E) Diagram showing that the methionine biosynthesis is tightly linked to SAM cycling in methylation reactions coordinated by methyltransferases. The genes that are transcriptionally different between cantharidin-resistant and sensitive mutant are shown in red. STR3 (P-value <0.0057), MET17 (P-value <0.0036), MET6 (P-value <0.012), SAM1 (P-value <0.02), SAH1 (P-value <0.03), MMP1 (P-value <0.04), SAM2 (P-value <0.056) (Table S2).
Because our data suggested that CRG1 responds to cantharidin in a gene dose-dependent manner, we next tested whether the transcription of CRG1 is induced in the presence of the drug. qRT-PCR analysis showed that the relative abundance of CRG1 transcripts increased drastically in the wild-type strain after 60 min of the drug treatment (250 µM) compared to the DMSO control (P-value <0.02; Figure 1B, left panel). Importantly, a gene-dose dependent effect in the response to cantharidin was also observed for CRG1 transcript levels in crg1Δ/CRG1 heterozygous and CRG1-overexpressing mutants (Figure S2A). In agreement with the qRT-PCR data, we also observed induction of Crg1 at the protein level. GFP-tagged Crg1 protein increased from undetectable levels prior to treatment and accumulated to high levels (restricted to the cytoplasm) following 1 hour of cantharidin treatment (Figure S1B).
Given the known high affinity of cantharidin towards Type 2A protein phosphatases (PP2A) and to a lesser degree towards Type 1 (PP1) [30], [31], we tested if CRG1 induction was mediated by chemical inhibition of protein phosphatase function. We phenocopied cantharidin treatment using a panel of protein phosphatase homozygous deletion strains. Consistent with the results of chemical inhibition of protein phosphatases with cantharidin, we found that the homozygous deletion strains sit4Δ/Δ (PP2A), ptc1Δ/Δ (PP2C) and the heterozygous deletion strain glc7Δ/GLC7 (PP1) also resulted in transcriptional upregulation of CRG1 in the absence of cantharidin (Figure 1B, right panel). It is important to note that perturbation of these protein phosphatases accounted for only ∼20% of the transcript induction observed by cantharidin. Furthermore, the treatment with calyculin A, a structurally distinct PP2 and PP1 inhibitor [35], known to interact with the yeast PP1 GLC7 [14], resulted in an increase of CRG1 transcript level to a similar degree as in glc7Δ/GLC7 mutant (∼2.5 fold; Figure S2C). This observation opens up the possibility that cantharidin acts independently of this PPase. This hypothesis is also supported by our observation that overexpression of GLC7 confers resistance to calyculin A, but not to cantharidin [14]. These results also suggest that these protein phosphatases are likely to be negative regulators of the cellular pathway regulating CRG1 induction.
We also observed that cantharidin-induced transcription of CRG1 follows a temporal pattern characteristic of diverse environmental stress responses [36], following a peak at 60 min of treatment the transcript levels began to decrease at 120 min (∼40 fold, P-value<0.01; Figure 1B). Indeed, a comprehensive genome-wide analysis of diverse environmental stresses from publicly available expression data [36] revealed that the transcription profile of CRG1 in diverse stress conditions correlates highly (r = 0.8) with a well-characterized stress-responsive gene, the heat shock protein SSE2 (Figure S2D), suggesting that CRG1 is also transcriptionally activated by other stress conditions in addition to cantharidin.
Because CRG1 is annotated (based on its amino acid sequence) as a putative SAM-dependent methyltransferase [18], we next asked whether its methyltransferase domain is required for cantharidin tolerance by mutating amino acids (D44A, D67A, E105A-D108A) within the conserved motifs (Figure 1C). These amino acids have previously been shown to be critical for activity of other methyltransferases [37]. Overexpression of these crg1 site-specific mutants in a crg1Δ/Δ strain failed to confer cantharidin resistance while, in contrast, mutation of a non-conserved residue (G96A) in the methyltransferase domain showed resistance equivalent to wild-type CRG1 (Figure 1C). The observed decrease in resistance to cantharidin was not due to reduced expression of the mutated Crg1 proteins (Figure S2E), suggesting that the methyltransferase domain of Crg1 is both functional and important for cellular survival in the presence of the drug.
To identify other potential cellular factors important for Crg1-mediated cantharidin resistance, we profiled the complete yeast transcriptome using whole-genome tiling microarrays. Transcriptional changes in wild type, crg1Δ/Δ deletion and CRG1-overexpressing crg1Δ/Δ strains were analyzed after 1 hour of exposure to the drug. To ensure that the transcriptome datasets for the different strains are comparable, the IC20 for wild type (250 µM) was applied to all strains. Even at this high dose, the hypersensitive crg1Δ/Δ strain is viable, after 1 hour of exposure (Figure S3A). When applied for an extended period, this dose is, in fact, inhibitory for growth of crg1Δ/Δ strains (Figure 1A and Figure S1A). Furthermore, the treatment of crg1Δ/Δ strain with a lower dose (30 µM, the IC20 for this mutant) resulted in quantitative difference in the transcriptome profile rather than in any qualitative differences, suggesting that the transcriptional changes are consistent across a range of concentrations. In particular, this observation was relevant to downregulated genes (Figure S3B and S3C). It is also worth noting that the expression of most genes was not affected by crg1 mutation. To uncover cantharidin-specific genes in our transcriptome analysis, we eliminated Environmental Stress Response (ESR) genes known to be activated by a large number of stresses, such as genes required for vacuole biogenesis, response to stress, ribosome biogenesis, and RNA processing [36]. We also eliminated those genes that did not demonstrate at least two-fold difference in the presence of cantharidin or if their differential expression failed to show statistical significance. To detect genes and biological processes that are differentially expressed among the strains and treatments, the enrichment of genes for Gene Ontology (GO) term Biological process in the transcriptomes of wild type, crg1Δ/Δ and CRG1-overexpressing crg1Δ/Δ strains in the presence and absence of cantharidin were compared (Table S1). We also clustered genes according to their expression pattern. The clustering and GO term comparative analysis revealed that significantly downregulated genes (log2 (drug/DMSO) <−1, P-value <0.05) in crg1Δ/Δ mutant and the wild type were enriched for the genes of amino acid process (multiple-testing corrected P-value <1.0×10−7 and P-value <7.0×10−7, respectively), while the transcriptional profile of cantharidin-resistant CRG1-overexpressing crg1Δ/Δ strain did not demonstrate a similar enrichment (Figure 1D and Figure S3D). Of particular interest, most of the genes that comprise methionine biosynthetic process (MET6, MET17, MMP1, STR3, ADE3, SAM1, SAM2, SAH1, MET22, MET31) were differentially expressed between cantharidin-resistant CRG1-overexpressing crg1Δ/Δ, wild type and cantharidin-sensitive crg1Δ/Δ strain in the presence of cantharidin (P-value <0.05; Table S2; Figure 1E). One noteworthy example is STR3, a cystathionine beta-lyase, the gene that demonstrated the most differential expression in the strains. STR3 was significantly induced by cantharidin in CRG1-overexpressing crg1Δ/Δ strain (log2 (drug/DMSO) = 4.7, P-value <0.022) and downregulated in the wild type (log2 = −0.6) and crg1Δ/Δ (log2 = −0.55). The observed differential expression of STR3 was further confirmed by qRT-PCR (Figure S3F). Str3 is of interest because it functions in methionine biosynthesis by converting cystathionine into homocysteine, a precursor for methionine, which is a substrate for the generation of SAM. SAM is required as a methyl donor for methylation reactions (Figure 1E). To further explore the role of the Crg1 SAM-dependent methyltransferase, we treated wild-type cells with a combination of cantharidin and S-adenosyl homocysteine (SAH), a non-specific methyltransferase inhibitor. We found that wild-type strains were more sensitive to the cantharidin/SAH combination compared to either single agent (Figure S3F). These observations confirm the requirement of SAM-dependent methyltransferase activity in response to cantharidin, and suggest that Crg1 is a functional methyltransferase that catalyzes a SAM-dependent methylation reaction important for cantharidin resistance.
Crg1 Methylates Cantharidin In Vitro
Given that CRG1 provides cantharidin resistance in a gene dose-dependent manner and because of its close sequence homology to TMT1, a small molecule methyltransferase that catalyzes the formation of methyl esters (Figure 2A), we hypothesized that Crg1 might methylate cantharidin because this drug bears some structural similarity to the substrates of Tmt1 (Figure 2B). To test this possibility, we performed in vitro biochemical assays with purified Crg1, cantharidin, and S-adenosyl-[methyl-14C]methionine (Figure 3A). These in vitro reactions were separated via reverse phase liquid chromatography and the radioactivity of the collected fractions was quantified with a scintillation counter. We detected a unique peak of radioactivity eluting in the 18–20 min fraction (Figure 3B). The appearance of this peak was both cantharidin and Crg1-dependent, suggesting that it could correspond to methylated cantharidin.
Figure 2. Small molecule methyltransferase TMT1 is a sequence homolog of Crg1.

(A) Tmt1, a sequence homolog of Crg1, modifies small molecule intermediates of TCA cycle (trans-aconitate) and leucine biosynthesis (3-isopropylmalate and isopropylmaleate) to form methyl esters. (B) Hypothetical methylation of cantharidin by Crg1.
Figure 3. Crg1 methylates cantharidin in vitro.

(A) Silver stained 12% SDS-PAGE of purified His-tagged Crg1. Wild-type cells (Y258) carrying empty vector BG1805 and BG1805-GAL1-CRG1 were grown in SD-URA and raffinose (2%) to mid-exponential phase. The expression of CRG1 was induced with galactose (2%) for 5 hours. His-tagged Crg1 was purified with Ni-sepharose resin. The diagram (right panel) shows the transfer of [methyl-14C] from S-adenosyl-[methyl-14C]methionine to a potential substrate by purified Crg1. (B) In vitro enzymatic reaction mixtures containing cantharidin, S-adenosyl-[methyl-14C]methionine, and Crg1 were separated by reverse phase chromatography. Radioactivity in the fractions was quantified with a scintillation counter, and a cantharidin and Crg1-dependent peak with a retention time of 18–20 minutes was identified (asterisk). (C) In vitro analysis of the reactions containing cantharidin and mutated forms of Crg1 by measurement of the amount of acid-labile volatile radioactivity (see Materials and Methods for details). (D) Additional in vitro reaction with unlabeled SAM were prepared in a similar manner and analyzed by liquid chromatography-mass spectrometry with positive ionization. The mass spectrum of the peak from the full reaction with an elution time of 18.6–18.8 minutes is shown. (E) The mass spectrum of the peak with an elution time of 19.2 minutes from the complete reaction with cantharidin, SAM and Crg1.
To confirm that the novel activity was catalyzed by Crg1 rather than by a co-purifying protein, we repeated the methylation reactions with mutant forms of Crg1 containing amino acid substitutions at critical residues within the methyltransferase domain. As described earlier, the D44A and E105A-D108A mutations abolished resistance to cantharidin (Figure 1C), so we assessed whether these mutated proteins were able to methylate the drug molecule. We prepared in vitro reactions containing varying concentrations of cantharidin and quantified the amount of acid-labile volatile radioactivity because methyl esters are known to readily hydrolyze in both strongly acidic and basic conditions to yield methanol [19], [38]. Unlike the reactions performed with wild-type Crg1, addition of cantharidin to the reactions with mutant forms of the enzyme showed no increase in acid-labile radioactivity (Figure 3C), strongly suggesting that a functional methyltransferase domain in Crg1 is required for cantharidin methylation.
To definitively determine whether cantharidin is a substrate of Crg1, we prepared and analyzed unlabeled reactions containing purified Crg1, cantharidin, and SAM by liquid chromatography-mass spectrometry. We first looked at the extracted ion chromatogram expected for unreacted cantharidin (C10H13O4 +; m/z = 197.0814±100 ppm) and found a large peak in cantharidin-containing reactions with an elution time of 18.6–18.8 min (Figure S4B). The combined spectra of this peak in the complete reaction mixture contained several species: m/z = 197.0942 and 215.1065, corresponding to the m/z for cantharidin and hydrated cantharidin (C10H15O5 +; m/z = 215.0919), respectively (Figure 3D). Because these species co-elute at 18.6–18.8 min in the complete reaction and the control reactions containing cantharidin (Figure S4B and S4C) and because sterically hindered anhydrides do not favor hydrolysis [39], a likely explanation is that cantharidin forms a co-eluting water adduct during ionization.
Next, we analyzed the full reaction to determine whether Crg1 methylates cantharidin. Specifically, we analyzed the extracted ion chromatogram expected for methyl cantharidin (C11H15O4 +; m/z = 211.0970±100 ppm). We identified a peak eluting after cantharidin at 19.2 min corresponding to the mass of methyl cantharidin in the complete reaction mixture (Figure S4D). Importantly, this species was absent in each of our control reactions lacking cantharidin, SAM, or Crg1 (Figure S4D). This is strong evidence that cantharidin is indeed methylated by Crg1. Finally we analyzed the combined spectra of the 19.2-min peak (Figure 3E). In addition to the m/z = 211.1105 species, we observed m/z = 229.1215 and m/z = 197.0930 species, corresponding to hydrated methyl cantharidin (C11H17O5 +; m/z = 229.1076) and cantharidin, respectively. Importantly, all of these species co-elute (Figure S4B, S4D and S4E).
Based on its close sequence homology to Tmt1, we suspect that Crg1 catalyzes the formation of a cantharidin methyl ester (Figure 2). In solution, this putative methyl ester is likely in equilibrium with its ring-closed methyl anhydride form. If the equilibrium favors the ester form, some fraction of the cantharidin methyl ester could undergo ring-closing elimination reactions during ionization to yield methyl cantharidin and cantharidin products. Likewise, if the equilibrium favors the methyl anhydride, it may possibly undergo in-source fragmentation to give cantharidin, and like cantharidin, it may simply form a water adduct during ionization. Although our mass spectrometry data strongly support our hypothesis that Crg1 methylates cantharidin in vitro, additional analysis is needed to determine the structure of the methylated drug molecule. It is possible that the observed methyl cantharidin and hydrated methyl cantharidin species are ionization products of another methylated cantharidin derivative that is the actual product of the Crg1-catalyzed reaction.
Revealing the Genetic Interactome of CRG1 in the Presence of Cantharidin
To identify cellular processes required for cantharidin resistance and to define the spectrum of genes that compensate for the absence of CRG1 in the presence of cantharidin, we used a chemogenomic approach to analyze genetic interactions between CRG1 and each of the ∼4800 non-essential yeast genes both in the presence and absence of the drug. A genetic interaction between two genes occurs when the phenotype of the double deletion mutant shows significant deviation in fitness compared with the expected (multiplicative) effect of combining two single mutants (e.g. sickness or synthetic lethality) [17], [40]. When synthetic lethality is observed, it suggests that the genes may have overlapping functions. By analogy, identification of drug-gene interactions will similarly uncover genes that act in parallel with a gene of interest and these interactions can illuminate a compound's effect on a cell. To perform this experiment, we generated double deletion mutants (with a crg1Δ strain as the query) using Synthetic Genetic Array (SGA) technology [40], pooled all viable double deletion mutants and analyzed their growth in a competitive fitness assay in the presence and absence of cantharidin (Figure 4A) [41]. Six highly reproducible and independent crg1ΔxxxΔ pools (r = 0.72, Figure S5A) were further averaged yielding 70 double deletion mutants (Table S3; Datasets S1 and S2) that showed significant growth defects (log2 (drug/DMSO) <−1, P-value <0.05) in the presence of an IC20 dose of cantharidin (30 µM) in YPD when grown in a pool. Noteworthy, these genes were not sensitive as single deletion mutants (P-value <0.025; Figure 4B and 4C, Table S3), and, thus, the effect was specific to the double mutant combination. To obtain a general overview of “aggravating” (negative) interactors of CRG1 in the presence of cantharidin, we categorized this set of genes according to their GO term Biological process (Figure 4B). This dataset comprised diverse biological processes, including vesicle-mediated transport (P-value <0.008), chromosome organization (P-value <0.001), response to chemical stimulus (P-value <0.019), lipid metabolic process (P-value <2.0×10−5), response to stress (P-value <0.018), and protein modification process (P-value <0.003).
Figure 4. Characterization of cantharidin-specific genetic interactome of CRG1.

(A) Experimental scheme for analysis of cantharidin-specific genetic interactors of CRG1. Double deletion mutants crg1ΔxxxΔ generated through Synthetic Genetic Array (SGA) were pooled together and treated with cantharidin (30 µM) for 20 generations in YPD. Genomic DNA was isolated, unique strain-representative barcodes were PCR amplified, and the PCR products were hybridized to TAG4 arrays for the quantitative analysis of fitness of the mutants (see Materials and Methods for details). (B) Scatter plot representing cantharidin-gene interactions obtained from the comparative analysis of ura3ΔxxxΔ (control single deletion pool) and crg1ΔxxxΔ pools. CRG1-dependent interactors are highlighted in the red square. The hits are obtained from the averaged datasets (n = 6 for crg1ΔxxxΔ pools and n = 4 for ura3ΔxxxΔ). Significant negative genetic interactors were categorized according to their biological processes (P-value <0.002 before multiple testing correction) (Table S3). (C) Representative growth curves for the top hits (sensitive and resistant) that genetically interact with CRG1 in the presence of cantharidin. Cells were grown in YPD media with and without cantharidin. met22 and dbf2 deletion strains were treated with cantharidin to test their sensitivity and resistance, respectively.
We also identified the serine/threonine kinase DBF2 as a strong suppressor of CRG1-dependent cantharidin toxicity (log2 (drug/DMSO) = 1.12, P-value <4.0×10−5; Figure 4B). This interaction was confirmed by evaluating the fitness of individual strains in liquid and on solid SD medium in the presence of 25 µM and 10 µM cantharidin, respectively (lethal doses for crg1Δ strain in these media conditions; Figure 4C and Figure S5B). Furthermore, the alleviating interaction between DBF2 and CRG1 was not observed at 37°C, indicating cantharidin-specific nature of this interaction (Figure S5B). In addition to the well-characterized roles of Dbf2 in the mitotic exit network [42], this newly uncovered interaction suggests that this kinase may have an opposing function to the protein phosphatases (PP2A and PP1), the primary targets of cantharidin [30], [31]. As independent evidence for an interaction with cantharidin, we found that DBF2 transcript levels were significantly decreased in a crg1Δ/Δ mutant in the presence of cantharidin (250 µM) (∼2-fold, P-value <0.013) compared to DMSO control, and that change was not detected in other strains (Figure S5C). This observation confirms the role of Crg1 in phosphorylation/dephosphorylation homeostasis during cantharidin stress.
CRG1 Is Important for Lipid Homeostasis during Cantharidin Treatment
To identify genes specifically required for growth in the presence of cantharidin, we removed the genes that behave as multidrug resistance genes (MDR) from our chemogenomic dataset described above. MDR genes are defined here as those that are required for growth in the presence of multiple stress conditions (at least 20% of tested conditions for homozygous deletion strains) [15]. This filtering removed apparent enrichment of genes involved in vesicle-mediated transport genes (P-value = 0.216), response to stress (P-value = 0.024), chromosome organization (P-value = 0.075) and protein modification process (P-value = 0.021). Following this, we found that cantharidin-specific CRG1 interactors are significantly enriched for genes required for lipid metabolic process (multiple testing corrected P-value <0.0003) (Figure S6A). In particular, lipid methyltransferases (CHO2, ERG6), glycosylphosphatidylinositol (GPI) lipid biosynthesis genes (ARV1, GUP1, PER1) and lipid-related genes (SAC1, MOT3, DEP1, RVS167, YTA7) are essential in the double deletion strains in the presence but not absence of cantharidin treatment (Figure 5A). Furthermore, we demonstrated that an increase in cantharidin concentration (10 µM) did not result in cantharidin sensitivity for these genes compared to wild type (Figure 5A), confirming the dependence of the detected interactions on the presence of CRG1.
Figure 5. Crg1 is important for lipid homeostasis during cantharidin stress.

(A) CRG1 is synthetically lethal with lipid-related genes in the presence of cantharidin. Cells were normalized to an equivalent OD600,10-fold diluted, spotted onto synthetic complete defined medium containing cantharidin and incubated at 30°C. (B) Comparative phospholipid profiles of wild type and crg1 mutants in response to cantharidin. Cells grown to mid-exponential phase in YPD were treated with cantharidin (250 µM) for 2 hours. Lipid standards were added to the cells, and extracted lipids were measured using ESI-MS/MS. The quantities of lipid species are expressed as ion intensities relative to the levels in DMSO, and converted to a log2 scale. Data are the average of three samples. Statistical significance in the abundance of lipid species in the presence of cantharidin between wild type and mutants was determined using Kruskal-Wallis test, *P-value <0.05 (Table S4). (C) A simplified diagram of phospholipid biosynthesis linked to sphingolipid biosynthesis. PIs species contribute to biosynthesis of complex sphingolipids, GPI anchors, and PIPs. (D) Comparison of sphingolipid profiles of wt, crg1Δ/Δ and CRG1-overexpressing crg1Δ/Δ mutants in the presence of cantharidin (250 µM). The suffixes -B, -C, and -D on IPC and MIPC denote hydroxylation states, having two, three, or four hydroxyl groups, respectively. Statistical significance in the abundance of lipid species in the presence of cantharidin between wild type and mutants was determined using Kruskal-Wallis test, *P-value <0.05 (Table S4).
To explore further the role of CRG1 in lipid metabolism and related processes, we compared the lipid content (or “lipidome”) of wild type, crg1Δ/Δ homozygous deletion and CRG1-overexpressing crg1Δ/Δ strains in the presence and absence of cantharidin (250 µM) using electrospray ionization tandem mass spectrometry (ESI-MS/MS) analysis [43]. We observed significant changes in the abundance of most glycerophospholipids and sphingolipids in both the wild type and crg1Δ/Δ strains after growth in cantharidin-containing medium (P-value <0.05, Kruskal-Wallis test). The strains with a CRG1-overexpressing construct did not exhibit significant cantharidin-induced lipid alterations (P-value >0.08, Kruskal-Wallis test; Figure 5B, Table S4, Dataset S3). Specifically, in both the wild type and crg1Δ/Δ strains, cantharidin measurably increased the levels of short chain phosphatidylcholine (PC), phosphatidylethanolamine (PE), and phosphatidylinositol (PI) species, while the levels of long-chain PCs and PIs were reduced (Figure 5B). In the crg1Δ/Δ strain we also noted a substantial decrease in the levels of mixed size phosphatidylserine (PS) species after cantharidin stress, while the wild type and crg1Δ/Δ strain had increased levels of saturated short chain (C16 and C18) PI species compared to mono-unsaturated short chain PIs in cantharidin (Table S4). Such abundance changes with respect to acyl chain length and saturation were not observed in the CRG1-overexpressing mutant, suggesting that extra copies of CRG1 complemented the cantharidin-induced defects.
It has been previously reported that phospholipid and sphingolipid biosynthetic pathways are interconnected (Figure 5C) [44]–[46]. One way in which this interconnection is seen is when, a single gene deletion or chemical perturbation of cells results in the so-called “ripple effect” [45] characterized by lipidome-wide perturbations. We see evidence of this effect: the amounts of the most abundant sphingolipid inositolphosphoceramide (IPC) and mannosyl-inositolphosphoceramide (MIPC) were also affected by cantharidin in a crg1Δ/Δ mutant (Figure 5D). To investigate if other lipid intermediates are affected by the drug in a similar manner in crg1 mutants, we analyzed both sterol content and the formation of lipid droplets, which serve as storage pools of triacylglycerols and steryl esters [47]. We found no obvious changes in these lipid species in the presence of drug (Figure S6B and S6C). Taken together, these results demonstrated that cantharidin's effect is specific towards phospholipids and sphingolipids in crg1 mutants.
To test if cantharidin-induced alterations in yeast lipidomes are evolutionally conserved, we examined the lipidome of the human fungal pathogen Candida albicans in response to cantharidin. A C. albicans homozygous crg1 deletion (orf19.633Δ/Δ) displayed similar growth defects to those observed in S. cerevisiae when challenged with cantharidin (Figure S7A). Lipidomic analysis demonstrated that cantharidin treatment (2 mM, IC20 for C. albicans wild type) resulted in the significant changes in most phospholipid species in both wild type and orf19.633Δ/Δ homozygous mutant (P-value <0.05). Furthermore, although to a more modest degree than seen in S. cerevisiae, we found that C. albicans CRG1 may account for some difference between wild type and a mutant strain (P-value <0.05; Figure S7B; Dataset S4). In addition, we have shown previously that the overexpression of C. albicans ORF orf19.633 restored cantharidin resistance in S. cerevisiae crg1Δ/Δ mutant [14], further suggesting that the lipid homeostasis functions of this C. albicans putative SAM-dependent methyltransferase are conserved.
Crg1-Dependent Effects of Cantharidin on Cytoskeleton Organization
One of the phospholipids manifesting substantial changes in our lipidome analysis was phosphatidylinositol (PI) (Figure S7C). PI is an essential phospholipid with multiple roles in the biosynthesis and metabolism of phosphoinositides (PIP), inositol polyphosphates (IPs), complex sphingolipids and glycerophosphoinositols (GPIs) (Figure 5D) [48]. It has been previously reported that phosphorylated derivatives of PI species (mainly PI(4,5)P) are well-conserved second messengers involved in the regulation of the actin cytoskeleton in Pkc1-dependent manner (Figure 6A) [48], [49]. Therefore, to examine one of the physiological consequences of altered levels of PI, we tested if cantharidin affects the actin cytoskeleton. Microscopy of FITC-phalloidin stained cells revealed that crg1Δ/Δ strain treated with 250 µM cantharidin for 1 hour lacked actin patches and displayed highly disorganized actin cables compared to wild type. Overexpression of CRG1 in crg1Δ/Δ strain restored the number of actin patches close to that seen in the wild-type strain without cantharidin (Figure 6B). These results demonstrate that Crg1 is critical for both actin patch and actin cytoskeleton integrity during cantharidin stress. Although, the observed role of Crg1 in cytoskeleton organization might be indirect, in our genome-wide screen (without cantharidin) we found that positive genetic interactions (alleviating) of CRG1 were significantly enriched for the genes involved in the actin cytoskeleton, bud emergence, and cell polarity (P-value <1.0×10−5; Figure S8A; Dataset S5). In particular, the deletion of RVS167, a well-characterized actin patch and lipid-interacting protein, manifested fitness defects that are suppressed by the deletion of CRG1 (Figure S8B) [50], [51]. These findings further support the role of Crg1 in actin-related biological process.
Figure 6. Crg1 is important for actin patch formation during cantharidin treatment.

(A) Diagram showing how PIP species are involved in Pkc1-dependent changes in actin cytoskeleton. (B) Crg1 is important for actin patch integrity during cantharidin stress. Wt, crg1Δ/Δ mutant and CRG1-overexpressing crg1Δ/Δ cells were grown to mid-exponential phase at 30°C in YPD in the presence of cantharidin (250 µM) or DMSO for 1 hour. Cells fixed with formaldehyde were stained for actin with FITC-phalloidin and visualized by fluorescence microscopy. Bar, 5 µm. The number of actin patches per cell in each sample was quantified. Values are the mean of three independent replicates (n = 270–1000), error bars are the standard deviation; * P-value <0.025, ** P-value <0.0002 (Student's t-test). (C) Cantharidin induces CRG1 transcription via the Cell Wall Integrity (CWI) pathway. Cells grown to mid-exponential phase were treated with cantharidin (250 µM) in YPD for indicated time. Total RNA was extracted, cDNA was prepared and analyzed by qRT-PCR. CRG1 transcript levels were normalized to ACT1. The simplified diagram of CWI pathway is shown. Slt2 is a kinase, Rlm1 is a transcriptional activator governed by the CWI pathway. (D) A preliminary model describing Crg1-cantharidin interaction. See text for details.
CRG1 Transcription Is Regulated via the Cell Wall Integrity (CWI) Pathway
Finally, to determine how Crg1 is regulated at the transcriptional level in response to cantharidin, we explored which pathways, if any, are required for cantharidin resistance. Based on our observation that the homozygous deletion strains slt2Δ/Δ and bck1Δ/Δ (both CWI kinases) are hypersensitive to cantharidin [14], [15], combined with the fact that the promoter region of CRG1 contains a binding site for Rlm1 (a transcriptional regulator of CWI pathway) [52], we asked if CRG1 expression is activated by cantharidin via the CWI pathway. We found that deletion of these genes blunted the increase of CRG1 transcript in response to cantharidin (250 µM) compared to the wild type (Figure 6C), indicating that CWI pathway components are required for CRG1 expression in the presence of cantharidin. The CRG1 promoter also contains a binding site for Yap1, a transcription factor required for cadmium tolerance and the oxidative stress response. In contrast to Rlm1 and Slt2, the relative amount of CRG1 transcript in the yap1Δ/Δ mutant was unchanged in the presence of cantharidin (Figure S9A). While these data suggest that Crg1 may be regulated via the CWI pathway and is transcriptionally responsive to numerous cell wall stressing agents (Figure S9B), we did not detect any drastic fitness defects when crg1 mutants were grown in the presence of cell wall perturbing agents (Figure S9B). However, overexpression of CRG1 in the crg1Δ/Δ mutant did confer resistance to lithium chloride and fenpropimorph, both of which are known perturbants of the cell membrane and other lipid processes (Figure S9C) [53]–[58]. Together these results supports a model in which Crg1 is involved in lipid-related processes that indirectly impinges on cell wall integrity.
Discussion
In this study we demonstrated that yeast genetic and chemical genome-wide approaches, when combined with rigorous biological follow-up, can effectively characterize a novel gene that, despite being subject to numerous large-scale phenotypic studies, had little functional annotation. Our previous work demonstrated that Crg1, a putative SAM-dependent methyltransferase, was a novel mediator of resistance to the protein phosphatase inhibitor, cantharidin [14]. Here we show that the mechanism of Crg1-cantharidin interaction is through direct methylation of the compound, and that, furthermore, Crg1 plays an essential role in the cellular response to cantharidin-induced lipid alterations.
Our initial observation that cantharidin cytotoxicity is suppressed by overexpression of CRG1 suggested a specific, although not necessarily direct, cantharidin-Crg1 interaction in vivo [14]. Here, we demonstrate that Crg1 is able to interact with cantharidin in vitro, resulting in the formation of a methylated cantharidin species. Modification by methylation is known to remove negative charges on diverse molecules which can alter hydrophobicity and modulate cellular pathways and processes. Given the clear phenotype of Crg1-deficient cells and the results from our in vitro biochemical characterization of Crg1, we hypothesize that methylation of cantharidin alters its physical properties such that it is no longer harmful to cells. In a manner similar to other methyltransferases that are known to detoxify small molecules [17], [59]–[62], chemical modification of cantharidin provides some insight regarding how its methylation may modify its activity. For example, endothall, an unmethylated, ring-opened form of cantharidin, has been assayed for protein phosphatase inhibition [26] and the methyl, ethyl, and propyl esters of endothall are still potent inhibitors of PP1 and PP2A. Several lactol derivatives of norcantharidin (the anhydride form of endothall) formed by reducing one of the carbonyl groups to a hydroxyl group have been synthesized and characterized. Modification of the free hydroxyl to form methyl, ethyl, and propyl ethers sharply reduced the ability of the drug derivatives to inhibit protein phosphatases. While the unmodified lactol form inhibited PP2A with an IC50 of 5 µM, the IC50 for the methyl ether lactol form was >1000 µM. Collectively, these observations suggest that methylation of closed-ring forms, not open-ring forms, reduces cellular toxicity. Cantharidin is more sterically hindered than norcantharidin, and as such, we would expect that its equilibrium would favor the closed-ring anhydride form more than that of norcantharidin. Accordingly, we are intrigued by the possibility that the methyl cantharidin product of the reaction catalyzed by Crg1 resembles the closed-ring lactol ether compounds that are less potent inhibitors of both growth and protein phosphatase activity. Further study to elucidate the structure of this product will enhance our understanding of how the methylation of cantharidin by Crg1 facilitates its detoxification.
Looking ahead, it will be interesting to explore if similar mechanisms of cellular detoxification in mammalian cells are mediated by methyltransferases, such as METTL7A or METTL7B, which both share sequence homology to CRG1. Interestingly, METTL7B, also known as ALDI, was reported to be highly expressed in kidney and liver, and associated with hepatic lipid droplets [63], providing a provocative link between Crg1-like methyltransferases cantharidin toxicity and lipid process.
In addition to characterizing the direct interaction of Crg1 with cantharidin, we investigated cellular pathways of Crg1-mediated cantharidin resistance using cells sensitized with a crg1 deletion allele. This analysis revealed that genes involved in lipid-related processes are required for survival under cantharidin-induced stress in the absence of CRG1 (CHO2, OPI3, ERG6, SAC1, ARV1, GUP1, PER1, MOT3, DEP1). Because CRG1 is both not essential and also shows very few genetic interactions under standard laboratory conditions, the identification of these genes required condition-specific assays. Our chemogenomic data are supported by lipidome-wide analysis, which demonstrated that cantharidin-induced alterations in glycerophospholipids and sphingolipids occur in a CRG1 gene dose-dependent manner. Specifically, we observed the accumulation of short chain phospholipids in the crg1Δ/Δ mutant, suggesting that the drug affects fatty acid elongation in a Crg1-dependent fashion. Consistent with this result, we also observed that overexpression of CRG1 confers resistance to lipid stressing agents such as lithium ions and the ergosterol inhibitor fenpropimorph (Figure S9C). Resistance to fenpropimorph is acquired by mutations in the fatty acid elongase FEN1 (ELO2), which is known to be involved in sphingolipid biosynthesis [64]. Thus, it will be informative to test if CRG1 and FEN1 have overlapping functions in lipid biosynthesis.
Another possible explanation for our observation that cantharidin-induced lipidome alterations can be suppressed by increasing the gene dose of CRG1 can be found in the transcriptional changes that occur in these strains. Genes involved in methionine biosynthesis are differentially expressed in CRG1-overexpressing strains in the presence of the drug compared to wild type and the crg1Δ/Δ mutant. This is of particular interest because changes in methionine metabolism can regulate methylation reactions by altering levels of SAM, a methyl donor [65], [66]. For example, Tehlivets et al. showed that defects in methionine cycling enzymes result in an imbalance of phospholipid and triacylglycerol synthesis [67], [68]. The mechanisms underlying these relationships are not yet clear, but it is possible that cells sense that the level of SAM is depleted via Crg1 activity, which results in transcriptional changes in methionine biosynthesis genes, in particular, the cystathionine beta-lyase Str3. These findings suggest that overexpression of Crg1 may buffer cantharidin-treated lipidome alterations in part through changes in the methionine cycle.
To define the ‘core’ buffering network to CRG1 in the presence of cantharidin, we compared the transcriptome and cantharidin SGA profiles. Although we did not find any obvious overlap in GO term biological processes between these datasets, in our cantharidin SGA one of the most sensitive mutants was MET22 (Figure 4C), a gene with a role in sulfur assimilation and methionine biosynthesis. This gene was also differentially expressed in CRG1-overexpressing mutant vs. wild-type strain (P-value <0.02; Table S2).
Our chemical genomics results were corroborated by traditional SGA analysis. This analysis demonstrated that CRG1 has an alleviating (suppressing) genetic interaction with RVS167. It is well established that a similar phenotype is observed when RVS167 is deleted in combination with genes involved sphingolipid biosynthesis (e.g. SUR1, SUR2, FEN1, ELO3 and IPT1), implicating sphingolipid biosynthesis in the regulation of the actin cytoskeleton [49], [50], [69]–[71]. Similarly to S. cerevisiae and C. albicans, studies in the ciliate Tetrahymena showed that cantharidin treatment also influences PI metabolism and the actin cytoskeleton [72], demonstrating the conservation of cantharidin-lipid-actin interactions.
Understanding the transcriptional regulation of CRG1 during cantharidin stress adds many layers to the picture of the complex physiological roles of this methyltransferase. CRG1 transcription is activated by cantharidin via the conserved MAPK family components of the CWI signaling pathway [52], [73]. Hoon et al. previously demonstrated that deletion of slt2 and bck1 results in cantharidin sensitivity, suggesting that this pathway is critical for cantharidin resistance [14]. In mammalian cells, several studies have reported that the MAP kinases ERK and JNK are also activated by cantharidin [27], [29], likely as a consequence of the inhibition of protein phosphatases. Moreover, other studies reported that an intact CWI cascade is essential for maintaining lipid homeostasis [74]. It remains to be determined what specific steps are involved in the activation of CRG1 by cantharidin. One possible scenario is that the CWI pathway is activated by the accumulation of aberrant lipid species in a manner analogous to previous reports that suggest that long chain bases induce the Pkc1-MAPK CWI pathway in yeast [75], [76].
Based on our observations, we propose the following mechanism for Crg1-cantharidin interaction (Figure 6D). Cantharidin treatment inhibits PP2A and PP1, resulting in the perturbation of both lipid homeostasis and actin cytoskeleton organization. This perturbation activates the CWI pathway, which in turn induces of CRG1 transcription. The resulting Crg1 protein directly methylates cantharidin, alleviating its cytotoxicity and restoring lipid homeostasis, actin cytoskeletal architecture, as well as other cantharidin-associated effects.
In summary, our study demonstrates the value of combining classic biology approaches and chemical genomics with other “omic”-based methods for de-orphaning proteins and elucidating previously unknown mechanisms of therapeutics action.
Materials and Methods
Strains, Plasmids, and Growth Conditions
Yeast strains and plasmids used in this study are described in Table S5 and Table S6. Unless otherwise stated, wild-type (wt) strain is BY4743; crg1Δ/Δ was derived from BY4743. Yeast cells were grown in YPD (2% yeast extract, 1% peptone, 2% glucose) or in synthetically defined medium, SD (0.67% yeast nitrogen base, 2% glucose, and amino acids). Cantharidin, norcantharidin, cantharidic acid, and fenpropimorph were purchased from Sigma Aldrich (Toronto, Canada). Lithium chloride was purchased from Teknova (Hollister, CA, USA). Cantharidin, cantharidic acid, norcantharidin, and fenpropimorph were dissolved in DMSO and stored at −20°C. The IC20 of cantharidin in YPD for wild-type is 250 µM, in SD it is 5 µM, both determined in liquid culture as described [77].
Site-Directed Mutagenesis of CRG1
CRG1 was amplified from wild-type strain using primers (Table S7) with homology to the vector p426-GAL1-TAP at the 5′ end. The amplified CRG1 and HindIII linearized vector were directly co-transformed into a crg1Δ/Δ mutant and transformant colonies were selected in synthetic defined media lacking uracil (SD-URA). CRG1 was cloned downstream of a GAL1 inducible promoter and in frame with the TAP coding sequence. Transformants were screened by PCR and for cantharidin resistance. CRG1 missense mutants were prepared using the QuickChange Lightning Site-directed mutagenesis kit (Stratagene - Agilent Technologies Company, La Jolla, CA, USA). Clones were sequenced to verify the mutations. To express Crg1, transformants were grown to mid-exponential phase in SD-URA and raffinose (2%), then induced by the addition of galactose to a final concentration of 2%. 30 µM of cantharidin was used to test sensitivity of mutants. Cells were harvested after 3 hours of induction, and Crg1 expression was verified by Western blots of 12% SDS-PAGE gels using anti-TAP antibodies (OpenBiosystems – Thermo Fisher Scientific, Huntsville, AL, USA).
RNA Isolation, cDNA Preparation, and Quantitative Real-Time PCR Analysis
Cells grown to mid-exponential phase in YPD medium were incubated with or without cantharidin (250 µM) for various amounts of time, harvested by centrifugation, frozen in liquid N2 and stored at −80°C. RNA was extracted with hot acidic phenol [78] and treated with the Turbo DNA-free kit (Ambion – Applied Biosystems, Austin, TX, USA). RNA purity was tested using a spectrophotometer and integrity was evaluated by denaturing gel electrophoresis. First-strand cDNA was synthesized from 1 µg of DNase-treated RNA with 0.5 µg of oligo(dT12–18) primers (Invitrogen, Burlington, ON, Canada) using 200 units of Superscript II Reverse transcriptase (Invitrogen, Burlington, ON, Canada). Real-time PCR analysis was conducted with Power SYBR Green PCR master mix (Applied Biosystems, Foster City, CA, USA) and gene-specific primers (Table S7) at a final concentration of 250 nM. qRT-PCR was carried out on a 7900HT Fast system (Applied Biosystems) using Sequence Detection System software version 2.3. Fold change in CRG1 transcript level normalized to ACT1 was calculated using the 2−ΔΔCt method. At least three independent replicates of each reaction were performed. Student's t-test was applied for statistical analysis (paired for drug vs. DMSO treatments, and unpaired for mutants vs. wild type).
Microarray Analysis
Cells grown to mid-exponential phase in YPD medium were incubated with or without cantharidin (250 µM) for 1 hour, harvested by centrifugation. Isolation of RNA and hybridization to the tiling arrays was performed as described [79], except that actinomycin D was added in a final concentration of 6 µg/mL during cDNA synthesis to prevent antisense artifacts [80]. Two independent replicates were used for the analysis. Hybridization to Affymetrix Tiling Arrays using the GeneChip Fluidics Station 450 (Affymetrix) was followed by the extraction of intensity values for the probes using the GeneChip Operating Software (Affymetrix). Acquisition and quantification of array images were performed using the Affymetrix tiling analysis software (http://www.affymetrix.com/support/developer/downloads/TilingArrayTools/index.affx). The resulting .BAR files containing probe position and intensities were further analyzed by aligning the probes that match the position of the S. cerevisiae Genome Database list of defined ORFs (http://downloads.yeastgenome.org/chromosomal_feature/saccharomyces_cerevisiae.gff). The log2 of signal intensity of each ORF was defined as the average across the probes associated with the ORF. Quantile normalized datasets were clustered with a correlation similarity metric and the average linkage method using Cluster 3.0 software (http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm). The cluster was visualized using TreeView software (http://jtreeview.sourceforge.net/). The significance for differential expression was set as log2 (drug/DMSO) >1 and <−1, P-value <0.05 as determined by Student's t test. Significantly up- and downregulated transcripts were further tested for Gene Ontology (GO) biological process enrichment using FunSpec (http://funspec.med.utoronto.ca/) with P-value cutoff of 0.01 and multiple testing correction (Bonferroni) (Table S1; Dataset S6). The probability was calculated using a test employing hypergeometric distribution (see below). To detect cantharidin-specific genes the genes involved in ESR [36] were eliminated from the gene-set.
Expression and Purification of Crg1 Fusion Protein
Purification of 6xHis-Crg1 was performed as previously described [81]. Wild-type yeast strain Y258 carrying a vector pBG1805-GAL1-CRG1 with a triple affinity tag at C-terminal (His6-HAepitope-3Cprotease site-ZZprotein A) was grown in 660 mL of synthetically defined medium (SD-URA and 2% raffinose) to mid-exponential phase at 30°C. To induce expression of CRG1 340 mL of 3x YP (yeast extract and peptone) and 6% galactose was added to a final concentration of 2%. Cells were harvested by centrifugation at 3,000 rpm for 5 min. All steps following harvest were performed at 4°C. Cells were washed with PBS buffer, resuspended in 7 mL of resuspension buffer (20 mM HEPES pH 7.5, 1 M NaCl, 5% glycerol), and lysed using an acid washed Zirconia beads in the presence of protease inhibitors (1 mM Pefablock, 2.5 µg/mL pepstatin A, 2.5 µg/mL leupeptin, 1 mM PMSF). The cell lysate was centrifuged at 20,000 rpm for 45 min, and diluted two fold with binding buffer (20 mM HEPES pH 7.5, 40 mM imidazole, 5% glycerol). 300 µL of Ni Sepharose 6 Fast Flow beads (50% slurry in 20% ethanol) was added to the sample and rotated for 1.5 hours, followed by three washes with 40 mL of wash buffer (20 mM HEPES pH 7.5, 40 mM imidazole, 5% glycerol, 0.5 M NaCl). To elute Crg1 the Ni beads were resuspended in 1 mL of elution buffer (20 mM HEPES pH 7.5, 250 mM imidazole pH 7.7, 5% glycerol, 0.5 M NaCl), and Crg1p was released by rotating the mixture for 15 min at 4°C. The protein was further concentrated with Amicon Ultra tubes (10 K) (Millipore, Etobicoke, ON, Canada) to 100 µL.
Purification of TAP-tagged wild-type and mutant forms of Crg1 was performed in BY4743 carrying p426-GAL1-CRG1-TAP as described in Rigaut et al. [82]. Cell growth, induction of Crg1 with galactose (2%), and preparation of cell lysates were performed as described for the Crg1-6xHis fusion. 300 µL IgG-agarose was added to the extract and incubated for 2 hours followed by a triple wash with 25 mL of Low Salt and High Salt Wash Buffer (50 mM HEPES pH 7.5, 10% glycerol, 150 mM/750 mM NaCl, 0.1% Tween20). The final wash was performed with 15 mL TEV Cleavage Buffer (10 mM, Tris pH 8.0, 150 mM NaCl, 0.05% Tween20, 10% glycerol, 0.5 mM EDTA, 1 mM DTT). The extract was incubated overnight with 100 U TEV protease (Invitrogen, #12575-015). 1.2 mL CaM Binding Buffer (10 mM Tris pH 8.0, 150 mM NaCl, 0.05% Tween20, 10% glycerol, 1 mM MgOAc, 1 mM imidazole pH 8.0, 2 mM CaCl2, 1 mM DTT) and 2.4 µL 1 M CaCl2 were added to the protein eluates. The eluates were then incubated with 400 µL (50% slurry) Calmodulin Sepharose in 5 mL CaM Binding Buffer for 2 hours, followed by a wash with 25 mL CaM Binding Buffer and elution with 5×200 µL Elution Buffer (10 mM Tris pH 8.0, 150 mM NaCl, 0.05% Tween20, 10% glycerol, 1 mM MgOAc, 1 mM imidazole pH 8.0, 2 mM EGTA, 1 mM DTT). Protein eluates were stored at −20°C in the presence of 50% glycerol. Recovery of Crg1 was determined using Bradford reagent (BioRad Laboratories, Mississauga, ON, Canada) and its integrity and purity was assessed with 12% silver-stained SDS-PAGE gel.
Preparation and Analysis of Radiolabeled In Vitro Crg1 Reactions
In vitro enzymatic reactions were prepared with 0.09 µg 6×His-tagged Crg1, 0.2 mM cantharidin dissolved in DMSO (Sigma Aldrich, St. Louis, MO, USA), and 20 µM S-adenosyl-[methyl-14C]methionine (55.8 mCi/mmol) (GE Healthcare, Piscataway, NJ, USA) in 0.1 M sodium phosphate, pH 7.4 with a final volume of 50 µL and 2% DMSO. The complete reactions and relevant controls were incubated at 30°C for 2 hours. Following incubation, the enzymatic reactions were separated by reverse-phase high-performance liquid chromatography (Series II 1090 Liquid Chromatograph, Hewlett Packard, Palo Alto, CA, USA). The chromatography gradient was adapted from [83], except that the flow rate was 1 mL/min. Mobile phase A contained 0.1% trifluoroacetic acid in water and mobile phase B was 0.1% trifluoroacetic acid in acetonitrile. A BetaBasic-18 column (250 mm×4.6 mm; 5-µm particle size) (Thermo, Waltham, MA) was used. 40 µL was injected, 2-minute fractions were collected, and 350 µl of each fraction was mixed with 5 mL of Safety-Solve (Research Products International, Mt. Prospect, IL, USA) before quantification of radioactivity with a LS6500 liquid scintillation counter (Beckman Coulter, La Brea, CA, USA). Each fraction was counted three times for 3 minutes.
Other in vitro reactions were prepared in an identical manner with 0.09 µg of either mutant or wild-type TAP-tagged Crg1 and varying concentrations of cantharidin (USB, Cleveland, OH, USA). After incubation at 30°C for 2 hours, 40 µL 2 N HCl was added to each 50 µL reaction. Immediately, 80 µL of this mixture was transferred to a 1.9-cm×9-cm folded piece of filter paper in the neck of a scintillation vial containing 5 mL of Safety-Solve and the vials were capped. After 4 hour incubation at room temperature, the pieces of filter paper were removed from the neck of each vial and the acid-labile volatile radioactivity was quantified with a liquid scintillation counter as described above [84].
Preparation of In Vitro Enzymatic Reactions for Analysis by Mass Spectrometry
In vitro enzymatic reactions using unlabeled SAM (Sigma Aldrich) were prepared in a similar manner with 0.09 µg of 6xHis-tagged Crg1, 200 µM cantharidin (Sigma Aldrich), and a SAM concentration of 1.6 mM. These reactions were quenched with addition of 200 µL acetonitrile, and 12.5 µL of 15% ammonium bicarbonate was added to reduce product degradation. After concentration with a vacuum centrifuge and resuspension in 50 µL of water, these reaction mixtures were analyzed by liquid chromatography-tandem mass spectrometry (1100 Series Liquid Chromatograph, Agilent, Santa Clara, CA; QSTAR Elite Mass Spectrometer, Applied Biosystems, Foster City, CA) in positive ionization mode with a Turbo Spray source. 8 µL of sample was injected onto a reverse-phase column (Luna C18 (2), 150 mm×1 mm, 5-µm particle size, Phenomenex, Aschaffenburg, Germany) with a flow rate of 50 µL/min and the following gradient: t = 0–1 min, 2% mobile phase B; t = 10 min, 35% B; t = 14–16 min, 90% B; t = 16.5–35 min, 2% B. Mobile phase A was 0.1% formic acid in 98% water and 2% acetonitrile, while mobile phase B was 0.1% formic acid in 98% acetonitrile and 2% water. Spectra were collected with the instrument information-dependent acquisition mode (full scan: m/z = 70–600, 1.000071 s; 3 product experiments: m/z = 65–500, <2 s per experiment). The following mass spectrometer parameters were used: declustering potential, 85 V; focusing potential, 300 V; declustering potential II, 15 V; ionspray voltage, 5500 V; ion source gas, 45 units; ion source gas II, 5 units; curtain gas, 45 units; collision gas, 7 units.
Fitness Profiling of Double Deletion Mutants with Cantharidin
A pool of double deletion mutants (crg1ΔxxxΔ) was prepared by generating viable double deletion mutants using Synthetic Genetic Array (SGA) technology with crg1Δ as a query strain [40] (Text S1). ∼4800 viable double deletion mutant colonies were collected, normalized to 50 OD's/mL and stored at −80°C in media containing 7% DMSO. Two independent pools were generated for the analysis. Each was tested in triplicate. The pooling of the strains was possible due to the presence of strain-specific sequence tags flanking each gene deletion region [5]. The double deletion pool was treated with 30 µM of cantharidin, a dose which inhibits growth of the crg1 double deletion pool by ∼20%. Fitness analysis using a tag-specific algorithm that takes into account the intensities of each tag in cantharidin-treated cells compared to non-treated cells was performed as described [41]. Hybridization to Affymetrix Gene Chips using GeneChip Fluidics Station 450 (Affymetrix) was followed by the extraction of intensity values for the probes using the GeneChip Operating Software (Affymetrix). The data was quantile normalized, outliers (one standard deviation off) were omitted, and fitness defect scores as the log2 ratio between the mean signal intensities of the control (DMSO) and the drug were calculated for each deletion strain in the pool as previously described [41]. As a control, the relative fitness of the double-gene deletion mutants exhibiting high sensitivity to cantharidin (log2 (drug/DMSO) <−1; P-value <0.05) was compared to the relative fitness of the corresponding single gene deletion mutants. For a given double deletion mutants, the resulting interaction was evaluated using comparison of the observed double mutant growth rate to the expected assuming that there is no interaction exists. Log2 ratio <−1 represents those strains with a measurable growth defect (or lethality) and log2 ratio >1 demonstrates resistance to the drug. Cantharidin-specific CRG1 negative genetic interactors were further verified as individual clones in liquid growth assays and/or by spot dilutions. To evaluate the chemogenomic dataset for statistical significant enrichment for general biological processes (Gene Ontology Slim mapper), we used a standard hypergeometric test, that asses the probability that the intersection of given list with any given functional category occurs by chance. Obtained P-values were corrected for multiple testing correction (Bonferroni) by multiplying P-values with the number of genes in the test. The probability was calculated as follows: the P-value of observing x genes, belonging to the same functional category, is:
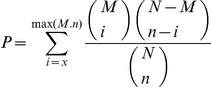 |
where M is the total number of genes involved in a functional category, n is the total number of genes in the cluster, and N is the total number of yeast ORFs.
Lipidome Analysis by Liquid Chromatography-Tandem Mass Spectrometry
Cells were grown to mid-exponential phase and treated with cantharidin (250 µM) for 2 hours. Lipids were extracted from 25 OD600-equivalent of cells and analyzed as described [43]. All lipid standards were obtained from Avanti Polar Lipids (Alabaster, AL, USA), with the exception of dioctanoyl glycerophosphoethanolamine, which was obtained from Echelon Biosciences (Salt Lake City, UT, USA). Quantification of individual molecular species was carried out using multiple reaction monitoring (MRM) with an Applied Biosystems 4000 Q-Trap mass spectrometer (Applied Biosystems, Foster City, CA, USA). 25 µl of samples were subjected to analysis as described previously [43], [85]. Lipid levels in each sample were normalized to internal standards. For each lipid species, the mean normalized signal from the wild type and mutant strains grown in the presence or absence of cantharidin were calculated. Three independent experiments were used for analysis. Lipid levels were calculated relative to relevant internal standards. The quantities of lipids are expressed as ion intensities relative to the levels without cantharidin, and then converted to a log2 (drug/DMSO) scale. The difference in levels of individual lipid species in DMSO vs. cantharidin was determined with Kruskal-Wallis test (Table S4). Similarly, the difference between wild type and mutants was assessed statistically using the Kruskal-Wallis test, with a P-value cutoff of 0.05.
Actin Staining
Cells were grown to mid-exponential phase in YPD media, and treated with and without cantharidin (250 µM) for 1 hour. Cells were then fixed by addition methanol-free formaldehyde (Polysciences, Warrington, PA) to 4% for 1 hour, centrifuged at 3,000 rpm 5 min and washed with PBS buffer three times. Cells were permeabilized with 0.2% Triton X-100 in PBS at 25°C for 15 min, washed with PBS three times and a normalized number of cells were stained with Alexa Fluor 488 phalloidin (Invitrogen, Burlington, ON) in the dark at 25°C for 1 hour. Cells were observed with 100× objective, and fluorescence images were acquired using AxioVision software on an Axiovert 200 M fluorescence microscope (Carl Zeiss) using a 1.5 s exposure for all images. The average number of actin patches per cell was determined by dividing the total number of actin patches per total number of cells (N = 3, n≥300 cells).
Supporting Information
Chemical SGA. Fitness profiling of wild-type double deletion mutants (ura3ΔxxxΔ) with cantharidin.
(XLS)
Chemical SGA. Fitness profiling of crg1 double deletion mutants (crg1ΔxxxΔ) with cantharidin.
(XLS)
Glycerophospholipid and sphingolipid analysis by liquid chromatography-tandem mass spectrometry in S. cerevisiae.
(XLS)
Glycerophospholipid and sphingolipid analysis by liquid chromatography-tandem mass spectrometry in C. albicans.
(XLS)
SGA scores for crg1 double deletion mutants in the absence of cantharidin.
(XLS)
Transcriptome profiling of crg1 mutants upon exposure to cantharidin treatment (250 µM for 1 hour).
(XLS)
(Related to Figure 1.) (A) CRG1 gene dose is important for cantharidin tolerance. Wt, crg1Δ/CRG1 heterozygous, crg1Δ/Δ homozygous and CRG1-overexpressing crg1Δ/Δ mutants were assessed in the presence of cantharidin in YPD. Growth curves were obtained by plotting OD600 vs. time at the tested concentrations of cantharidin. At least three independent replicates were analyzed and the representative growth curves are shown. (B) Cantharidin is more potent in SD media than in YPD. Wt, crg1Δ/Δ and CRG1-overexpressing crg1Δ/Δ mutants were assessed in the presence of cantharidin in SD medium. Dose-response curves were obtained by plotting OD600 at saturation point vs. tested drug concentrations. Values are means of three independent replicates, and error bars represent standard deviation. (C) Crg1 is important for resistance to the cantharidin analogues cantharidic acid and norcantharidin. Growth of wt, crg1Δ/Δ mutant and crg1Δ/Δ cells overexpressing CRG1 was assessed in the presence of cantharidin analogues in YPD. Dose-response curves were obtained by plotting OD600 at saturation vs. drug concentration. Values are the mean of three independent replicates, and error bars represent the standard deviation.
(PDF)
(Related to Figure 1.) (A) Cantharidin induces CRG1 transcription in a gene-dose dependent manner. Wt, crg1Δ/CRG1 heterozygous, crg1Δ/Δ homozygous deletion mutants and CRG1-overexpressing crg1Δ/Δ mutant grown to mid-exponential phase were incubated with or without cantharidin (250 µM) for 1 hour. Total RNA was extracted, cDNA synthesized and the relative abundance of CRG1 transcript was analyzed by qRT-PCR. Data are the mean of at least three independent experimental replicates, and error bars are the standard deviation. (B) GFP-tagged Crg1 is localized to the cytosol after 1 hour treatment with cantharidin (in low fluorescence medium at 4 µM). Bar, 2.5 µm (see Text S1). (C) Chemical inhibition of protein phosphatases with calyculin A results in the transcriptional induction of CRG1. Wt cells grown to mid-exponential phase in YPD were treated with calyculin A (2 µM) for 30, 60 and 120 min. Total RNA was extracted, cDNA synthesized and the relative abundance of CRG1 transcript was analyzed by qRT-PCR. Data represent the mean of at least three independent experimental replicates, and error bars are the standard deviation. (D) CRG1 is a stress-responsive methyltransferase. Expression profile of CRG1 was compared with other yeast genes during 174 diverse environmental stresses [36]. Expression profiles of CRG1, ACT1 and SSE2 are shown. (E) Crg1 protein level was not altered by mutations in methyltransferase domain. Cells were collected after induction with galactose (2%) for 3 hours. The cell lysates were analyzed by western blotting with anti-TAP antibody. Tubulin was used as an internal loading control and detected by an anti-tubulin antibody.
(PDF)
(Related to Figure 1.) (A) Viability of crg1 mutants treated with cantharidin for 1 hour. Wt, crg1Δ/CRG1 heterozygous, crg1Δ/Δ homozygous deletion mutants and CRG1-overexpressing crg1Δ/Δ mutants grown to mid-exponential phase were incubated with or without cantharidin (30 µM and 250 µM, the IC20 for crg1Δ/Δ and the IC20 for wt, respectively) for 1 hour. Cells were normalized to an equivalent OD600, 10-fold diluted, spotted onto YPD solid medium and incubated at 30°C. (B) The comparison of GO term Biological processes for the genes that are upregulated and downregulated (log2 ratio >1 and <−1) in a crg1Δ/Δ mutant treated with cantharidin (30 µM and 250 µM) for 1 hour. (C) Heat map of the transcriptional profiles of wt, crg1Δ/Δ and CRG1-overexpressing crg1Δ/Δ mutants in response to cantharidin were compared. (D) Hierarchical clustering was used to group all significantly expressed genes (at least two fold) in at least one of the strains in the presence of cantharidin (250 µM). Clusters of genes exhibiting highly similar profiles across the strains are boxed. The overrepresentation of GO Biological processes in these gene clusters are indicated on the right. (E) Cantharidin significantly induces STR3 transcript levels in CRG1-overexpressing crg1Δ/Δ mutant. Wt, crg1Δ/CRG1 heterozygous, crg1Δ/Δ homozygous deletion mutants and CRG1-overexpressing crg1Δ/Δ mutants grown to mid-exponential phase were incubated with or without cantharidin (250 µM) for 1 hour. Total RNA was extracted, cDNA synthesized and the relative abundance of STR3 transcript was analyzed by qRT-PCR. Data are the mean of at least three independent experimental replicates, and error bars are the standard deviation. (F) Treatment of cells with the non-specific methyltransferase inhibitor SAH increases sensitivity to cantharidin. Wt cells were grown in SC media with or without cantharidin (4 µM) and SAH (50 µM).
(PDF)
(Related to Figure 3.) (A) Silver stained 12% SDS-PAGE of purified TAP-tagged wild-type and mutated Crg1 (see Materials and Methods for details). (B-E) Single ion chromatograms of the major species identified in the spectra shown in Figure 3.
(PDF)
(Related to Figure 4.) (A) Two independent replicates for crg1Δ/xxxΔ pools are highly correlated. Only strains with significant log2 (drug/DMSO) (P-value <0.05) are included in the analysis. (B) The deletion of DBF2 suppresses CRG1 sensitivity to cantharidin. Cells were normalized to an equivalent OD600,10-fold diluted, spotted onto synthetic complete medium containing cantharidin (10 µM) and incubated at 30°C. (C) CRG1-dependent changes of DBF2 transcript levels in the presence of cantharidin. Wt, crg1Δ/CRG1 heterozygous, crg1Δ/Δ homozygous deletion mutants and CRG1-overexpressing crg1Δ/Δ mutants grown to mid-exponential phase were incubated with or without cantharidin (250 µM) for 1 hour. Total RNA was extracted, cDNA synthesized and the relative abundance of DBF2 transcript was analyzed by qRT-PCR. Data are the mean of at least three independent experimental replicates, and error bars are the standard deviation.
(PDF)
(Related to Figure 5.) (A) GO term enrichment analysis of cantharidin-specific CRG1-interactors before and after MDR gene filtering. Only the terms with significant P-values (<0.05) are shown. Bonferroni correction was applied to the MDR-filtered terms. (B) Representative images of cells stained with Nile Red for lipid droplets. Cells were grown at 30°C for 42 hours to reach stationary phase, and inoculated into fresh medium with cantharidin (250 µM) for 2 hours. Cells were fixed and stained with Nile Red. Bar, 2.5 µm. (C) Sterol species are not affected by the deletion of CRG1 and cantharidin treatment (see Materials and Methods for details).
(PDF)
(Related to Figure 5.) The C. albicans functional homolog of CRG1, orf19.633, has a role in lipid homeostasis. (A) orf19.633, a putative methyltransferase, is required for cantharidin resistance. Fitness of wt (SN87) and orf19.633Δ/Δ mutant were measured in liquid YPD medium in the presence of cantharidin (2 mM, the IC20 for wt). Dose-response growth curves were obtained by plotting OD600 at saturation point in liquid versus tested drug concentrations. (B) Comparative lipidomics of cantharidin-treated C. albicans wild type and orf19.633Δ/Δ mutant. The cells were treated with 2 mM cantharidin (2 hours) and further prepared as described in Figure 5. Only those lipid species with significant changes in their abundance between wt and orf19.633Δ/Δ mutant are shown (P-value <0.05, Student's t-test). (C) PI species are significantly affected by cantharidin in Crg1-dependent manner. Cells were prepared as described in Figure 5. Only those PI species with significant changes in their abundance between wt and crg1Δ/Δ mutant are shown (P-value <0.05, Student's t-test). The PI content of CRG1-overexpressing mutant is not significantly different form the one in wild-type strain (P-value >0.05).
(PDF)
(Related to Figure 5.) The genetic interactome of CRG1 reveals functional networks required for buffering the absence of CRG1 in standard laboratory condition. (A) Genetic interactors of CRG1 identified through SGA analysis. The double deletion mutants crg1ΔxxxΔ were generated by SGA as previously described [86]. The double and single mutant fitness (based on colony sizes) from two independent SGA screens were used to quantify the strength of genetic interaction between CRG1 and other gene. Quantification was performed using the quantitative SGA scoring algorithm as described in [87]. The significant genetic interactors (P-value <0.05) common between two SGA screens were considered as hits. The mutants with SGA score >0.08 are significant positive or alleviating interactors, the ones with <0.08 are significant negative or aggravating interactors. (B) Deletion of CRG1 suppresses fitness defect of rvs167Δ mutant. Double and single mutants were grown in synthetic complete (SC) medium to the saturation. ura3Δ his3Δ is used as the control strain.
(PDF)
(Related to Figure 6.) (A) Transcriptional activator Yap1 is not required to activate CRG1 transcription during cantharidin stress. Sample treatment and qRT-PCR analysis were performed as described in Figure 1B. (B) CRG1 is transcriptionally activated by cell wall perturbing agents, however, it is not required for the growth in their presence. qRT-PCR analysis was performed as described in Figure 1B. Cells normalized to equal OD600 were 10-fold diluted, spotted onto solid YPD medium containing various cell wall and membrane perturbing agents, and incubated at 30°C for 2–3 days. (C) Overexpression of CRG1 confers resistance to fenpropimorph. Mid-exponentially grown cells were normalized to equal OD600 were 10-fold diluted, spotted onto solid YPD medium containing fenpropimorph (250 µM), and incubated at 30°C for 2–3 days.
(PDF)
Significantly enriched (P-value <1.0×10−6) Gene Ontology Biological processes for significantly up- and downregulated genes (log2 >1 and <−1) in the presence of cantharidin.
(XLSX)
Differentially expressed genes of methionine biosynthesis in crg1 mutants under cantharidin stress.
(XLSX)
Cantharidin-specific genetic interactors of CRG1.
(XLSX)
Statistical analysis (Kruskal-Wallis test) of lipid abundance in crg1 mutants exposed to cantharidin.
(XLSX)
Yeast strains used in this study.
(DOC)
Plasmids used in this study.
(DOC)
Primers used in this study.
(DOC)
Supporting Materials and Methods.
(DOC)
Acknowledgments
We thank Kyle Tsui (University of Toronto) for assistance with microarray analysis, Cheuk Hei Ho (University of Toronto) for providing plasmid YEp351-CRG1, Marinella Gebbia (University of Toronto) for experimental assistance, Larry Heisler and Anu Surendra (University of Toronto) for assistance in computational analysis, and Ilia Tikhomirov for critical reading and discussion of the manuscript. We are also grateful to Michael Jung and Kym Faull (UCLA) for their advice on cantharidin chemistry. Finally, we thank Gregg Czerwieniec at the Molecular Instrumentation Center at UCLA for his help with mass spectrometry.
Footnotes
The authors have declared that no competing interests exist.
EL is supported by an Ontario Graduate Scholarship. GG and CN are supported by grants from the NIH (HG003317) and CIHR (81340 to GG) and (84305 to CN). GG is a CRC chair in Chemical Genetics and is also supported by the Canadian Cancer Society (020380). MRW is supported by grants from the Singapore National Research Foundation under CRP Award No. 2007-04, the Biomedical Research Council of Singapore (R-183-000-211-035), the National Medical Research Council (R-183-000-224-213), and the SystemsX.ch RTD project LipidX. SGC is supported by NIH grant GM026020. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Katz JE, Dlakis M, Clarke S. Automated identification of putative methyltransferases from genomic open reading frames. Mol Cell Proteomics. 2003;2:525–540. doi: 10.1074/mcp.M300037-MCP200. [DOI] [PubMed] [Google Scholar]
- 2.Chiang PK, Gordon RK, Tal J, Zeng GC, Doctor BP, et al. S-Adenosylmethionine and methylation. FASEB J. 1996;10:471–480. [PubMed] [Google Scholar]
- 3.Schubert HL, Blumenthal RM, Cheng X. Many paths to methyltransfer: a chronicle of convergence. Trends Biochem Sci. 2003;28:329–335. doi: 10.1016/S0968-0004(03)00090-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loenen WAM. S-Adenosylmethionine: jack of all trades and master of everything? Biochem Soc Trans. 2006;34:330–333. doi: 10.1042/BST20060330. [DOI] [PubMed] [Google Scholar]
- 5.Giaever G, Chu AM, Ni L, Connelly C, Riles L, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- 6.Hughes TR, Marton MJ, Jones AR, Roberts CJ, Stoughton R, et al. Functional discovery via a compendium of expression profiles. Cell. 2000;102:109–126. doi: 10.1016/s0092-8674(00)00015-5. [DOI] [PubMed] [Google Scholar]
- 7.Giaever G. A chemical genomics approach to understanding drug action. Trends Pharmacol Sci. 2003;24:444–446. doi: 10.1016/S0165-6147(03)00225-6. [DOI] [PubMed] [Google Scholar]
- 8.Giaever G, Flaherty P, Kumm J, Proctor M, Nislow C, et al. Chemogenomic profiling: identifying the functional interactions of small molecules in yeast. Proc Natl Acad Sci USA. 2004;101:793–798. doi: 10.1073/pnas.0307490100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lum PY, Armour CD, Stepaniants SB, Cavet G, Wolf MK, et al. Discovering modes of action for therapeutic compounds using a genome-wide screen of yeast heterozygotes. Cell. 2004;116:121–137. doi: 10.1016/s0092-8674(03)01035-3. [DOI] [PubMed] [Google Scholar]
- 10.Lee W, St Onge RP, Proctor M, Flaherty P, Jordan MI, et al. Genome-wide requirements for resistance to functionally distinct DNA-damaging agents. PLoS Genet. 2005;1:e24. doi: 10.1371/journal.pgen.0010024. doi: 10.1371/journal.pgen.0010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kung C, Kenski DM, Dickerson SH, Howson RW, Kuyper LF, et al. Chemical genomic profiling to identify intracellular targets of a multiplex kinase inhibitor. Proc Natl Acad Sci USA. 2005;102:3587–3592. doi: 10.1073/pnas.0407170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Butcher RA, Bhullar BS, Perlstein EO, Marsischky G, LaBaer J, et al. Microarray-based method for monitoring yeast overexpression strains reveals small-molecule targets in TOR pathway. Nat Chem Biol. 2006;2:103–109. doi: 10.1038/nchembio762. [DOI] [PubMed] [Google Scholar]
- 13.Parsons AB, Lopez A, Givoni IE, Williams DE, Gray CA, et al. Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell. 2006;126:611–625. doi: 10.1016/j.cell.2006.06.040. [DOI] [PubMed] [Google Scholar]
- 14.Hoon S, Smith AM, Wallace IM, Suresh S, Miranda M, et al. An integrated platform of genomic assays reveals small-molecule bioactivities. Nat Chem Biol. 2008;4:498–506. doi: 10.1038/nchembio.100. [DOI] [PubMed] [Google Scholar]
- 15.Hillenmeyer ME, Fung E, Wildenhain J, Pierce SE, Hoon S, et al. The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science. 2008;320:362–365. doi: 10.1126/science.1150021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsvetanova NG, Klass DM, Salzman J, Brown PO. Proteome-wide search reveals unexpected RNA-binding proteins in Saccharomyces cerevisiae. PLoS ONE. 2010;5:e12671. doi: 10.1371/journal.pone.0012671. doi: 10.1371/journal.pone.0012671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, et al. The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niewmierzycka A, Clarke S. S-Adenosylmethionine-dependent methylation in Saccharomyces cerevisiae. Identification of a novel protein arginine methyltransferase. J Biol Chem. 1999;274:814–824. doi: 10.1074/jbc.274.2.814. [DOI] [PubMed] [Google Scholar]
- 19.Cai H, Dumlao D, Katz JE, Clarke S. Identification of the gene and characterization of the activity of the trans-aconitate methyltransferase from Saccharomyces cerevisiae. Biochemistry. 2001;40:13699–13709. doi: 10.1021/bi011380j. [DOI] [PubMed] [Google Scholar]
- 20.Katz JE, Dumlao DS, Wasserman JI, Lansdown MG, Jung ME, et al. 3-Isopropylmalate is the major endogenous substrate of the Saccharomyces cerevisiae trans-aconitate methyltransferase. Biochemistry. 2004;43:5976–5986. doi: 10.1021/bi049784+. [DOI] [PubMed] [Google Scholar]
- 21.Dumlao DS, Hertz N, Clarke S. Secreted 3-isopropylmalate methyl ester signals invasive growth during amino acid starvation in Saccharomyces cerevisiae. Biochemistry. 2007;47:698–709. doi: 10.1021/bi7018157. [DOI] [PubMed] [Google Scholar]
- 22.Petrossian TC, Clarke S. Multiple motif scanning to identify methyltransferases from the yeast proteome. Mol Cel Proteomics. 2009;8:1516–1526. doi: 10.1074/mcp.M900025-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang GS. Medical uses of mylabris in ancient China and recent studies. J Ethnopharmacol. 1989;26:147–162. doi: 10.1016/0378-8741(89)90062-7. [DOI] [PubMed] [Google Scholar]
- 24.Laidley CW, Cohen E, Casida JE. Protein phosphatase in neuroblastoma cells: [3H]cantharidin binding site in relation to cytotoxicity. J Pharmacol Exp Ther. 1997;280:1152–1158. [PubMed] [Google Scholar]
- 25.Moed L, Shwayder TA, Chang MW. Cantharidin revisited: a blistering defense of an ancient medicine. Arch Dermatol. 2001;137:1357–1360. doi: 10.1001/archderm.137.10.1357. [DOI] [PubMed] [Google Scholar]
- 26.Sakoff JA, Ackland SP, Baldwin ML, Keane MA, McCluskey A. Anticancer activity and protein phosphatase 1 and 2A inhibition of a new generation of cantharidin analogues. Invest New Drugs. 2002;20:1–11. doi: 10.1023/a:1014460818734. [DOI] [PubMed] [Google Scholar]
- 27.Huh JE, Kang KS, Chae C, Kim HM, Ahn KS, et al. Roles of p38 and JNK mitogen-activated protein kinase pathways during cantharidin-induced apoptosis in U937 cells. Biochem Pharmacol. 2004;67:1811–1818. doi: 10.1016/j.bcp.2003.12.025. [DOI] [PubMed] [Google Scholar]
- 28.Efferth T, Rauh R, Kahl S, Tomicic M, Bochzelt H, et al. Molecular modes of action of cantharidin in tumor cells. Biochem Pharmacol. 2005;69:811–818. doi: 10.1016/j.bcp.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 29.Li W, Xie L, Chen Z, Zhu Y, Sun Y, et al. Cantharidin, a potent and selective PP2A inhibitor, induces an oxidative stress-independent growth inhibition of pancreatic cancer cells through G2/M cell-cycle arrest and apoptosis. Cancer Science. 2010:1–8. doi: 10.1111/j.1349-7006.2010.01523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y, Casida JE. Cantharidin-binding protein: identification as protein phosphatase 2A. Proc Natl Acad Sci USA. 1992;89:11867–11870. doi: 10.1073/pnas.89.24.11867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Honkanen RE. Cantharidin, another natural toxin that inhibits the activity of serine/threonine protein phosphatases types 1 and 2A. FEBS Lett. 1993;330:283–286. doi: 10.1016/0014-5793(93)80889-3. [DOI] [PubMed] [Google Scholar]
- 32.Tsauer W, Lin JG, Lin RY, Hsu FL, Chiang HC. The effect of cantharidin analogues on xanthine oxidase. Anticancer Res. 1997;17:2095–2098. [PubMed] [Google Scholar]
- 33.Wu LT, Chung JG, Chen JC, Tsauer W. Effect of norcantharidin on N-acetyltransferase activity in HepG2 cells. American Journal of Chinese Medicine. 2001;32:161–172. doi: 10.1142/S0192415X01000186. [DOI] [PubMed] [Google Scholar]
- 34.Wang CC, Wu CH, Hsieh KJ, Yen KY, Yang LL. Cytotoxic effects of cantharidin on the growth of normal and carcinoma cells. Toxicology. 2000;147:77–87. doi: 10.1016/s0300-483x(00)00185-2. [DOI] [PubMed] [Google Scholar]
- 35.Ishihara H, Martin BL, Brautigan DL, Karaki H, Ozaki H, et al. Calyculin A and okadaic acid: inhibitors of protein phosphatase activity. Biochem Biophys Res Commun. 1989;159:871–877. doi: 10.1016/0006-291x(89)92189-x. [DOI] [PubMed] [Google Scholar]
- 36.Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, et al. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu M, Turner RJ, Winstone TL, Saetre A, Dyllik-Brenzinger M, et al. Escherichia coli TehB requires S-Adenosylmethionine as a cofactor to mediate tellurite resistance. J Bacteriol. 2000;182:6509–6513. doi: 10.1128/jb.182.22.6509-6513.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kleene SJ, Toews ML, Adler J. Isolation of glutamic acid methyl ester from an Escherichia coli membrane protein involved in chemotaxis. J Biol Chem. 1977;252:3214–3218. [PubMed] [Google Scholar]
- 39.Eberson L, Welinder H. Cyclic anhydrides. III. Equilibrium constants for the acid-anhydride equilibrium in aqueous solutions of certain vicinal diacids. J Am Chem Soc. 1971;93:5821–5826. [Google Scholar]
- 40.Tong AHY, Evangelista M, Parsons AB, Xu H, Bader GD, et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294:2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- 41.Pierce SE, Davis RW, Nislow C, Giaever G. Genome-wide analysis of barcoded Saccharomyces cerevisiae gene-deletion mutants in pooled cultures. Nat Protoc. 2007;2:2958–2974. doi: 10.1038/nprot.2007.427. [DOI] [PubMed] [Google Scholar]
- 42.Hergovich A, Stegert MR, Schmitz D, Hemmings BA. NDR kinases regulate essential cell processes from yeast to humans. Nature Rev Mol Cell Biol. 2006;7:253–264. doi: 10.1038/nrm1891. [DOI] [PubMed] [Google Scholar]
- 43.Guan XL, Wenk MR. Mass spectrometry-based profiling of phospholipids and sphingolipids in extracts from Saccharomyces cerevisiae. Yeast. 2006;23:465–477. doi: 10.1002/yea.1362. [DOI] [PubMed] [Google Scholar]
- 44.Sims KJ, Spassieva SD, Voit EO, Obied LM. Yeast sphingolipid metabolism: clues and connections. Biochem Cell Biol. 2004;82:45–61. doi: 10.1139/o03-086. [DOI] [PubMed] [Google Scholar]
- 45.Ejsing CS, Sampaio JL, Surendranath V, Duchoslav E, Ekroos K, et al. Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc Natl Acad Sci USA. 2009;106:2136–2141. doi: 10.1073/pnas.0811700106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guan XL, Souza CM, Pichler H, Dewhurst G, Schaad O, et al. Functional interactions between sphingolipids and sterols in biological membranes regulating cell physiology. Mol Biol Cell. 2009;20:2083–2095. doi: 10.1091/mbc.E08-11-1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Czabany T, Wagner A, Zweytick D, Lohner K, Leitner E, et al. Structural and biochemical properties of lipid particles from the yeast Saccharomyces cerevisiae. J Biol Chem. 2008;283:17065–17074. doi: 10.1074/jbc.M800401200. [DOI] [PubMed] [Google Scholar]
- 48.Gardocki ME, Jani N, Lopes JM. Phosphatidylinositol biosynthesis: biochemistry and regulation. Bioch Biophyscia Acta. 2005:89–100. doi: 10.1016/j.bbalip.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 49.Yin HL, Janmey PA. Phosphoinositide regulation of the actin cytoskeleton. Annu Rev Physiol. 2003;65:761–789. doi: 10.1146/annurev.physiol.65.092101.142517. [DOI] [PubMed] [Google Scholar]
- 50.Janmey PA, Linberg U. Cytoskeletal regulation: rich in lipids. Nature Mol Cell Biol. 2004;5:658–666. doi: 10.1038/nrm1434. [DOI] [PubMed] [Google Scholar]
- 51.Ren G, Vajjhala P, Lee JS, Winsor B, Munn AL. The BAR domain proteins: molding membranes in fission, fusion, and phagy. Microbiol Mol Biol Rev. 2006;70:37–120. doi: 10.1128/MMBR.70.1.37-120.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Levin DE. Cell wall integrity signaling in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 2005;69:262–291. doi: 10.1128/MMBR.69.2.262-291.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ding D, Greenberg ML. Lithium and valproate decrease the membrane phosphatidylinositol/phosphatidylcholine ratio. Mol Microbiol. 2003;47:373–381. doi: 10.1046/j.1365-2958.2003.03284.x. [DOI] [PubMed] [Google Scholar]
- 54.Sengupta N, Datta SC, Sengupta D. Platelet and erythrocyte membrane lipid and phospholipid patterns in different types of mental patients. Biochem Med. 1981;25:267–275. doi: 10.1016/0006-2944(81)90084-3. [DOI] [PubMed] [Google Scholar]
- 55.Strunecka A, Kolesarova J, Haskovec L. Lithium increases turnover of phospholipids in flight muscles of Periplaneta Americana. L Physiol Bohemoslov. 1985;34:543–547. [PubMed] [Google Scholar]
- 56.Rahier A, Schmitt P, Huss B, Benveniste P, Pommer EH. Chemical structure activity relationships of the inhibition of sterol biosynthesis by N-substituted morpholines in higher plant cells. Pestic Biochem Physiol. 1986;25:112–124. [Google Scholar]
- 57.Marcireau C, Guilloton M, Karst F. In vivo effects of fenpropimorph on the yeast Saccharomyces cerevisiae and determination of the molecular basis of the antifungal property. Antimicrob Agents Chemother. 1990;34:989–993. doi: 10.1128/aac.34.6.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liepkalns VA, Leli U, Hauser G. Alterations in the metabolism of choline-containing phospholipids by lithium and carbachol in SH-SY5Y neuroblastoma cells. Biol Psychiatry. 1993;34:51–58. doi: 10.1016/0006-3223(93)90256-d. [DOI] [PubMed] [Google Scholar]
- 59.Jancova P, Anzenbacher P, Anzenbacherova E. Phase II drug metabolizing enzymes. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2010;154:103–116. doi: 10.5507/bp.2010.017. [DOI] [PubMed] [Google Scholar]
- 60.Weinshilboum R. Pharmacogenetics of methylation: relationship to drug metabolism. Clin Biochem. 1988;21:201–210. doi: 10.1016/s0009-9120(88)80002-x. [DOI] [PubMed] [Google Scholar]
- 61.Gros L, Delaporte C, Frey S, Decesse J, de Saint-Vincent BR, et al. Identification of new drug sensitivity genes using genetic suppressor elements: protein arginine N-methyltransferase mediates cell sensitivity to DNA-damaging agents. Cancer Res. 2003;63:164–171. [PubMed] [Google Scholar]
- 62.Mishra MV, Bisht KS, Sun L, Muldoon-Jacobs K, Awward R, et al. DNMT1 as a molecular target in a multimodality-resistant phenotype in tumor cells. Mol Cancer Res. 2008;6:243–249. doi: 10.1158/1541-7786.MCR-07-0373. [DOI] [PubMed] [Google Scholar]
- 63.Turro S, Inglemo-Torres M, Estanyol JM, Tebar F, Fernandez MA, et al. Identification and characterization of associated with lipid droplet protein 1: a novel membrane-associated protein that resides on hepatic lipid droplets. Traffic. 2006;7:1254–1269. doi: 10.1111/j.1600-0854.2006.00465.x. [DOI] [PubMed] [Google Scholar]
- 64.Lorenz RT, Parks LW. Physiological effects of fenpropimorph on wild-type Saccharomyces cerevisiae and fenpropimorph-resistant mutants. Antimicrob Agents Chemother. 1991;35:1532–1537. doi: 10.1128/aac.35.8.1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Banerjee R, Zou C. Redox regulation and reaction mechanism of human cystathionine-≤-synthase: a PLP-dependent hemesensor protein. Arch Biochem Biophys. 2005;43:144–156. doi: 10.1016/j.abb.2004.08.037. [DOI] [PubMed] [Google Scholar]
- 66.Grillo MA, Colombatto S. S-adenosylmethionine and its products. Amino Acids. 2008;34:187–193. doi: 10.1007/s00726-007-0500-9. [DOI] [PubMed] [Google Scholar]
- 67.Tehlivets O, Hasslacher M, Kohlwein SD. S-Adenosyl-L-homocysteine hydrolase in yeast: key enzyme of methylation metabolism and coordinated regulation with phospholipid synthesis. FEBS Lett. 2004;577:501–506. doi: 10.1016/j.febslet.2004.10.057. [DOI] [PubMed] [Google Scholar]
- 68.Malanovic N, Streith I, Wolinski H, Rechberger G, Kohlwein SD, et al. S-Adenosyl-L-homocysteine hydrolase, key enzyme of methylation metabolism, regulates phosphatidylcholine synthesis and triacylglycerol homeostasis in yeast. J Biol Chem. 2008;283:23989–23999. doi: 10.1074/jbc.M800830200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Desfarges L, Durrens P, Juguelin H, Cassagne C, Bonneu M, et al. Yeast mutants affected in viability upon starvation have a modified phospholipid composition. Yeast. 1993;9:267–277. doi: 10.1002/yea.320090306. [DOI] [PubMed] [Google Scholar]
- 70.Balguerie A, Bagnat M, Bonneu M, Aigle M, Breton AM. Rvs161p and sphingolipids are required for actin repolarization following salt stress. Eukaryot Cell. 2002;1:1021–1031. doi: 10.1128/EC.1.6.1021-1031.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Germann M, Swain E, Bergman L, Nickels JT. Characterizing the sphingolipid signaling pathway that remediates defects associated with loss of the yeast amphiphysin-like orthologs, Rvs161p and Rvs167p. J Biol Chem. 2005;280:4270–4278. doi: 10.1074/jbc.M412454200. [DOI] [PubMed] [Google Scholar]
- 72.Kovacs P, Pinter M. Effects of phosphoprotein phosphatase inhibitors (phenylarsine oxide and cantharidin) on Tetrahymena. Cell Biochem Func. 2001;19:197–205. doi: 10.1002/cbf.913. [DOI] [PubMed] [Google Scholar]
- 73.Gustin MC, Albertyn J, Alexander M, Davenport K. MAP kinase pathways in the yeast Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1998;62:1264–1300. doi: 10.1128/mmbr.62.4.1264-1300.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nunez LR, Jesch SA, Gaspar ML, Almaguer C, Villa-Garcia M, et al. Cell Wall Integrity MAPK pathway is essential for lipid homeostasis. J Biol Chem. 2008;283:34204–34217. doi: 10.1074/jbc.M806391200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang X, Skrzypek MS, Lester RL, Dickson RC. Elevation of endogenous sphingolipid long-chain base phosphates kills Saccharomyces cerevisiae cells. Curr Genet. 2001;40:221–233. doi: 10.1007/s00294-001-0259-6. [DOI] [PubMed] [Google Scholar]
- 76.Zhang X, Lester RL, Dickson RC. Pil1p and Lsp1 negatively regulate the 3-phosphoinositide-dependent protein kinase-like kinase Pkh1p and downstream signaling pathways Pck1p and Ypk1p. J Biol Chem. 2004;279:22030–22038. doi: 10.1074/jbc.M400299200. [DOI] [PubMed] [Google Scholar]
- 77.Ericson E, Hoon S, St Onge RP, Giaever G, Nislow C. Exploring gene function and drug action using chemogenomic dosage assays. Methods Enzymol. 2010;470:233–255. doi: 10.1016/S0076-6879(10)70010-0. [DOI] [PubMed] [Google Scholar]
- 78.Collart MA, Oliviero S. Preparation of yeast RNA. Current Protocols in Molecular Biology. 1993:13.12.1–13.12.5. doi: 10.1002/0471142727.mb1312s23. [DOI] [PubMed] [Google Scholar]
- 79.Juneau K, Palm C, Miranda M, Davis RW. High-density yeast-tiling array reveals previously undiscovered introns and extensive regulation of meiotic splicing. Proc Natl Acad Sci USA. 2007;104:1522–1527. doi: 10.1073/pnas.0610354104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Perocchi F, Xu Z, Clauder-Munster S, Steinmetz LM. Antisense artifacts in transcriptome microarray experiments are resolved by actinomycin D. Nuc Acids Research. 2007;35:e128. doi: 10.1093/nar/gkm683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gelperin DM, White MA, Wilkinson ML, Kon Y, Kung LA, et al. Biochemical and genetic analysis of the yeast proteome with a movable ORF collection. Gen Dev. 2005;19:2816–2826. doi: 10.1101/gad.1362105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, et al. A generic protein purification method for protein complex characterization and proteome exploration. Nature. 1999;17:1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- 83.Bennett BD, Kimball EH, Gao M, Osterhout R, Van Dien SJ, Rabinowitz JD. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat Chem Biol. 2009;5:593–599. doi: 10.1038/nchembio.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Murray ED, Jr, Clarke S. Metabolism of a synthetic L-isoaspartyl-containing hexapeptide in erythrocyte extracts. Enzymatic methyl esterification is followed by nonenzymatic succinimide formation. J Biol Chem. 1986;261:306–312. [PubMed] [Google Scholar]
- 85.Guan XL, Riezman I, Wenk MR, Riezman H. Yeast lipid analysis and quantification by mass spectrometry. Meth Enzym. 2010;470:369–391. doi: 10.1016/S0076-6879(10)70015-X. [DOI] [PubMed] [Google Scholar]
- 86.Tong AH, Boone C. Synthetic genetic array analysis in Saccharomyces cerevisiae. Methods Mol Biol. 2006;313:171–192. doi: 10.1385/1-59259-958-3:171. [DOI] [PubMed] [Google Scholar]
- 87.Baryshnikova A, Costanzo M, Kim Y, Ding H, Koh J, et al. Quantitative analysis of fitness and genetic interactions in yeast on a genome scale. Nat Methods. 2010;7:1017–1024. doi: 10.1038/nmeth.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vizeacoumar FJ, Vreden WN, Fagarasanu M, Eitzen GA, Aitchison JD, et al. The dynamin-like protein Vps1p of the yeast Saccharomyces cerevisiae associates with peroxisomes in a Pex19p-dependent manner. J Biol Chem. 2006;281:12817–12823. doi: 10.1074/jbc.M600365200. [DOI] [PubMed] [Google Scholar]
- 89.Gietz RD, Woods RA. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002;350:87–96. doi: 10.1016/s0076-6879(02)50957-5. [DOI] [PubMed] [Google Scholar]
- 90.Robinson MD, Grigull J, Mohammad N, Hughes TR. FunSpec: a web-based cluster interpreter for yeast. BMC Bioinformatics. 2002;3:35. doi: 10.1186/1471-2105-3-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Connerth M, Grillitsch K, Köfeler H, Daum G. Analysis of lipid particles from yeast. Methods Mol Biol. 2009;579:359–374. doi: 10.1007/978-1-60761-322-0_18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Chemical SGA. Fitness profiling of wild-type double deletion mutants (ura3ΔxxxΔ) with cantharidin.
(XLS)
Chemical SGA. Fitness profiling of crg1 double deletion mutants (crg1ΔxxxΔ) with cantharidin.
(XLS)
Glycerophospholipid and sphingolipid analysis by liquid chromatography-tandem mass spectrometry in S. cerevisiae.
(XLS)
Glycerophospholipid and sphingolipid analysis by liquid chromatography-tandem mass spectrometry in C. albicans.
(XLS)
SGA scores for crg1 double deletion mutants in the absence of cantharidin.
(XLS)
Transcriptome profiling of crg1 mutants upon exposure to cantharidin treatment (250 µM for 1 hour).
(XLS)
(Related to Figure 1.) (A) CRG1 gene dose is important for cantharidin tolerance. Wt, crg1Δ/CRG1 heterozygous, crg1Δ/Δ homozygous and CRG1-overexpressing crg1Δ/Δ mutants were assessed in the presence of cantharidin in YPD. Growth curves were obtained by plotting OD600 vs. time at the tested concentrations of cantharidin. At least three independent replicates were analyzed and the representative growth curves are shown. (B) Cantharidin is more potent in SD media than in YPD. Wt, crg1Δ/Δ and CRG1-overexpressing crg1Δ/Δ mutants were assessed in the presence of cantharidin in SD medium. Dose-response curves were obtained by plotting OD600 at saturation point vs. tested drug concentrations. Values are means of three independent replicates, and error bars represent standard deviation. (C) Crg1 is important for resistance to the cantharidin analogues cantharidic acid and norcantharidin. Growth of wt, crg1Δ/Δ mutant and crg1Δ/Δ cells overexpressing CRG1 was assessed in the presence of cantharidin analogues in YPD. Dose-response curves were obtained by plotting OD600 at saturation vs. drug concentration. Values are the mean of three independent replicates, and error bars represent the standard deviation.
(PDF)
(Related to Figure 1.) (A) Cantharidin induces CRG1 transcription in a gene-dose dependent manner. Wt, crg1Δ/CRG1 heterozygous, crg1Δ/Δ homozygous deletion mutants and CRG1-overexpressing crg1Δ/Δ mutant grown to mid-exponential phase were incubated with or without cantharidin (250 µM) for 1 hour. Total RNA was extracted, cDNA synthesized and the relative abundance of CRG1 transcript was analyzed by qRT-PCR. Data are the mean of at least three independent experimental replicates, and error bars are the standard deviation. (B) GFP-tagged Crg1 is localized to the cytosol after 1 hour treatment with cantharidin (in low fluorescence medium at 4 µM). Bar, 2.5 µm (see Text S1). (C) Chemical inhibition of protein phosphatases with calyculin A results in the transcriptional induction of CRG1. Wt cells grown to mid-exponential phase in YPD were treated with calyculin A (2 µM) for 30, 60 and 120 min. Total RNA was extracted, cDNA synthesized and the relative abundance of CRG1 transcript was analyzed by qRT-PCR. Data represent the mean of at least three independent experimental replicates, and error bars are the standard deviation. (D) CRG1 is a stress-responsive methyltransferase. Expression profile of CRG1 was compared with other yeast genes during 174 diverse environmental stresses [36]. Expression profiles of CRG1, ACT1 and SSE2 are shown. (E) Crg1 protein level was not altered by mutations in methyltransferase domain. Cells were collected after induction with galactose (2%) for 3 hours. The cell lysates were analyzed by western blotting with anti-TAP antibody. Tubulin was used as an internal loading control and detected by an anti-tubulin antibody.
(PDF)
(Related to Figure 1.) (A) Viability of crg1 mutants treated with cantharidin for 1 hour. Wt, crg1Δ/CRG1 heterozygous, crg1Δ/Δ homozygous deletion mutants and CRG1-overexpressing crg1Δ/Δ mutants grown to mid-exponential phase were incubated with or without cantharidin (30 µM and 250 µM, the IC20 for crg1Δ/Δ and the IC20 for wt, respectively) for 1 hour. Cells were normalized to an equivalent OD600, 10-fold diluted, spotted onto YPD solid medium and incubated at 30°C. (B) The comparison of GO term Biological processes for the genes that are upregulated and downregulated (log2 ratio >1 and <−1) in a crg1Δ/Δ mutant treated with cantharidin (30 µM and 250 µM) for 1 hour. (C) Heat map of the transcriptional profiles of wt, crg1Δ/Δ and CRG1-overexpressing crg1Δ/Δ mutants in response to cantharidin were compared. (D) Hierarchical clustering was used to group all significantly expressed genes (at least two fold) in at least one of the strains in the presence of cantharidin (250 µM). Clusters of genes exhibiting highly similar profiles across the strains are boxed. The overrepresentation of GO Biological processes in these gene clusters are indicated on the right. (E) Cantharidin significantly induces STR3 transcript levels in CRG1-overexpressing crg1Δ/Δ mutant. Wt, crg1Δ/CRG1 heterozygous, crg1Δ/Δ homozygous deletion mutants and CRG1-overexpressing crg1Δ/Δ mutants grown to mid-exponential phase were incubated with or without cantharidin (250 µM) for 1 hour. Total RNA was extracted, cDNA synthesized and the relative abundance of STR3 transcript was analyzed by qRT-PCR. Data are the mean of at least three independent experimental replicates, and error bars are the standard deviation. (F) Treatment of cells with the non-specific methyltransferase inhibitor SAH increases sensitivity to cantharidin. Wt cells were grown in SC media with or without cantharidin (4 µM) and SAH (50 µM).
(PDF)
(Related to Figure 3.) (A) Silver stained 12% SDS-PAGE of purified TAP-tagged wild-type and mutated Crg1 (see Materials and Methods for details). (B-E) Single ion chromatograms of the major species identified in the spectra shown in Figure 3.
(PDF)
(Related to Figure 4.) (A) Two independent replicates for crg1Δ/xxxΔ pools are highly correlated. Only strains with significant log2 (drug/DMSO) (P-value <0.05) are included in the analysis. (B) The deletion of DBF2 suppresses CRG1 sensitivity to cantharidin. Cells were normalized to an equivalent OD600,10-fold diluted, spotted onto synthetic complete medium containing cantharidin (10 µM) and incubated at 30°C. (C) CRG1-dependent changes of DBF2 transcript levels in the presence of cantharidin. Wt, crg1Δ/CRG1 heterozygous, crg1Δ/Δ homozygous deletion mutants and CRG1-overexpressing crg1Δ/Δ mutants grown to mid-exponential phase were incubated with or without cantharidin (250 µM) for 1 hour. Total RNA was extracted, cDNA synthesized and the relative abundance of DBF2 transcript was analyzed by qRT-PCR. Data are the mean of at least three independent experimental replicates, and error bars are the standard deviation.
(PDF)
(Related to Figure 5.) (A) GO term enrichment analysis of cantharidin-specific CRG1-interactors before and after MDR gene filtering. Only the terms with significant P-values (<0.05) are shown. Bonferroni correction was applied to the MDR-filtered terms. (B) Representative images of cells stained with Nile Red for lipid droplets. Cells were grown at 30°C for 42 hours to reach stationary phase, and inoculated into fresh medium with cantharidin (250 µM) for 2 hours. Cells were fixed and stained with Nile Red. Bar, 2.5 µm. (C) Sterol species are not affected by the deletion of CRG1 and cantharidin treatment (see Materials and Methods for details).
(PDF)
(Related to Figure 5.) The C. albicans functional homolog of CRG1, orf19.633, has a role in lipid homeostasis. (A) orf19.633, a putative methyltransferase, is required for cantharidin resistance. Fitness of wt (SN87) and orf19.633Δ/Δ mutant were measured in liquid YPD medium in the presence of cantharidin (2 mM, the IC20 for wt). Dose-response growth curves were obtained by plotting OD600 at saturation point in liquid versus tested drug concentrations. (B) Comparative lipidomics of cantharidin-treated C. albicans wild type and orf19.633Δ/Δ mutant. The cells were treated with 2 mM cantharidin (2 hours) and further prepared as described in Figure 5. Only those lipid species with significant changes in their abundance between wt and orf19.633Δ/Δ mutant are shown (P-value <0.05, Student's t-test). (C) PI species are significantly affected by cantharidin in Crg1-dependent manner. Cells were prepared as described in Figure 5. Only those PI species with significant changes in their abundance between wt and crg1Δ/Δ mutant are shown (P-value <0.05, Student's t-test). The PI content of CRG1-overexpressing mutant is not significantly different form the one in wild-type strain (P-value >0.05).
(PDF)
(Related to Figure 5.) The genetic interactome of CRG1 reveals functional networks required for buffering the absence of CRG1 in standard laboratory condition. (A) Genetic interactors of CRG1 identified through SGA analysis. The double deletion mutants crg1ΔxxxΔ were generated by SGA as previously described [86]. The double and single mutant fitness (based on colony sizes) from two independent SGA screens were used to quantify the strength of genetic interaction between CRG1 and other gene. Quantification was performed using the quantitative SGA scoring algorithm as described in [87]. The significant genetic interactors (P-value <0.05) common between two SGA screens were considered as hits. The mutants with SGA score >0.08 are significant positive or alleviating interactors, the ones with <0.08 are significant negative or aggravating interactors. (B) Deletion of CRG1 suppresses fitness defect of rvs167Δ mutant. Double and single mutants were grown in synthetic complete (SC) medium to the saturation. ura3Δ his3Δ is used as the control strain.
(PDF)
(Related to Figure 6.) (A) Transcriptional activator Yap1 is not required to activate CRG1 transcription during cantharidin stress. Sample treatment and qRT-PCR analysis were performed as described in Figure 1B. (B) CRG1 is transcriptionally activated by cell wall perturbing agents, however, it is not required for the growth in their presence. qRT-PCR analysis was performed as described in Figure 1B. Cells normalized to equal OD600 were 10-fold diluted, spotted onto solid YPD medium containing various cell wall and membrane perturbing agents, and incubated at 30°C for 2–3 days. (C) Overexpression of CRG1 confers resistance to fenpropimorph. Mid-exponentially grown cells were normalized to equal OD600 were 10-fold diluted, spotted onto solid YPD medium containing fenpropimorph (250 µM), and incubated at 30°C for 2–3 days.
(PDF)
Significantly enriched (P-value <1.0×10−6) Gene Ontology Biological processes for significantly up- and downregulated genes (log2 >1 and <−1) in the presence of cantharidin.
(XLSX)
Differentially expressed genes of methionine biosynthesis in crg1 mutants under cantharidin stress.
(XLSX)
Cantharidin-specific genetic interactors of CRG1.
(XLSX)
Statistical analysis (Kruskal-Wallis test) of lipid abundance in crg1 mutants exposed to cantharidin.
(XLSX)
Yeast strains used in this study.
(DOC)
Plasmids used in this study.
(DOC)
Primers used in this study.
(DOC)
Supporting Materials and Methods.
(DOC)