

Abstract
Excessive excitability or hyperexcitability of glutamate-containing neurons in the brain has been proposed as a possible explanation for anxiety, stress-induced disorders, epilepsy, and some neurodegenerative diseases. However, direct measurement of glutamate on a rapid time scale has proven to be difficult. Here we adapted enzyme-based microelectrode arrays (MEA) capable of detecting glutamate in vivo, to assess the effectiveness of hyperexcitability modulators on glutamate release in brain slices of the rat neocortex. Using glutamate oxidase coated ceramic MEAs coupled with constant voltage amperometry, we measured resting glutamate levels and synaptic overflow of glutamate after K+ stimulation in brain slices. MEAs reproducibly detected glutamate on a second-by-second time scale in the brain slice preparation after depolarization with high K+ to evoke glutamate release. This stimulus-evoked glutamate release was robust, reproducible, and calcium dependent. The K+-evoked glutamate release was modulated by ligands to the a2δ subunit of voltage sensitive calcium channels (PD-0332334 and PD-0200390). Meanwhile, agonists to Group II metabotropic glutamate (mGlu) receptors (LY379268 and LY354740), which are known to alter hyperexcitability of glutamate neurons, attenuated K+-evoked glutamate release but did not alter resting glutamate levels. This new MEA technology provides a means of directly measuring the chemical messengers involved in glutamate neurotransmission and thereby helping to reveal the role multiple glutamatergic system components have on glutamate signaling.
Keywords: L-glutamate, voltammetry, metabotropic glutamate receptors, voltage sensitive calcium channels, a2δ subunits, anxiety
1. Introduction
Glutamate neurotransmission contributes to normal neural function in the central nervous system. Normally, glutamate release and signaling are tightly regulated by an ensemble of presynaptic and postsynaptic receptors in association with glutamate transporters. However, when glutamate neurotransmission falters, an aberrant functioning glutamatergic system may develop that becomes associated with several neural and psychiatric disorders. Disorders such as anxiety and stress involve an alteration in communication among neurons in various regions of the brain including the thalamus, amygdala, hippocampus, and the neocortex (Holmes and Wellman, 2009; Rodrigues et al., 2009; Shin and Liberzon, 2009). Aberrant glutamate neurotransmission can take the form of excessive excitability as increased or uncontrolled excitation alters the normal communication among a neural network (Swanson et al., 2005). The need to identify clinically effective approaches to modulating glutamate neurotransmission remains challenging and largely unmet, in part, because of the temporal and spatial constraints of existing analytical methodologies (Burmeister et al., 2002; Juranyi et al., 2003; Timmerman and Westerink, 1997; van der Zeyden et al., 2008).
One common aspect of neuronal signaling is that calcium entry into the presynaptic terminals is critical for the release of neurotransmitters from synaptic vesicles into the synaptic cleft. With this in mind, ligands, such as gabapentin and pregabalin, to the α2δ-auxiliary subunit of voltage-sensitive calcium channels (VSCC) provide a new alternative to modulating glutamate release (Dooley et al., 2000; Dooley et al., 2007). There are four variants of the a2δ subunits that interact with the channel-forming primary α1 subunit of VSCC and other subunits to form a functional VSCC (Catterall et al., 2005; Wolf et al., 2003). In particular, the α2δ-type1 subunit appears to be the major binding protein for gabapentin and pregabalin (Bian et al., 2006; Thorpe and Offord, 2010). Ligands acting on the α2δ subunit appear to have the added attribute that while normal, resting levels of neurotransmitters are not affected, stimulus-evoked release is.
An additional regulatory point for glutamate neurotransmission is the role of metabotropic glutamate receptors. Glutamate signaling is mediated by two families of glutamate receptors: the ionotropic glutamate receptors and the G-protein coupled metabotropic glutamate (mGlu) receptors. The eight subtypes of the mGlu receptor family are divided among three groups (Group I, Group II, Group III) with members of each group found both on the presynaptic and postsynaptic terminals of glutamatergic synapses. In particular, the mGlu2 receptors are located perisynapticaly on presynaptic neurons and thus are activated only when glutamate overflows the synapse, such as in cases of hyperexcitability or interruptions to the glutamate transporters (Swanson et al., 2005). The mGlu2/3 receptors are distributed throughout the brain including areas linked to anxiety and stress disorder such as the frontal cortex area (Ohishi et al., 1993a; Ohishi et al., 1993b; Petralia et al., 1996; Shigemoto et al., 1997). mGlu Group II agonists can inhibit excitatory postsynaptic potentials (Kilbride et al., 1998) and long-term potentiation induction (Huang et al., 1997) through a negative feedback action on glutamate release. In addition, mGlu2/3 receptor agonists show an anxiolytic effect in the treatment of generalized anxiety disorder (Schoepp et al., 2003).
Here we report methodology using enzyme-based microelectrode arrays (MEA) to directly measure, on a second-by-second basis, glutamate release from living brain slices of the rat neocortex. These MEAs have been shown to selectively detect glutamate, have a rapid response time (~600 msec), and measure TTX-dependent glutamate release in anesthetized or freely-moving rodents (Day et al., 2006). We first characterized the basic properties of glutamate release in brain slices of rat neocortex, determining the basic characteristics, reproducibility, and calcium dependence. Next, we examined the role two components of the glutamatergic system, VSCC and mGlu2/3 receptors, have on glutamate neurotransmission, especially stimulus-evoked glutamate release, in order to assess their potential effectiveness at modulating excessive or uncontrolled excitation.
2. Results
2.1 Using glutamate oxidase-based ceramic microelectrodes to measure glutamate
Ceramic-based MEAs capable of measuring glutamate have been used in anesthetized and freely moving preparations (Burmeister et al., 2002; Rutherford et al., 2007) but have not been tested in the in vitro conditions of a perfusion slice chamber or in brain slices. The ceramic-based MEAs (130 μm thick) used for these studies consisted of four, 50 μm × 150 μm platinum recording sites arranged in a row with 50 μm spacing between sites with an approximate cross sectional dimension of 0.015 mm2 for the MEA (Fig 1a). To verify the effectiveness of the MEA to identify glutamate in the chamber, we placed a glutamate-oxidase (GluOx) coated MEA in the chamber and superfused a solution of 60 μM glutamate dissolved in artificial cerebral spinal fluid (aCSF) through the chamber. The enzyme-coated electrode site showed a I+GluOx (current on GluOx-coated MEA site) response to the addition of glutamate to the chamber (Fig. 1C). Meanwhile, an uncoated MEA electrode site did not show a change in I−GluOx (current on MEA site without GluOx) response during a glutamate addition to the chamber. Therefore, MEA sites coated with glutamate-oxidase show a response to glutamate that is absent in the uncoated electrode site.
Figure 1.
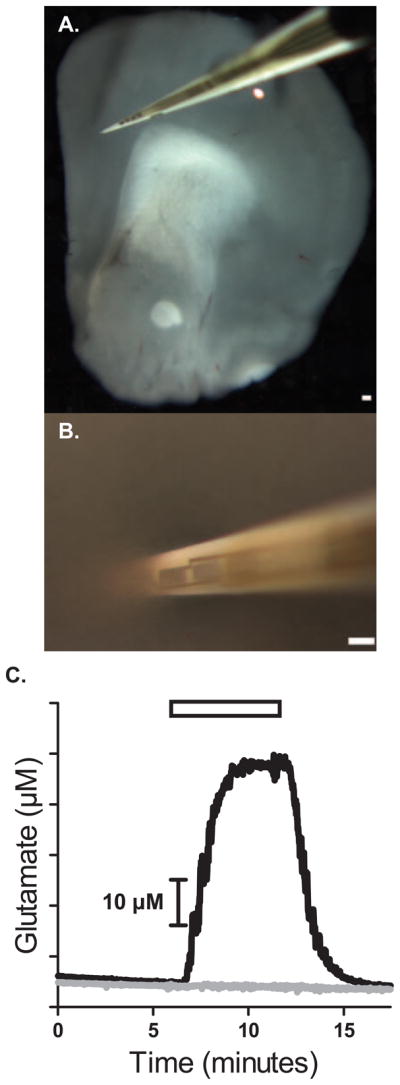
MEAs directly measure glutamate in brain slices. A) Four Pt recording sites are 150×50 μm with 50 μm spacing in between each other in a brain slice of the frontal cortex. B) The Pt site closest to the tip was inserted into the brain slice and used for measuring glutamate neurotransmission. C) MEAs coated with GluOx enzyme selectively detect glutamate. Glutamate (▭) perfusion of the recording chamber in the absence of a slice using a self-referencing microelectrode. (—) represents the Pt site coated with the GluOx solution; (—) Pt site with no enzyme coating and shows no responses to glutamate Bars: 150 μm.
2.2 K+ -evoked glutamate release in brain slices
High, extracellular K+ stimulation is commonly used to depolarize neurons and evoke neurotransmitter release. Previously, we have used high potassium stimulation to evoke glutamate release in anesthetized animals (Day et al., 2006; Quintero et al., 2007). To verify the MEA’s capability to detect dynamic glutamate neurotransmission, we superfused a high K+ (70 mM) aCSF solution to depolarize the neural network and stimulate glutamate release. Delivery of high K+ repeatedly evoked glutamate release from brain slices of the neocortex (Fig. 2) with a difference in amplitude between the first stimulation (S1) and the second stimulation (S2) of 85.5 ± 5.0 % (N = 28). Using the pre-experiment calibration of the MEA to calculate glutamate concentration, the average glutamate peak size was 4.4 ± 0.6 μM (n=28). These results indicate the utility of these MEAs to measure K+-evoked glutamate release from living brain slices.
Figure 2.

Perfusion of high K+ produced repeated increases in extracellular glutamate in brain slices. Stimulation (▲) with high K+ solution evoked repeated glutamate release. A moving average (5 point half-width) was used, and the non-faradaic current component was corrected by subtracting a moving average (1K point half-width).
2.3 K+-evoked glutamate release is calcium dependent
Synaptic neurotransmission is dependent on the entry of calcium into the presynaptic nerve terminals to enable neurotransmitter release. To determine if the glutamate release we observed was calcium mediated, we replaced extracellular calcium with the voltage-sensitive calcium channel blocker, cadmium chloride (CdCl2). Control slices receiving multiple high K+ stimulations showed a S2/S1 ratio of 0.88 ± 0.11(Fig 3A). When extracellular calcium was replaced with 500 μM CdCl2 for at least 15 minutes, glutamate release from brain slices showed an attenuated amplitude to K+-stimulation (S2/S1 ratio of 0.14 ± 0.02, t(12) = 6.386, p < 0.001, unpaired- t test; Fig 3B). After a washout period where Ca++ was reintroduced to the extracellular solution, the K+-evoked glutamate response returned (Fig. 3A).
Figure 3.

CdCl2 attenuates high K+ -evoked glutamate release. A) Repeated perfusion of the brain slice with high K+ (▭) depolarized the tissue and evoked the release of glutamate. Substitution of extracellular CaCl2 with the VSCC blocker CdCl2 (▬) attenuated the stimulus-evoked glutamate release. B) Evoked glutamate responses were significantly lower with CdCl2 in the bath than in control conditions. *: p<0.05.
2.4 Voltage-dependent calcium channel α2δ subunit ligands attenuate K+-evoked glutamate release
With the recent identification of compounds capable of binding to the α2δ subunit of VSCC (Corrigan et al., 2009; Dooley et al., 2007), we determined if these compounds would have an effect on K+-evoked glutamate release in brain slices. We used the pregabalin-like compounds PD-0332334 and PD-0200390 to assess the potential role of the α2δ subunit on stimulus-evoked glutamate release. The S2/S1 ratios were significantly lower in PD-0332334 treated slices (0.1 mM: 0.44 ± 0.15 and 1.0mM: 0.49 ± 0.12) than in control slices (0.98 ± 0.15; F(4,16) = 2.7, p = 0.035; Fig. 4A). In PD-0200390 treated slices, S2/S1 ratios were significantly lower at 1.0 μM (0.42 ± 0.08) than control slices (0.70 ± 0.07; F(4,28) = 4.19, p = 0.009; Fig. 4B). PD-0332334 and PD-0200390 decreased the size of K+-evoked glutamate release in rat brain slices likely by interfering with calcium entry. Neither PD-0332334 nor PD-0200390 significantly changed basal glutamate levels (data not shown).
Figure 4.

Ligands to the α2δ subunit of VSCC attenuated glutamate release. A) The S2/S1 ratios were significantly lower in PD-0332334 treated than in control slices. B) In PD-0200390 treated slices, S2/S1 ratios were significantly lower than control slices. *: p < 0.05.
2.5 Group II metabotropic glutamate autoreceptors attenuate K+-evoked glutamate release
Presynaptic terminals in the neocortex contain Group II mGlu receptors (mGluR) capable of providing negative feedback to reduce glutamate release from the terminals (Cahusac and Wan, 2007; Tamaru et al., 2001). To determine if Group II mGluR autoreceptors could modulate the K+ evoked glutamate release that we previously measured, we tested the effects of mGluR2/3 agonists and antagonists on glutamate neurotransmission. First, we examined if a mGluR2/3 receptor agonist and a selective antagonist would have an effect on resting glutamate levels. Baseline resting glutamate levels were not significantly changed by the mGlu2/3 receptor agonist LY379268 (10 nM to 10 μM), 2.7 ± 0.3 μM glutamate before treatment to 2.1 ± 0.4 μM glutamate after 1 μM LY379268 treatment, (F(4, 20) = 1.945, p = 0.1421; N = 5 each group). The mGlu2/3 receptor antagonist (LY341495; 1 μM) had no significant effects on resting (glutamate levels 2.3 ± 0.5 μM before treatment vs. 2.2 ± 0.9 μM after treatment; t(3) = 0.5486, p = 0.6215, N=4)
On the other hand, when the slices were superfused with 10 μM LY379268, repeated stimulation with high K+ (Figures 5b and 5c) showed a significant 36% lower S2/S1 ratio of the glutamate release peak (0.57 ± 0.08) than vehicle alone (Fig. 5a) (0.89 ± 0.09) (one-way ANOVA: F(2, 18) = 7.155, p =0.0052; Dunnett’s test: control vs. LY379268, p < 0.05, N=7). The attenuation of glutamate release was replicated with the mGlu2/3 receptor agonist, LY354740. Slices superfused with LY354740 (0.5 μM) showed a significantly attenuated high K+ -evoked glutamate release by 55% (S2/S1 = 0.40 ± 0.11; p < 0.01, N = 7) (Figure 5c), but did not significantly change baseline levels (data not shown). The mGluR2/3 antagonist LY341495 did not have a significant effect on high K+ -evoked glutamate release (control S2/S1 = 0.76 ± 0.06 vs. LY341495 S2/S1 =0.72 ± 0.07; t(6) = 0.4185; p = 0.69; N =4).
Figure 5.

The mGlu2/3 receptor agonists attenuated KCl-evoked glutamate release in rat frontal cortex brain slices. Superfusion of 70 mM KCl (□) stimulated a reproducible increase in extracellular glutamate release (A) that was attenuated (B) in the presence of 10μM LY379268 (■). LY379268 was applied at least 10 minutes before a second 70 mM KCl stimulation. C) In the presence of LY379268 and LY354740, the S2/S1 ratio of KCl-evoked glutamate release amplitudes was significantly less than control slices. *: p < 0.05; **: p < 0.01.
2.6 Local, direct K+ delivery stimulates glutamate release
In anesthetized animals, we have used pressure ejection delivery of high potassium solution to evoke glutamate release from localized areas (Nickell et al., 2006; Stephens et al., 2009). We used rapid, locally applied, pressure ejections of isotonic 70 mM potassium solution to depolarize a more focal area, measure glutamate release, and determine if activation of mGlu2/3 receptors could also affect glutamate release in this paradigm (Figure 6a). Application of 10 μM LY379268 before local 70 mM potassium stimulation decreased stimulus-evoked glutamate release by 45% (average amplitude of 1.1 ± 0.4 μM without vs. 0.6 ± 0.1 μM with 10 μM LY379268, t(5) = 7.685; p = 0.0006; N = 6, Figure 6b) similar to our observations with superfusion with the isotonic 70 mM KCl solution (see above) in brain slices. While we observed a slight shift in baseline glutamate levels (Figure 6a), there was no significant change to basal glutamate levels.
Figure 6.

Glutamate release after focal potassium stimulation was attenuated in brain slices of the frontal cortex by the selective mGlu2/3 receptor agonist LY379268 (A). Local, pressure ejection of an isotonic 70 mM potassium solution (shown by the arrowheads) produced repeated, rapid glutamate signals. B) Signal amplitudes decreased after superfusion with 10 μM LY379268 (■). *: p < 0.05.
3. Discussion
In these studies we adapted the enzyme-based microelectrode array, first developed for whole-animal recordings, for use in neocortical brain slices to directly measure extracellular glutamate at 1 Hz. Our studies revealed two different pathways by which the glutamate signals may be modulated. Brain slice recordings provide a different model for understanding glutamate function in the nervous system because the brain slice preparation contains a neural network and, at the same time, allows for the complete manipulation of discrete synaptic pathways. While there have been other methods for directly measuring extracellular glutamate such as sampling the perfusate or media of the slice chamber and then analyzing the sample with HPLC or radioactive isotopes (Barnes et al., 1988; Maneuf et al., 2001; Shinohara et al., 1998), the drawback of these techniques is that they sample the whole slice content of neurotransmitters released into the extracellular milieu, thus limiting the ability to localize the intended response, especially in a heterogeneous tissue. Additionally, the sampling rate of these techniques is slow compared to the rapid nature of neurotransmission from chemical messengers, like glutamate. Instead, the faster response time (< 2 Hz) of these GluOx-coated MEAs and the micrometer platinum (Pt) recording site surface areas enabled us to readily measure from the thin section of a brain slice. These attributes allowed the MEAs to quantify the rapid nature of extracellular glutamate signaling in brain slices in a more precise manner. For example, we were able to measure the effects of glutamate system modulators on resting glutamate levels, whole slice depolarization (70 mM KCl superfusion) and local, rapid (pressure ejection delivery of 70 mM potassium solution) depolarization-evoked glutamate release.
While we measured resting glutamate levels and stimulus-evoked glutamate release and uptake, the absence of an effect by CdCl2 on resting glutamate levels is surprising given the findings of Day et al. (2006) that showed that blocking TTX-dependent neurotransmission reduced the resting glutamate levels in vivo. However, the difference between this study and the Day et al. (2006) study may simply be a case of the difference between a slice preparation and a whole animal recording as Oldenziel et al. (2007) also did not observe a significant change in resting glutamate levels in hippocampal slices in the presence of CdCl2. Thus, it is possible that resting levels of glutamate in brain slices are less dependent on calcium as compared to the phasic release produced, in this case, by excess K+.
To examine transient glutamate signals using the MEAs, we stimulated slices with high K+ depolarization to measure glutamate neurotransmission. Glutamate signals showed a rapid rise and decay after K+ stimulation. We elected to use K+ stimulation to continue the approach we have used previously to describe glutamate and catecholamine signaling in brain slices and anesthetized animal recordings (Hoffman and Gerhardt, 1999; Quintero et al., 2007; Su et al., 1990). We found that high K+ stimulation produced consistent, robust glutamate release in brain slices. Additionally, depolarization with high K+, as shown here, can also be used either to stimulate a whole slice or only a more localized region. While we limited our observations to K+-evoked stimulation, glutamate-sensitive MEAs have been used to measure behaviorally induced glutamate release (Rutherford et al., 2007) or rapid, transient glutamate signals (Hascup et al., 2011) in behaving animals. Future studies could incorporate electrical stimulation combined with the enzyme-based MEAs to assess glutamate neurotransmission and compare the dynamics of glutamate transmission between high K+-evoked glutamate release and electrically-evoked glutamate release.
We measured glutamate signals that were calcium dependent. These results indicate that the glutamate release we measured was most likely of neuronal origin because calcium is necessary for vesicular docking and neurotransmitter release in the presynaptic terminals. In the presence of cadmium chloride, the amplitude of K+ evoked glutamate signals was reduced by 84%. While future studies may help determine which specific calcium channels are involved in this process, the general effect of calcium blockade highlights the relevance of examining modulators of calcium channels for modulating glutamate release, especially in instances of excessive or inappropriate excitation.
With this in mind, we examined the role of a relatively new class of compounds, ligands to the α2δ subunit of the voltage sensitive calcium channel. Both α2δ ligands, PD-0332334 and PD-0200390 decreased K+ evoked glutamate release in slices. While cadmium chloride reduced glutamate release amplitudes by almost 85% from controls, these two α2δ ligands reduced the glutamate release amplitudes by nearly 50% from controls. Compared with cadmium chloride, the attenuation from these compounds may have also reached a floor effect based on the results with PD-020039 where we found a significant decrease at 1 μM that was not extended to the higher dose (100 μM). That α2δ ligands can attenuate stimulus-evoked neurotransmission appears to be a hallmark of these drugs as they may provide a means of decreasing the intensity of an excitatory response without fully blocking the promulgation of excitatory communication (Cunningham et al., 2004; Dooley et al., 2007). The P/Q-type VSCC often found on the terminal region of presynaptic neurons and thought to be strongly involved in calcium dependent neurotransmission are implicated as one of the sites of action for α2δ ligands.
While previous studies have used glutamate-release-associated responses to examine the effects of mGlu2/3 activation (Marek et al., 2000), here we directly measured glutamate release and showed how mGlu2/3 receptor activation affects the release. The mGlu2/3 receptors are located in the frontal cortex, an area associated with neurological and psychiatric disorders (Ohishi et al., 1998; Petralia et al., 1996). Specifically, these receptors are localized to neurons in the extrasynaptic area and acting as autoreceptors (Conn and Pin, 1997) to provide negative feedback on glutamate release. Our results with the mGlu2/3 receptor agonists support this modulatory role of mGlu2/3 receptors to function as a presynaptic mediator of feedback signal to prevent excessive glutamate release. Still, we cannot rule out the role of glia in mediating, at least in part, some effect on glutamate release as the mGlu3 receptor expressed on glial cells (Liu et al., 1998; Shigemoto et al., 1997; Tamaru et al., 2001) may enhance glutamate uptake transport such that after a potassium stimulation, enhanced glutamate uptake might remove glutamate in sufficient amounts from the synaptic area so as to minimize synaptic overflow (D’Antoni et al., 2008).
The addition of the mGlu2/3 receptor antagonist LY341495 or the mGlu2/3 receptor agonists LY379268 or LY354740 to the slices did not change resting glutamate levels. Meanwhile, LY369268 and LY354740 did decrease K+ evoked glutamate release. The results are in line with the proposed mechanism by which mGlu2/3 receptors mediate glutamate release. Namely, only when sufficient synaptic glutamate spillover takes place would mGlu2/3 receptors exert their effects to re-establish glutamate levels to normal physiological concentrations. The implication is that mGlu2 receptors may not interfere with normal excitatory synaptic transmission, but instead may serve to prevent a hyperexcitability condition from interfering with normal brain function (Imre, 2007). Based on our results and the results of others showing a lack of an effect on resting glutamate levels by mGlu2/3 receptor agonists (Lorrain et al., 2003), normal, tonic glutamate release levels are unaltered by mGlu2/3 receptor activation, whereas the phasic or stimulus-evoked glutamate signals are modulated by mGlu2/3 receptor activation. In fact, activation of mGlu2/3 receptors can suppress the signaling of other neurotransmitters possibly through the activation of presynaptic K+ channels, suppression of a presynaptic Ca2+ conductance, and/or direct inhibition via transmitter release proteins (see Anwyl, 1999). Accordingly, in the presence of the general voltage-gated calcium channel blocker, cadmium chloride, high K+-evoked glutamate release was significantly lower in brain slices. Future studies should help clarify the pathways mediating the mGlu2/3 receptor action by which activation of the mGlu2/3 receptors can suppresses glutamate release and the role Ca2+ entry into the glutamatergic presynaptic terminals plays in the mGlu receptor mediated-suppression of neurotransmitter release.
In conclusion, we used an enzyme-based MEA to measure calcium-dependent glutamate release from neocortical brain slices and then assess the effects of pharmacological compounds to modulate glutamate neurotransmission. We think this methodology has the great potential to be used to screen compounds that not only can affect glutamate neurotransmission but other neurotransmitter systems as well (Burmeister et al., 2005; Burmeister et al., 2008). The ability to directly measure glutamate signaling provides a long sought means to a better understanding of the process of neurochemical signaling that occurs in normal states and neurological and psychiatric disorders.
4. Experimental Procedures
4.1 Glutamate-oxidase coated microelectrode arrays
MEAs were manufactured as previously described (Burmeister et al., 2002). To selectively measure glutamate, MEAs were first dip coated in a Nafion® stock solution and baked for 4 minutes in a 170°C oven which resulted in a coating that repels anionic interferents or plated for 20 minutes with a 5 mM meta-phenylenediamine solution to create a molecular size exclusion layer. Next, to measure glutamate, the MEA site closest to the tip was coated with three applications of approximately 0.1 μL of a glutamate oxidase solution [containing 1% (w/v) GluOx, 1% (w/v) bovine serum albumin, and 0.125% (w/w) glutaraldehyde] each time. The MEAs were then allowed to cure at room temperature at least 24 hours before use in an experiment. The same coated MEAs were often used multiple times, and in one occasion, for 35 different slices across 24 days. In all cases, MEAs were calibrated for glutamate sensitivity in the morning before the start of experiments.
4.2 MEA calibration
On the day of the experiment, coated MEAs were connected to a headstage amplifier (2 pA/mV) and a FAST16 mkII potentiostat (Quanteon LLC, Nicholasville, KY) to perform constant voltage amperometry (+0.7 V versus Ag/AgCl reference) at 1 Hz. Enzyme-based MEAs measure glutamate through the enzymatic breakdown of glutamate to yield a reporter molecule of hydrogen peroxide. Then, the hydrogen peroxide molecule is oxidized at the platinum recording surface and the resulting current (I+GluOX) is measured by the FAST system. The MEAs were calibrated with glutamate in vitro at 31–34°C in a phosphate buffered solution (0.05 M, pH 7.4) to (a) generate a standard curve (sensitivity > 2 pA/μM), (b) determine the limit of detection (= 3 times the signal to noise ratio; < 2.0 μM), and (c) assess the selectivity of glutamate relative to an endogenous electroactive compound, ascorbic acid (> 30:1, glutamate:ascorbic acid).
4.3 Brain slice preparation
Male Sprague Dawley rats (2–8 weeks old) were housed in AAALAC-accredited facilities. Animals had ad libitum access to food and water and were housed under a 12-hr light-dark cycle with recordings occurring during the light phase of the light-dark cycle. The brains were removed by blunt dissection and placed in ice-cold buffer until slice preparation. Coronal brain slices of the neocortex from AP: +3.7 to +1.7 mm (Paxinos and Watson, 2005) were sectioned to 350 – 400 μm thickness using a Vibratome®. All animal protocols were approved by the Animal Care and Use Committee of the University of Kentucky.
4.4 Measurements of extracellular glutamate levels in cortical brain slices
Brain slices were maintained for at least 1 h at room temperature in aCSF before starting glutamate measurements [aCSF; composition: NaCl (124 mM), KCl (5 mM), CaCl2 (2.5 mM), MgCl2 (1.5 mM), NaHCO3 (26 mM), NaH2PO4 (1.4 mM), D-glucose (10 mM); saturated with 95% O2/5% CO2; pH = 7.2–7.4]. The slices were transferred to an immersion-style chamber (one slice/chamber) and superfused at a rate of 1.5 – 2.0 ml/min with aCSF (31–33°C). Each chamber was fitted with a Ag/AgCl reference electrode. Calibrated MEAs were lowered at approximately a 60° angle into the superficial layers of the neocortex (frontal and parietal cortex) to ensure that the glutamate-oxidase coated Pt electrode site closest to the tip was completely inserted in the slice (Fig. 1b). Once baseline glutamate levels stabilized (~ 10 min.), extracellular glutamate levels were measured.
Glutamate release was evoked by stimulation through one of two means: 1) bath application of high K+, i.e. increase of KCl to depolarize slices [70 mM, except for cadmium chloride experiments (40 mM), in aCSF with a corresponding decrease of NaCl to maintain iso-osmolarity] or 2) direct, local application of 70 mM potassium solution to depolarize the local glutamatergic network via pressure ejection (70 mM KCl, 79 mM NaCl, 2.5 mM CaCl2, pH 7.0–7.4) (Gerhardt and Palmer, 1987). Bath applications of high K+ were given as 50 sec. pulses at least 15–20 min. apart.
For local application, micropipettes (10–15 μm i.d.) were pulled and bumped from 1 mm o.d., 0.58 mm i.d. glass (A-M Systems, Everett, WA) to yield the desired i.d. Micropipettes were attached and the tip was centered over the MEA recording site closest to the end (the one inserted in the slice) at a tip-to-MEA distance of 70–110 μm. The 70 mM potassium solution was applied at one-minute intervals until at least two to five reproducible glutamate responses were recorded. Delivery of solution volumes (25 nL up to 400 nL; 0.1 sec – 3 sec, 2–12 psi) was controlled by a pressure-ejection system (Picospritzer II, Parker Hannifin Corp., Cleveland, OH) and volume ejected was monitored using a stereomicroscope fitted with a reticule.
In experiments involving cadmium chloride, extracellular calcium was substituted with 0.5 mM cadmium chloride and NaH2PO4 was removed to prevent precipitants forming in the aCSF.
4.5 Materials
Substances were obtained from Sigma-Aldrich unless otherwise noted: Glutamate oxidase (Associates of Cape Cod, Falmouth, MA), meta-phenylenediamine (Acros Organics, New Jersey, USA), LY341495 (Tocris Bioscience, Ellisville, MO), LY379268 and LY354740 (donated from Eli Lilly & Co.), and PD-0332334 and PD-0200390 (donated from Pfizer Co.)
4.6 Analysis and statistical comparison
Extracellular glutamate levels were analyzed offline by custom Excel®- or MatLab®- based software. Glutamate release amplitudes were calculated as the difference between maximum K+-evoked glutamate release values and basal values. Values given are mean ± S.E.M. If appropriate, the results were analyzed using the t-statistic for group means, or one-way analysis of variance followed by post-hoc comparisons using Dunnett’s multiple comparison statistic (Prism® 5.0, GraphPad Software Inc., San Diego, CA). The minimal level of significance was p ≤ 0.05 (two-tail criterion).
Highlights.
Glutamate signals in brain slices measured with enzyme-based microelectrode arrays.
The measured high K+-evoked glutamate signals are calcium dependent.
mGlu2/3 receptor and α2δ subunits of calcium channels modulate glutamate release.
Acknowledgments
Support provided USPHS grants AG00242, NS39787, DA017186; NSF EEC-0310723, Eli Lilly and Co., and Pfizer Inc.
Abbreviations
- VSCC
voltage sensitive calcium channels
- mGlu
metabotropic glutamate
- MEA
microelectrode array
- GluOX
glutamate oxidase
- mPD
meta-phenylenediamine
- S1
first stimulus
- S1
second stimulus
- aCSF
artificial cerebrospinal fluid
Footnotes
Disclosures: GAG is the principal owner of Quanteon LLC. JEQ, FP, and PH have served as consultants to Quanteon LLC. KWJ is an employee of Eli Lilly and Co. JO is an employee of Pfizer Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anwyl R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Research Reviews. 1999;29:83–120. doi: 10.1016/s0165-0173(98)00050-2. [DOI] [PubMed] [Google Scholar]
- Barnes S, Leighton GE, Davies JA. A novel superfusion chamber for the measurement of endogenous glutamate release from cerebellar slices. Journal of Neuroscience Methods. 1988;23:57–61. doi: 10.1016/0165-0270(88)90022-2. [DOI] [PubMed] [Google Scholar]
- Bian F, Li Z, Offord J, Davis MD, McCormick J, Taylor CP, Walker LC. Calcium channel alpha2-delta type. 1 subunit is the major binding protein for pregabalin in neocortex, hippocampus, amygdala, and spinal cord: an ex vivo autoradiographic study in alpha2-delta type 1 genetically modified mice. Brain Res. 2006;1075:68–80. doi: 10.1016/j.brainres.2005.12.084. [DOI] [PubMed] [Google Scholar]
- Burmeister J, Pomerleau F, Palmer M, Day B, Huettl P, Gerhardt G. Improved ceramic-based multisite microelectrode for rapid measurements of L-glutamate in the CNS. J Neurosci Methods. 2002;119:163–171. doi: 10.1016/s0165-0270(02)00172-3. [DOI] [PubMed] [Google Scholar]
- Burmeister JJ, Palmer M, Gerhardt GA. L-lactate measures in brain tissue with ceramic-based multisite microelectrodes. Biosens Bioelectron. 2005;20:1772–1779. doi: 10.1016/j.bios.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Burmeister JJ, Pomerleau F, Huettl P, Gash CR, Werner CE, Bruno JP, Gerhardt GA. Ceramic-based multisite microelectrode arrays for simultaneous measures of choline and acetylcholine in CNS. Biosens Bioelectron. 2008;23:1382–9. doi: 10.1016/j.bios.2007.12.013. [DOI] [PubMed] [Google Scholar]
- Cahusac PMB, Wan H. Group II metabotropic glutamate receptors reduce excitatory but not inhibitory neurotransmission in rat barrel cortex in vivo. Neuroscience. 2007;146:202–212. doi: 10.1016/j.neuroscience.2007.01.049. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev. 2005;57:411–25. doi: 10.1124/pr.57.4.5. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–37. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Corrigan B, Feltner DE, Ouellet D, Werth JL, Moton AE, Gibson G. Effect of renal impairment on the pharmacokinetics of PD 0200390, a novel ligand for the voltage-gated calcium channel alpha-2-delta subunit. British Journal of Clinical Pharmacology. 2009;68:174–180. doi: 10.1111/j.1365-2125.2009.03444.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham MO, Woodhall GL, Thompson SE, Dooley DJ, Jones RS. Dual effects of gabapentin and pregabalin on glutamate release at rat entorhinal synapses in vitro. Eur J Neurosci. 2004;20:1566–1576. doi: 10.1111/j.1460-9568.2004.03625.x. [DOI] [PubMed] [Google Scholar]
- D’Antoni S, Berretta A, Bonaccorso CM, Bruno V, Aronica E, Nicoletti F, Catania MV. Metabotropic glutamate receptors in glial cells. Neurochem Res. 2008;33:2436–43. doi: 10.1007/s11064-008-9694-9. [DOI] [PubMed] [Google Scholar]
- Day BK, Pomerleau F, Burmeister JJ, Huettl P, Gerhardt GA. Microelectrode array studies of basal and potassium-evoked release of L-glutamate in the anesthetized rat brain. J Neurochem. 2006;96:1626–1635. doi: 10.1111/j.1471-4159.2006.03673.x. [DOI] [PubMed] [Google Scholar]
- Dooley DJ, Mieske CA, Borosky SA. Inhibition of K(+)-evoked glutamate release from rat neocortical and hippocampal slices by gabapentin. Neurosci Lett. 2000;280:107–110. doi: 10.1016/s0304-3940(00)00769-2. [DOI] [PubMed] [Google Scholar]
- Dooley DJ, Taylor CP, Donevan S, Feltner D. Ca2+ channel alpha2delta ligands: novel modulators of neurotransmission. Trends Pharmacol Sci. 2007;28:75–82. doi: 10.1016/j.tips.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Gerhardt GA, Palmer MR. Characterization of the techniques of pressure ejection and microiontophoresis using in vivo electrochemistry. J Neurosci Methods. 1987;22:147–159. doi: 10.1016/0165-0270(87)90009-4. [DOI] [PubMed] [Google Scholar]
- Hascup KN, Hascup ER, Stephens ML, Glaser PE, Yoshitake T, Mathe AA, Gerhardt GA, Kehr J. Resting Glutamate Levels and Rapid Glutamate Transients in the Prefrontal Cortex of the Flinders Sensitive Line Rat: A Genetic Rodent Model of Depression. Neuropsychopharmacology. 2011 doi: 10.1038/npp.2011.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman AF, Gerhardt GA. Differences in pharmacological properties of dopamine release between the substantia nigra and striatum: an in vivo electrochemical study. J Pharmacol Exp Ther. 1999;289:455–463. [PubMed] [Google Scholar]
- Holmes A, Wellman CL. Stress-induced prefrontal reorganization and executive dysfunction in rodents. Neuroscience & Biobehavioral Reviews. 2009;33:773–783. doi: 10.1016/j.neubiorev.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LQ, Rowan MJ, Anwyl R. mGluR II agonist inhibition of LTP induction, and mGluR II antagonist inhibition of LTD induction, in the dentate gyrus in vitro. Neuroreport. 1997;8:687–93. doi: 10.1097/00001756-199702100-00022. [DOI] [PubMed] [Google Scholar]
- Imre G. The preclinical properties of a novel group II metabotropic glutamate receptor agonist LY379268. CNS Drug Rev. 2007;13:444–64. doi: 10.1111/j.1527-3458.2007.00024.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juranyi Z, Zigmond MJ, Harsing LG., Jr [3H]Dopamine release in striatum in response to cortical stimulation in a corticostriatal slice preparation. J Neurosci Methods. 2003;126:57–67. doi: 10.1016/s0165-0270(03)00066-9. [DOI] [PubMed] [Google Scholar]
- Kilbride J, Huang LQ, Rowan MJ, Anwyl R. Presynaptic inhibitory action of the group II metabotropic glutamate receptor agonists, LY354740 and DCG-IV. Eur J Pharmacol. 1998;356:149–57. doi: 10.1016/s0014-2999(98)00526-3. [DOI] [PubMed] [Google Scholar]
- Liu XB, Munoz A, Jones EG. Changes in subcellular localization of metabotropic glutamate receptor subtypes during postnatal development of mouse thalamus. J Comp Neurol. 1998;395:450–65. doi: 10.1002/(sici)1096-9861(19980615)395:4<450::aid-cne3>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Lorrain DS, Baccei CS, Bristow LJ, Anderson JJ, Varney MA. Effects of ketamine and N-methyl-D-aspartate on glutamate and dopamine release in the rat prefrontal cortex: modulation by a group II selective metabotropic glutamate receptor agonist LY379268. Neuroscience. 2003;117:697–706. doi: 10.1016/s0306-4522(02)00652-8. [DOI] [PubMed] [Google Scholar]
- Maneuf YP, Hughes J, McKnight AT. Gabapentin inhibits the substance P-facilitated K(+)-evoked release of [(3)H]glutamate from rat caudial trigeminal nucleus slices. Pain. 2001;93:191–6. doi: 10.1016/S0304-3959(01)00316-5. [DOI] [PubMed] [Google Scholar]
- Marek GJ, Wright RA, Schoepp DD, Monn JA, Aghajanian GK. Physiological antagonism between 5-hydroxytryptamine(2A) and group II metabotropic glutamate receptors in prefrontal cortex. J Pharmacol Exp Ther. 2000;292:76–87. [PubMed] [Google Scholar]
- Nickell J, Salvatore MF, Pomerleau F, Apparsundaram S, Gerhardt GA. Reduced plasma membrane surface expression of GLAST mediates decreased glutamate regulation in the aged striatum. Neurobiol Aging. 2006 doi: 10.1016/j.neurobiolaging.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Ohishi H, Shigemoto R, Nakanishi S, Mizuno N. Distribution of the mRNA for a metabotropic glutamate receptor (mGluR3) in the rat brain: an in situ hybridization study. J Comp Neurol. 1993a;335:252–66. doi: 10.1002/cne.903350209. [DOI] [PubMed] [Google Scholar]
- Ohishi H, Shigemoto R, Nakanishi S, Mizuno N. Distribution of the messenger RNA for a metabotropic glutamate receptor, mGluR2, in the central nervous system of the rat. Neuroscience. 1993b;53:1009–1018. doi: 10.1016/0306-4522(93)90485-x. [DOI] [PubMed] [Google Scholar]
- Ohishi H, Neki A, Mizuno N. Distribution of a metabotropic glutamate receptor, mGluR2, in the central nervous system of the rat and mouse: an immunohistochemical study with a monoclonal antibody. Neurosci Res. 1998;30:65–82. doi: 10.1016/s0168-0102(97)00120-x. [DOI] [PubMed] [Google Scholar]
- Oldenziel WH, van der Zeyden M, Dijkstra G, Ghijsen WE, Karst H, Cremers TI, Westerink BH. Monitoring extracellular glutamate in hippocampal slices with a microsensor. J Neurosci Methods. 2007;160:37–44. doi: 10.1016/j.jneumeth.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Elsevier Academic Press; Amsterdam; Boston: 2005. [Google Scholar]
- Petralia RS, Wang YX, Niedzielski AS, Wenthold RJ. The metabotropic glutamate receptors, MGLUR2 and MGLUR3, show unique postsynaptic, presynaptic and glial localizations. Neuroscience. 1996;71:949–976. doi: 10.1016/0306-4522(95)00533-1. [DOI] [PubMed] [Google Scholar]
- Quintero JE, Day BK, Zhang Z, Grondin R, Stephens ML, Huettl P, Pomerleau F, Gash DM, Gerhardt GA. Amperometric measures of age-related changes in glutamate regulation in the cortex of rhesus monkeys. Exp Neurol. 2007;208:238–246. doi: 10.1016/j.expneurol.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Rodrigues SM, LeDoux JE, Sapolsky RM. The influence of stress hormones on fear circuitry. Annu Rev Neurosci. 2009;32:289–313. doi: 10.1146/annurev.neuro.051508.135620. [DOI] [PubMed] [Google Scholar]
- Rutherford EC, Pomerleau F, Huettl P, Stromberg I, Gerhardt GA. Chronic second-by-second measures of L-glutamate in the central nervous system of freely moving rats. J Neurochem. 2007;102:712–22. doi: 10.1111/j.1471-4159.2007.04596.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoepp DD, Wright RA, Levine LR, Gaydos B, Potter WZ. LY354740, an mGlu2/3 receptor agonist as a novel approach to treat anxiety/stress. Stress. 2003;6:189–97. doi: 10.1080/1025389031000146773. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Kinoshita A, Wada E, Nomura S, Ohishi H, Takada M, Flor PJ, Neki A, Abe T, Nakanishi S, Mizuno N. Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J Neurosci. 1997;17:7503–22. doi: 10.1523/JNEUROSCI.17-19-07503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin LM, Liberzon I. The Neurocircuitry of Fear, Stress, and Anxiety Disorders. Neuropsychopharmacology. 2009;35:169–191. doi: 10.1038/npp.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara K, Honma S, Katsuno Y, Abe H, Honma K. Circadian release of amino acids in the suprachiasmatic nucleus in vitro. Neuroreport. 1998;9:137–140. doi: 10.1097/00001756-199801050-00027. [DOI] [PubMed] [Google Scholar]
- Stephens ML, Quintero JE, Pomerleau F, Huettl P, Gerhardt GA. Age-related changes in glutamate release in the CA3 and dentate gyrus of the rat hippocampus. Neurobiol Aging. 2009 doi: 10.1016/j.neurobiolaging.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su MT, Dunwiddie TV, Mynlieff M, Gerhardt GA. Electrochemical characterization of stimulated norepinephrine overflow in locus coeruleus-hippocampus double brain grafts grown in oculo. Neurosci Lett. 1990;110:186–192. doi: 10.1016/0304-3940(90)90809-n. [DOI] [PubMed] [Google Scholar]
- Swanson CJ, Bures M, Johnson MP, Linden AM, Monn JA, Schoepp DD. Metabotropic glutamate receptors as novel targets for anxiety and stress disorders. Nat Rev Drug Discov. 2005;4:131–44. doi: 10.1038/nrd1630. [DOI] [PubMed] [Google Scholar]
- Tamaru Y, Nomura S, Mizuno N, Shigemoto R. Distribution of metabotropic glutamate receptor mGluR3 in the mouse CNS: differential location relative to pre- and postsynaptic sites. Neuroscience. 2001;106:481–503. doi: 10.1016/s0306-4522(01)00305-0. [DOI] [PubMed] [Google Scholar]
- Thorpe AJ, Offord J. The alpha2-delta protein: an auxiliary subunit of voltage-dependent calcium channels as a recognized drug target. Curr Opin Investig Drugs. 2010;11:761–70. [PubMed] [Google Scholar]
- Timmerman W, Westerink BH. Brain microdialysis of GABA and glutamate: what does it signify? Synapse. 1997;27:242–261. doi: 10.1002/(SICI)1098-2396(199711)27:3<242::AID-SYN9>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- van der Zeyden M, Oldenziel WH, Rea K, Cremers TI, Westerink BH. Microdialysis of GABA and glutamate: analysis, interpretation and comparison with microsensors. Pharmacol Biochem Behav. 2008;90:135–47. doi: 10.1016/j.pbb.2007.09.004. [DOI] [PubMed] [Google Scholar]
- Wolf M, Eberhart A, Glossmann H, Striessnig J, Grigorieff N. Visualization of the domain structure of an L-type Ca2+ channel using electron cryo-microscopy. Journal of Molecular Biology. 2003;332:171–182. doi: 10.1016/s0022-2836(03)00899-4. [DOI] [PubMed] [Google Scholar]