

Abstract
Background
A pressing clinical issue in prostate cancer (PCa) is to distinguish which men will have an indolent or aggressive course of disease. Clinical variables such as Gleason grade and stage are useful predictors of lethal cancer; however, the low predictive values of the common Gleason scores, changes in grading over time, and earlier diagnosis of patients due to screening limits their clinical utility. Identifying genetic variants associated with lethal PCa could inform clinical decision making.
Methods
We conducted a genome-wide association study comparing lethal PCa cases to cases surviving at least ten years beyond their initial diagnosis. Genotyping was performed with the Affymetrix 5.0 chip (~500,000 single nucleotide polymorphisms (SNPs) and 1483 copy number variants (CNVs)) on DNA from participants in the Physicians’ Health Study and Health Professionals Follow-up Study (196 lethal cases, 368 long-term survivors). After excluding SNPs and individuals based on quality control criteria, logistic regression assuming an additive model was performed using PLINK software.
Results
No SNP reached genome-wide significance (p≤1×10−7), however three independent SNPs had p<1×10−5. One top-ranked SNP replicated (p=0.05) in an independent follow-up study. While no CNV had genome-wide significance, 14 CNVs showed nominal association with PCa mortality (p<0.05).
Conclusions
No variants were significantly associated at a genome-wide level with PCa mortality. Common genetic determinants of lethal PCa are likely to have odds ratios <2.0.
Impact
Genetic markers identified could provide biological insight to improve therapy for men with potentially fatal cancer. Larger studies are necessary to detect genetic causes of PCa mortality.
Keywords: genome scan, prostate cancer, mortality
Introduction
One of the most urgent clinical questions in prostate cancer (PCa) is how to predict an individual’s course of disease at the time of diagnosis. PCa is the most common incident cancer (other than non-melanoma skin cancer) and the second leading cause of cancer mortality in men in the United States (1). However, the vast majority of PCa patients will not die from their cancer. While early detection and treatment play a role in cancer survival, some treated individuals still succumb to PCa while many survive without medical intervention. A recent large trial found that men randomized to prostatectomy had only a small (though significant) absolute reduction in PCa death compared to those randomized to watchful-waiting (2). Albertsen et al. (3) followed 767 men with conservatively treated localized PCa for over twenty years and observed that the majority of men (70%) did not die of PCa.
What causes one PCa patient to develop metastases or die from their cancer while others survive with the disease for many years? At present, the most utilized predictors of outcome at diagnosis are age, clinical stage, PSA level, and Gleason score. Gleason score, a measure based on the histological patterns of prostate tumors, is currently one of the best predictors. In a study using re-reviewed Gleason score from prostatectomy specimens, those with Gleason 8 cancers had a hazard of lethal cancer (dying from PCa or developing distant metastases) that was 7.4 (95% confidence interval (CI): 2.5–22) times higher than those with Gleason 3+4; cases with Gleason 9–10 had an even higher risk of lethal cancer (hazard ratio (HR)=19.1; 95% CI: 7.4–49.7) (4). However, the positive predictive value (PPV) for mortality of a higher Gleason score, including the most common Gleason 7 as well as 8–10, is only 29% (5), and therefore far from optimal. Gleason score has additional limitations as a predictor because of scoring changes over time (6, 7) and inter-observer variability (8, 9).
Epidemiological and experimental evidence supports the hypothesis that aggressive cancer has an inherited component. A recent study showed concordance of survival and PCa mortality among fathers and sons with PCa, implying that prognosis itself may have a hereditary component (10). Laboratory experiments using a highly metastatic mouse mammary model crossed with several different strains showed that the genetic background of an animal can influence the metastatic efficiency (11). Further quantitative trait mapping work identified regions on chromosome 19 that were significantly associated with metastatic efficiency, suggesting that inherited variation may influence metastasis (12). Thus far, genetic studies in humans have focused on Gleason score as a proxy for aggressive disease. Several regions have been implicated in linkage scans, but three of the regions (5q31–33, 7q31–33 and 19q12-q13.3) were strongly significantly associated with high-grade cancer (p<0.001) and replicated in at least two independent studies suggesting a locus may be present under these peaks (13–16).
In PCa genetic association studies for risk, a combined analysis of two GWAS identified a variant at chromosome 9q33.2 in a putative tumor suppressor gene (DABP2IP) that was associated with risk of aggressive PCa, defined by Gleason grade and clinical stage (17). Another study found that the TT genotype of rs4054823 at 17p12 that increased risk of aggressive cancer compared to non-aggressive cancer, again defined by clinical variables (18). A germ-line deletion at 2p24.3 was more strongly associated with the risk of aggressive cancer than non-aggressive cancer (19).
Substantial longitudinal follow-up is required to capture information on PCa mortality, so this outcome is studied less frequently than Gleason score. However, we believe that a large-scale genetic study for the most important PCa outcome is crucial to improve our understanding of PCa aggressiveness. We therefore performed a genome-wide association study (GWAS) for PCa mortality in the Physicians’ Health Study and Health Professionals Follow-up Study, with a replication study in the Dana-Farber Harvard Cancer Center SPORE (Gelb Center) case-series. In addition to examining the association of genotypes, we also evaluated whether copy number variants (CNVs) were associated with PCa mortality.
Methods
Study Population
Physicians’ Health Study (PHS)
The PHS began as a randomized, double-blind trial of aspirin and β-carotene in the prevention of cardiovascular disease and cancer among 22,071 healthy US physicians; written consent was obtained from each participant at the time of initial enrollment and the investigation was approved by the Human Subjects Committee at Brigham and Women’s Hospital. Men were excluded if they had any serious medical conditions at baseline including all cancers (except non-melanoma skin cancer). Blood samples were collected from 68% of the physicians in 1982–1984, as described previously (20). Participants are followed through annual questionnaires to collect data on diet, health and lifestyle behaviors, and medical history, and biannually through postcards to ascertain health endpoints, including PCa. All self-reported PCa cases are verified through medical record and pathology review. Through this systematic medical record review we also abstract data on clinical information, such as Gleason score. Cause of death is determined by review of death certificates, medical records, and information from the family by a panel of three physicians. There is a high follow-up rate for both cancer incidence (96%) and mortality (98%). Metastases are reported on follow-up questionnaires sent to all men living with PCa and are confirmed through medical record review.
For the current study, we included incident PCa diagnosed between 1982 and 2003, and restricted participants to self-reported Caucasians to reduce potential population stratification. Due to cost restraints, we were unable to genotype all PHS PCa cases on whom blood had been collected. We therefore examined the two extremes of PCa cases: long-term survivors (patients who survived a minimum of 10 years after diagnosis until death or end of follow-up (March 1, 2008) and did not develop metastases to bone or organs or die from PCa; n=415) and lethal PCa cases (patients who developed metastases to bone or organs after diagnosis or died from PCa; n=176).
Health Professionals Follow-up Study (HPFS)
The HPFS, an ongoing prospective cohort study on the causes of cancer and heart disease in men, consists of 51,529 U.S. health professionals who were aged 40–75 years in 1986 (21). At baseline and then biennially participants responded to a mailed questionnaire that included questions on demographics, lifestyle, and medical history. Between 1993 and 1995, 18,018 of the men provided a blood specimen. When a participant reports a PCa diagnosis medical and pathology records are obtained. Study investigators review these records to confirm the diagnosis and to abstract stage at diagnosis and Gleason grade. Deaths among cohort members were identified by reports from next-of-kin, the postal service, or searches of the National Death Index. In order to increase the number of lethal cancers in this study, we included 46 PCa deaths from the HPFS (self-reported Caucasian) among cases diagnosed between 1993 and 2000; these were selected from a larger nested case-control study and had the most available DNA from a total of 53 PCa deaths.
Dana-Farber Harvard Cancer Center SPORE (Gelb Center)
The Gelb Center is a case series of PCa patients diagnosed between 1976 and 2007. A detailed description of this study has been published previously (22). The study captures detailed clinical information from multiple sources, including medical records and patient registration, and a blood sample collected after diagnosis. Follow-up of the participants occurs at clinic visits to the Dana-Farber Cancer Institute and by searching the National Death Index. Because cause of death is not always available or known, if an individual was known to have metastases before their death they were assumed to have died from PCa. For this study, after restricting to self-reported Caucasians, we included 155 long-term survivors (end of follow-up July 1, 2007) and 500 lethal cases as a replication set.
GWAS
Affymetrix Scan
The samples from the PHS and HPFS were included in the genome scan. DNA was extracted from peripheral blood samples for all participants. Genotyping was performed with the Affymetrix 5.0 SNP chip, which contains probes for 500,568 SNPs. Briefly, approximately 500 ng of DNA from each sample is digested with Nsp and Sty restriction enzymes. The digested segments were ligated to enzyme specific adaptors which incorporate a universal PCR priming sequence; PCR amplification using universal primers was performed in a reaction optimized to amplify fragments. The products are fragmented, end-labeled with biotinylated nucleotides, and hybridized to a chip and detected (23). The resulting intensities for each allele are used to make a genotype call. The “Birdseed” calling algorithm, an updated version of the Robust Linear Modeling using Mahalanobis distance (RLMM) calling algorithm developed at the Broad Institute of Harvard and MIT, was used for this study (24). More information on the technology, calling algorithm, and SNP coverage can be found at (25).
Samples and Quality Control
A total of 637 unique samples from PHS and HPFS were included in this study; deaths and long-term survivors were interspersed across seven 96-well plates and laboratory personnel were blinded to outcome. Each plate had two empty wells (negative controls) as well as two duplicates to be used for quality control.
We assessed the genotype concordance of 458 SNPs from 500 kb regions of chromosomes 1, 5, 10, 15, and 20 for the 14 duplicate pairs (concordance=99.9%). We also compared the genotype calls for 31 SNPs that had previously been genotyped on a subset of these PHS participants; concordance was 99.3% for >14,000 genotypes.
Data Analysis
The PLINK program(26) was used to analyze these genome scan data (27). Forty-six individuals (33 long-term survivors, 13 deaths) with <90% of genotype calls made were removed from the analysis; the average call rate in the remaining individuals was 98.8%. Of the SNPs genotyped, SNPs missing >10% of genotypes (14,704), with minor allele frequency (MAF) <1% (68,603), or with Hardy-Weinberg Equilibrium p<1×10−6 (1,979) were excluded, leaving 419,613 SNPs for analysis.
To address potential remaining population stratification, we utilized the Eigenstrat program (28). We ran this program for all participants with the default parameters (5 outlier iterations across the top 10 eigenvectors, with outliers exceeding 6 standard deviations along a top principal component excluded), and output the first two eigenvector values. Several individuals were not assigned values along these eigenvectors due to missing data (as described above) or were designated outliers (14 long-term survivors, 12 deaths); these individuals were excluded from further analysis. Using PLINK, for the main analysis we ran an unconditional logistic regression model adjusting for the first two eigenvectors (excluding 1 HPFS death missing age at diagnosis), outputting the additive model results for the association of each SNP with lethal PCa (n=196) versus long-term survival (n=368). We then ran secondary analyses additionally adjusting for age at diagnosis and restricting to men with Gleason score of 7.
Follow-up Study
The Gelb Center samples were utilized for a genetic replication study. We selected and designed assays for SNPs with p<1×10−3 that fell in previously identified linkage peaks for Gleason score (chr5q31–33 n=6, chr7q31–33 n=1); we then selected markers to capture the independent variation with p<5×10−4 (n=72). Genotyping was performed with Sequenom iPLEX matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometry technology; see (29) for reaction details. The association of the additive model of these SNPs with lethal PCa versus long-term survival was performed using unconditional logistic regression. SNPs were excluded from analysis if they had <90% genotyping success rate. Of the 79 SNPs genotyped in the Gelb samples, 11 failed genotyping quality control. Replication was declared only if p≤0.05 and the direction of the effect was the same as in the GWAS; for the replicated SNP, a joint analysis with the original GWAS data was performed as a meta-analysis with a random-effects model. Analysis was performed with SAS v9.1 statistical software.
Copy Number Polymorphism analysis
We analyzed SNP chip based copy number polymorphism data as generated by the CNV detection software Canary (30) in the form of summarized intensity scores for 1483 CNVs and 565 subjects. We followed the subject filtering criteria as described above in our genotype analysis; individuals were excluded who were missing considerable data or were found to be outliers by Eigenstrat. Then we followed a likelihood ratio approach for testing association between each CNV and the binary status of mortality considered as a trait. The approach jointly fits two linear models, as outlined in Barnes et al. (31), and is described as follows. The first model classifies the summarized intensities for each CNV by fitting a finite mixture of Gaussian densities via an EM based algorithm that uses Bayesian Information Criterion (BIC) to select the optimal number of classes. Upon convergence, the classification assigns every individual subject to a copy number genotype, and given an optimal model with multiple copy number classes, we tested for its association with the subject’s trait with this joint model. This is done by fitting of a generalized logit linear model to test the null hypothesis H0 that there is no association between a subject’s copy number genotype and his binary PCa mortality trait (in this case, lethal/indolent). If the fitting is correct and there is indeed no association, then the computed likelihood ratio (LR) statistic is χ2 distributed with 1 degree of freedom, which leads to a corresponding p-value of association. The plots and statistics for CNV classification and the associated distribution of trait were generated with the BioConductor package CNVtools.
Results
GWAS results
A description of the PHS and HPFS participants is provided in Table 1. Although participants were restricted to self-reported Caucasians, residual population stratification was addressed with the Eigenstrat program (28). The correlation of eigenvectors 1 and 2 with outcome status was 0.046 and 0.009, respectively, demonstrating that overall population structure was not strongly related to outcome; the first two eigenvectors for the lethal PCa cases and long-term survivors are shown in Supplementary Figure 1.
Table 1.
Description of genome-wide association study and replication study participants
| PHS† | HPFS† | Gelb Center | |||
|---|---|---|---|---|---|
| Clinical Characteristics | lethal (n=176) | long-term survivors (n=415) | lethal (n=46) | lethal (n=500) | long-term survivors (n=155) |
| age at diagnosis, mean (s.d.) | 70.4 (8.4) | 67.8 (6.6) | 71.8 (7.3) | 62.4 (8.3) | 61.8 (7.4) |
| Gleason score*, % | (n=130) | (n=362) | (n=36) | (n=433) | (n=134) |
| 2–6 | 26.2 | 58.3 | 19.4 | 18.2 | 53.7 |
| 7 | 36.2 | 32.3 | 47.2 | 32.8 | 34.3 |
| 8–10 | 37.7 | 9.3 | 33.3 | 49.0 | 11.9 |
| Clinical stage, % | (n=161) | (n=404) | (n=38) | (n=360) | (n=97) |
| T1, T2 | 58.4 | 91.6 | 47.4 | 65.8 | 99.0 |
| T3, T4, N1, M1 | 41.6 | 8.4 | 52.6 | 34.2 | 1.0 |
| follow-up, median yrs (range) | 5.5 (0.1–17.9) | 15.4 (10–25.3) | 4.9 (0.1–11.1) | 5.7 (0.3–24.4) | 13.7 (10.5–21.8) |
values for all cases and the subset included in the final analysis are comparable
Gleason score was only from biopsy for Gelb Center; preferentially from prostatectomy then from biopsy for PHS and HPFS
A set of 419,613 SNPs passed quality control and were used for subsequent analyses (see Methods). A q-q plot of the results compares the chi-square values obtained in this study with the expected distribution under the null hypothesis of no association between genetic variation and mortality (Figure 1). Although no SNPs reached genome-wide significance (p≤1×10−7), three independent SNPs had p<1×10−5; the plot of p-values (Figure 2) shows that there are peaks on chromosome 2q31.2, 11q12.2 and 11q14.1. The results for all SNPs with p<1×10−3 (n=277) are provided in Supplementary Table 1.
Figure 1.
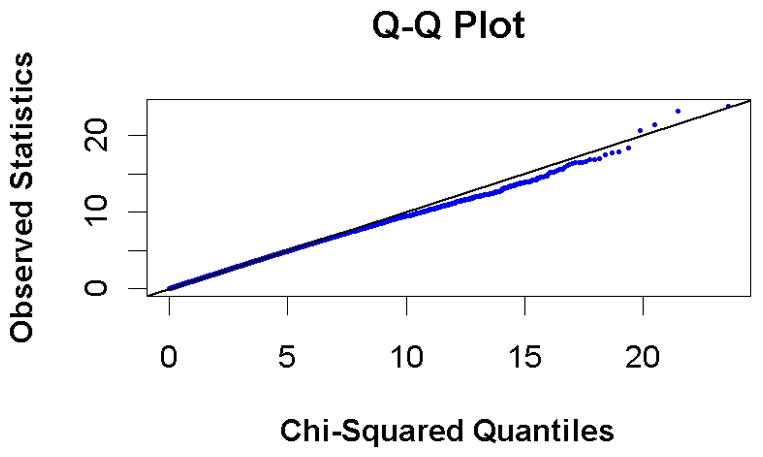
quantile-quantile plot, comparing observed statistics for all results to those expected based on the null distribution
Figure 2.
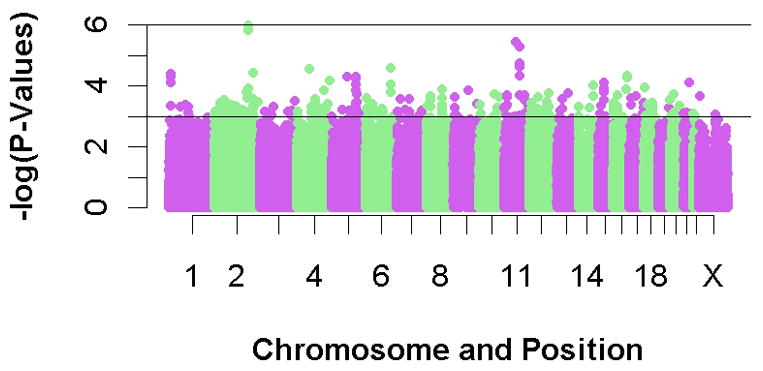
p-values for the association of SNPs with PCa mortality plotted by chromosome and position
To determine the associations of SNPs on mortality independent of their possible associations with age at diagnosis, we ran the analysis adjusting for age at diagnosis (continuous) in addition to the top 2 eigenvectors. When adjusting for age at diagnosis, there are 3,767 results with p<0.01; 19% of these results are not among the 3,803 results with p<0.01 from our main analysis. However, the top SNPs from the non-age adjusted results (Supplementary Table 1) all have p<0.005 in the adjusted analysis, suggesting the overall effect of SNPs on mortality through age at diagnosis may not be substantial. We also examined the association of SNPs with lethal cancer restricting to cases with Gleason 7; again, no SNPs reached genome-wide significance. With this much smaller number of participants, half of the SNPs with p<0.001 had p<0.05 in the main analysis.
We examined the results for previously identified PCa risk SNPs in our scan. Sixteen of the 31 confirmed risk SNPs compiled by Varghese and Easton (32) were either directly genotyped in our scan or had a proxy with R2>0.8. The most significant finding was for rs16901979 where the risk allele decreased the probability of lethal cancer (OR=0.35, p=0.006); all results are reported in Supplementary Table 2.
Replication study results
Since the majority of the top ranked SNPs from the scan will be false positives, we performed a replication study in the Dana-Farber Harvard Cancer Center SPORE Gelb Center (500 lethal cases, 155 indolent). We selected top ranked SNPs (p<10−3) that were located in previously identified Gleason linkage peaks (n=7). We also then selected markers to capture the independent variation with p<5×10−4 (n=72). Of the seventy-nine SNPs selected, 68 were successfully genotyped. Six of these had p≤0.05, but for five the direction of the effect was not consistent with the scan. The one SNP that replicated with the effect in the same direction, rs6973814 (odds ratio=1.95, 95% CI: 1.01, 3.79; p=0.05), was ranked 66th in the original GWAS (odds ratio=3.07; p=0.0003) and is located on chromosome 7q11.2 (nearest gene, AUTS2, 600kb away). In a joint analysis with the scan results, the combined odds ratio was 2.50 (95% CI: 1.60, 3.90; p=6×10−5). All Gelb Center results are in Supplementary Table 3.
CNV results
The model fitting results and number of classes for all 1483 CNVs are provided in Supplementary Table 4. For the copy number variants where the classification (based on iterative EM modeling) converged and produced more than one CNV genotype class (N=341), we examined the association between the number of copies an individual carries and lethal PCa. Fourteen CNVs had a nominal p<0.05; however, none remained significant after correction for multiple testing (Supplementary Figure 2 and Table 2).
Table 2.
Significant associations (p<0.05) between CNVs and PCa mortality
| CNV id | Chromosome | Start | Stop | Size (kb) | P-value | Gene (Location) |
|---|---|---|---|---|---|---|
| CNP11198 | 7p21 | 17060348 | 17062853 | 2.51 | 0.001 | |
| CNP11500 | 8q23 | 107607836 | 107615669 | 7.83 | 0.002 | OXR1 (chr8:107351648-107834097) |
| CNP399 | 3p22 | 37957108 | 37961932 | 4.82 | 0.002 | CTDSPL (chr3:37878672-38000964) |
| CNP10373 | 2q14 | 125368402 | 125374832 | 6.43 | 0.005 | CNTNAP5 (chr2:124499333-125389333) |
| CNP10161 | 1q24 | 166355735 | 166358038 | 2.30 | 0.007 | GPR161 (chr1:166320620-166372248) |
| CNP147 | 1q31 | 194997658 | 195068695 | 71.04 | 0.008 | CFHR3 (chr1:195010552-195029496) |
| CNP10030 | 1p36 | 15665011 | 15683808 | 18.80 | 0.012 | ELA2A (chr1:15655810-15671169) |
| CNP2113 | 15q24 | 74678296 | 74682830 | 4.53 | 0.014 | SCAPER (chr15:74427591-74963247) |
| CNP2430 | 19q13 | 56834427 | 56840009 | 5.58 | 0.021 | SIGLEC14 (chr19:56837617-56841944) |
| CNP207 | 2p22 | 34552819 | 34590561 | 37.74 | 0.023 | |
| CNP10834 | 4q32 | 162413794 | 162424561 | 10.77 | 0.031 | |
| CNP2150 | 16p12 | 19853151 | 19874863 | 21.71 | 0.033 | |
| CNP211 | 2p22 | 35831294 | 35841451 | 10.16 | 0.036 | |
| CNP11018 | 6p25 | 5978930 | 5979435 | 0.51 | 0.038 |
Discussion
A number of recent GWAS and follow-up replication studies have identified over twenty bona fide genetic PCa risk loci (33–40). Importantly, these studies have provided a new look into the biology of developing the disease. Some of these variants have been tested for association with aggressiveness, typically using the Gleason grade as a proxy for aggressive disease. However, identifying genetic determinants of lethal cancer could improve on the current clinical predictive ability at diagnosis. Understanding who would and who would not benefit from intervention could impact the selection of appropriate medical therapy for the individual, preventing unnecessary treatments and the physical and psychological side effects. In addition, the markers themselves may provide biological insight that could lead to improved therapy for those with potentially fatal cancer.
In this GWAS for lethal cancer, although no SNPs reached genome-wide significance, we identified one top-ranked SNP that replicated in an independent population. The closest gene to the one SNP that was replicated is AUTS2. A recent study based on mRNA expression data reported that this gene was included in the top 100 potential genetic mediators for non-recurrent primary PCa (41), suggesting a possible biological function.
As noted by McCaroll (42), it is increasingly possible to extend GWAS to examine CNVs and their association with disease phenotypes. In recent years, the SNP arrays have been redesigned to contain probes at the majority of CNVs, which in turn take advantage of the recent high-resolution maps of the CNV locations (43, 44). In this direction, the present GWAS was extended to study CNV in the same SNP array data based on 1483 mapped CNVs using a robust statistical modeling algorithm for classification. While no CNV achieved genome-wide significance, we identified 14 CNVs nominally associated with PCa mortality. Subsequent data mining with alternate modeling strategies or larger studies may reveal further associations.
PCa mortality is one of the most important phenotypes of this disease. Unfortunately, due to the long follow-up time and the cost necessary to obtain this information, few studies have information on survival and cause of death or the numbers of lethal cases necessary to study this outcome. A major strength of this study is its ability to examine the primary prostate cancer endpoint, lethal disease, with a substantial number of participants from cohorts that have been followed for decades. The top results were then evaluated in a large case-series that also captures survival data.
Figure 3 demonstrates we are only powered to detect relatively strong effects (e.g., OR>2 with MAF>20%). While this is a limitation of our study, it also provides insight into the genetic variants involved in PCa aggressiveness. Based on our data, no common variant will have a large effect on aggressiveness, but rather will most likely have the same magnitude of effect as the alleles that have previously been identified for risk. Although our one SNP that did replicate had a larger combined OR of 2.5, in the replication dataset alone the OR=1.95, suggesting that the initial finding is likely overestimating the magnitude of the effect.
Figure 3.

Power for genome scan
Power was calculated using the number of cases included in the final analysis (196 lethal PCa, 368 long-term survivors) with an alpha-level of 1×10−7 across a range of allele frequencies and additive model odds ratios
Another possible limitation (albeit one that exists in all studies of PCa mortality that are conducted in screened and treated populations) is misclassification of the outcome. Individuals who were labeled as having indolent cancer because they survived at least ten years without developing metastases or dying of cancer may only be in this category because they received aggressive medical treatment, without which they would have died. However, as the results of the Swedish randomized trial of prostatectomy versus watchful waiting suggest, the number needed to treat to save the life of one man with PCa is 19 (45); thus, the potential impact of misclassification is likely to be minimal. Additionally, it is important to investigate if these genetic variants predict PCa mortality independent of clinical variables such as treatment or Gleason score; however, missing data limits our ability to conduct these analyses. We did perform an analysis restricting to the most common Gleason score of 7; while results were somewhat similar to the overall analysis, a larger future study examining these associations among men with Gleason 7 would be interesting and could identify SNPs that are associated with lethal cancer independent of their effects on Gleason. A limitation in the CNV analysis is the number of probes included on this Affymetrix chip; a more comprehensive study of CNVs with PCa mortality should be performed.
Although several SNPs have been identified that are associated with risk of prostate cancer, these SNPs in general have not been found to confer an increased risk of aggressive compared to indolent disease. If lethal prostate cancer does indeed have a genetic component, this suggests that genetic variants determining aggressive disease are different from those that confer overall risk. It would be of clinical utility if future studies specifically focused on attempting to differentiate lethal from indolent cancer using germline genetic scans and follow-up studies.
Supplementary Material
Acknowledgments
This work was supported by a grant from the Doris Duke Clinical Scientist Development Award. The laboratory work at the Broad Institute Center for Genotyping and Analysis was supported by grant U54 RR020278 from the National Center for Research Resources. The PHS was supported by grants CA-34944, CA-40360, and CA-097193 from the National Cancer Institute and grants HL-26490 and HL-34595 from the National Heart, Lung, and Blood Institute, Bethesda, Maryland. The HPFS was supported by CA-55075 from the National Cancer Institute. Additional funding provided by the Dana-Farber/Harvard Cancer Center Prostate Cancer SPORE (P50 CA090381-08). The genotyping was performed by the Broad Institute Center for Genotyping and Analysis, which is supported by U54 RR020278 from the National Center for Research Resources. KLP was supported by a National Research Service Awards (T32 CA009001-34).
References
- 1.American Cancer Society. Cancer Facts and Figures, 2008. Atlanta, Georgia: American Cancer Society; 2008. [Google Scholar]
- 2.Bill-Axelson A, Holmberg L, Ruutu M, Haggman M, Andersson SO, Bratell S, et al. Radical prostatectomy versus watchful waiting in early prostate cancer. N Engl J Med. 2005;352:1977–84. doi: 10.1056/NEJMoa043739. [DOI] [PubMed] [Google Scholar]
- 3.Albertsen PC, Hanley JA, Fine J. 20-year outcomes following conservative management of clinically localized prostate cancer. Jama. 2005;293:2095–101. doi: 10.1001/jama.293.17.2095. [DOI] [PubMed] [Google Scholar]
- 4.Stark JR, Perner S, Stampfer MJ, Sinnott JA, Finn S, Eisenstein AS, et al. Gleason score and lethal prostate cancer: does 3 + 4 = 4 + 3? J Clin Oncol. 2009;27:3459–64. doi: 10.1200/JCO.2008.20.4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andren O, Fall K, Franzen L, Andersson SO, Johansson JE, Rubin MA. How well does the Gleason score predict prostate cancer death? A 20-year followup of a population based cohort in Sweden. J Urol. 2006;175:1337–40. doi: 10.1016/S0022-5347(05)00734-2. [DOI] [PubMed] [Google Scholar]
- 6.Albertsen PC, Hanley JA, Barrows GH, Penson DF, Kowalczyk PD, Sanders MM, et al. Prostate cancer and the Will Rogers phenomenon. J Natl Cancer Inst. 2005;97:1248–53. doi: 10.1093/jnci/dji248. [DOI] [PubMed] [Google Scholar]
- 7.Mitchell RE, Shah JB, Desai M, Mansukhani MM, Olsson CA, Benson MC, et al. Changes in prognostic significance and predictive accuracy of Gleason grading system throughout PSA era: impact of grade migration in prostate cancer. Urology. 2007;70:706–10. doi: 10.1016/j.urology.2007.06.1084. [DOI] [PubMed] [Google Scholar]
- 8.Allsbrook WC, Jr, Mangold KA, Johnson MH, Lane RB, Lane CG, Amin MB, et al. Interobserver reproducibility of Gleason grading of prostatic carcinoma: urologic pathologists. Hum Pathol. 2001;32:74–80. doi: 10.1053/hupa.2001.21134. [DOI] [PubMed] [Google Scholar]
- 9.Allsbrook WC, Jr, Mangold KA, Johnson MH, Lane RB, Lane CG, Epstein JI. Interobserver reproducibility of Gleason grading of prostatic carcinoma: general pathologist. Hum Pathol. 2001;32:81–8. doi: 10.1053/hupa.2001.21135. [DOI] [PubMed] [Google Scholar]
- 10.Hemminki K, Ji J, Forsti A, Sundquist J, Lenner P. Concordance of survival in family members with prostate cancer. J Clin Oncol. 2008;26:1705–9. doi: 10.1200/JCO.2007.13.3355. [DOI] [PubMed] [Google Scholar]
- 11.Lifsted T, Le Voyer T, Williams M, Muller W, Klein-Szanto A, Buetow KH, et al. Identification of inbred mouse strains harboring genetic modifiers of mammary tumor age of onset and metastatic progression. Int J Cancer. 1998;77:640–4. doi: 10.1002/(sici)1097-0215(19980812)77:4<640::aid-ijc26>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 12.Hunter KW, Broman KW, Voyer TL, Lukes L, Cozma D, Debies MT, et al. Predisposition to efficient mammary tumor metastatic progression is linked to the breast cancer metastasis suppressor gene Brms1. Cancer Res. 2001;61:8866–72. [PubMed] [Google Scholar]
- 13.Witte JS, Goddard KA, Conti DV, Elston RC, Lin J, Suarez BK, et al. Genomewide scan for prostate cancer-aggressiveness loci. Am J Hum Genet. 2000;67:92–9. doi: 10.1086/302960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Witte JS, Suarez BK, Thiel B, Lin J, Yu A, Banerjee TK, et al. Genome-wide scan of brothers: replication and fine mapping of prostate cancer susceptibility and aggressiveness loci. Prostate. 2003;57:298–308. doi: 10.1002/pros.10304. [DOI] [PubMed] [Google Scholar]
- 15.Slager SL, Schaid DJ, Cunningham JM, McDonnell SK, Marks AF, Peterson BJ, et al. Confirmation of linkage of prostate cancer aggressiveness with chromosome 19q. Am J Hum Genet. 2003;72:759–62. doi: 10.1086/368230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Slager SL, Zarfas KE, Brown WM, Lange EM, McDonnell SK, Wojno KJ, et al. Genome-wide linkage scan for prostate cancer aggressiveness loci using families from the University of Michigan Prostate Cancer Genetics Project. Prostate. 2006;66:173–9. doi: 10.1002/pros.20332. [DOI] [PubMed] [Google Scholar]
- 17.Duggan D, Zheng SL, Knowlton M, Benitez D, Dimitrov L, Wiklund F, et al. Two genome-wide association studies of aggressive prostate cancer implicate putative prostate tumor suppressor gene DAB2IP. J Natl Cancer Inst. 2007;99:1836–44. doi: 10.1093/jnci/djm250. [DOI] [PubMed] [Google Scholar]
- 18.Xu J, Zheng SL, Isaacs SD, Wiley KE, Wiklund F, Sun J, et al. Inherited genetic variant predisposes to aggressive but not indolent prostate cancer. Proc Natl Acad Sci U S A. 107:2136–40. doi: 10.1073/pnas.0914061107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu W, Sun J, Li G, Zhu Y, Zhang S, Kim ST, et al. Association of a germ-line copy number variation at 2p24.3 and risk for aggressive prostate cancer. Cancer Res. 2009;69:2176–9. doi: 10.1158/0008-5472.CAN-08-3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Final report on the aspirin component of the ongoing Physicians’ Health Study. Steering Committee of the Physicians’ Health Study Research Group. N Engl J Med. 1989;321:129–35. doi: 10.1056/NEJM198907203210301. [DOI] [PubMed] [Google Scholar]
- 21.Giovannucci E, Pollak M, Liu Y, Platz EA, Majeed N, Rimm EB, et al. Nutritional predictors of insulin-like growth factor I and their relationships to cancer in men. Cancer Epidemiol Biomarkers Prev. 2003;12:84–9. [PubMed] [Google Scholar]
- 22.Oh WK, Hayes J, Evan C, Manola J, George DJ, Waldron H, et al. Development of an integrated prostate cancer research information system. Clin Genitourin Cancer. 2006;5:61–6. doi: 10.3816/CGC.2006.n.019. [DOI] [PubMed] [Google Scholar]
- 23.Matsuzaki H, Loi H, Dong S, Tsai YY, Fang J, Law J, et al. Parallel genotyping of over 10,000 SNPs using a one-primer assay on a high-density oligonucleotide array. Genome Res. 2004;14:414–25. doi: 10.1101/gr.2014904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rabbee N, Speed TP. A genotype calling algorithm for affymetrix SNP arrays. Bioinformatics. 2006;22:7–12. doi: 10.1093/bioinformatics/bti741. [DOI] [PubMed] [Google Scholar]
- 25.Affymetrix. Genome-Wide Human SNP Array 5.0. 2009 [cited 2010; Available from: http://www.affymetrix.com/estore/browse/products.jsp?productId=131532&categoryId=35906#1_3.
- 26.PLINK. 2009 [cited 2010; Available from: http://pngu.mgh.harvard.edu/purcell/plink/
- 27.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 29.Ross RW, Oh WK, Xie W, Pomerantz M, Nakabayashi M, Sartor O, et al. Inherited variation in the androgen pathway is associated with the efficacy of androgen-deprivation therapy in men with prostate cancer. J Clin Oncol. 2008;26:842–7. doi: 10.1200/JCO.2007.13.6804. [DOI] [PubMed] [Google Scholar]
- 30.Korn JM, Kuruvilla FG, McCarroll SA, Wysoker A, Nemesh J, Cawley S, et al. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat Genet. 2008;40:1253–60. doi: 10.1038/ng.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barnes C, Plagnol V, Fitzgerald T, Redon R, Marchini J, Clayton D, et al. A robust statistical method for case-control association testing with copy number variation. Nat Genet. 2008;40:1245–52. doi: 10.1038/ng.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Varghese JS, Easton DF. Genome-wide association studies in common cancers-what have we learnt? Curr Opin Genet Dev. doi: 10.1016/j.gde.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 33.Freedman ML, Haiman CA, Patterson N, McDonald GJ, Tandon A, Waliszewska A, et al. Admixture mapping identifies 8q24 as a prostate cancer risk locus in African-American men. Proc Natl Acad Sci U S A. 2006;103:14068–73. doi: 10.1073/pnas.0605832103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amundadottir LT, Sulem P, Gudmundsson J, Helgason A, Baker A, Agnarsson BA, et al. A common variant associated with prostate cancer in European and African populations. Nat Genet. 2006;38:652–8. doi: 10.1038/ng1808. [DOI] [PubMed] [Google Scholar]
- 35.Gudmundsson J, Sulem P, Manolescu A, Amundadottir LT, Gudbjartsson D, Helgason A, et al. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet. 2007;39:631–7. doi: 10.1038/ng1999. [DOI] [PubMed] [Google Scholar]
- 36.Gudmundsson J, Sulem P, Steinthorsdottir V, Bergthorsson JT, Thorleifsson G, Manolescu A, et al. Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nat Genet. 2007;39:977–83. doi: 10.1038/ng2062. [DOI] [PubMed] [Google Scholar]
- 37.Yeager M, Orr N, Hayes RB, Jacobs KB, Kraft P, Wacholder S, et al. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet. 2007;39:645–9. doi: 10.1038/ng2022. [DOI] [PubMed] [Google Scholar]
- 38.Gudmundsson J, Sulem P, Rafnar T, Bergthorsson JT, Manolescu A, Gudbjartsson D, et al. Common sequence variants on 2p15 and Xp11.22 confer susceptibility to prostate cancer. Nat Genet. 2008;40:281–3. doi: 10.1038/ng.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eeles RA, Kote-Jarai Z, Giles GG, Olama AA, Guy M, Jugurnauth SK, et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet. 2008;40:316–21. doi: 10.1038/ng.90. [DOI] [PubMed] [Google Scholar]
- 40.Thomas G, Jacobs KB, Yeager M, Kraft P, Wacholder S, Orr N, et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet. 2008;40:310–5. doi: 10.1038/ng.91. [DOI] [PubMed] [Google Scholar]
- 41.Ergun A, Lawrence CA, Kohanski MA, Brennan TA, Collins JJ. A network biology approach to prostate cancer. Mol Syst Biol. 2007;3:82. doi: 10.1038/msb4100125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCarroll SA. Extending genome-wide association studies to copy-number variation. Hum Mol Genet. 2008;17:R135–42. doi: 10.1093/hmg/ddn282. [DOI] [PubMed] [Google Scholar]
- 43.McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, Wysoker A, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet. 2008;40:1166–74. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- 44.Cooper GM, Zerr T, Kidd JM, Eichler EE, Nickerson DA. Systematic assessment of copy number variant detection via genome-wide SNP genotyping. Nat Genet. 2008;40:1199–203. doi: 10.1038/ng.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bill-Axelson A, Holmberg L, Filen F, Ruutu M, Garmo H, Busch C, et al. Radical prostatectomy versus watchful waiting in localized prostate cancer: the Scandinavian prostate cancer group-4 randomized trial. J Natl Cancer Inst. 2008;100:1144–54. doi: 10.1093/jnci/djn255. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.