

Abstract
Background & Aims
Crohn’s disease (CD) can develop in any region of the gastrointestinal tract, including the stomach. The etiology and pathogenesis of Crohn’s gastritis are poorly understood, treatment approaches are limited, and there are not many suitable animal models for study. We characterized the features and mechanisms of chronic gastritis in SAMP1/YitFc (SAMP) mice, a spontaneous model of CD-like ileitis, along with possible therapeutic approaches.
Methods
Stomachs from specific pathogen-free and germ-free SAMP and AKR mice (controls) were evaluated histologically; the presence of Helicobacter spp. was tested in fecal pellets by PCR analysis. In vivo gastric permeability was quantified by fractional excretion of sucrose and epithelial tight junction protein expression was measured by quantitative reverse transcription PCR analysis. The effects of a proton pump inhibitor (PPI) or corticosteroids were measured and the ability of pathogenic immune cells to mediate gastritis was assessed in adoptive transfer experiments.
Results
SAMP mice developed Helicobacter-negative gastritis, characterized by aggregates of mononuclear cells, diffuse accumulation of neutrophils, and disruption of epithelial architecture; SAMP mice also had increased in gastric permeability compared with controls, without alterations in expression of tight junction proteins. The gastritis and associated permeability defect observed in SAMP mice were independent of bacterial colonization and reduced by administration of corticosteroids but not a PPI. CD4+ T cells isolated from draining mesenteric lymph nodes of SAMP mice were sufficient to induce gastritis in recipient SCID mice.
Conclusions
In SAMP mice, gastritis develops spontaneously and has many features of CD-like ileitis. These mice are a useful model to study Helicobacter-negative, immune-mediated Crohn’s gastritis.
Keywords: inflammatory bowel disease, stomach, epithelial permeability, germ-free
Inflammatory bowel disease (IBD) is a chronic inflammatory condition of the gastrointestinal tract that is mainly comprised of two disorders, CD and ulcerative colitis (UC). Anatomically, UC is limited to the colon, while CD may affect any portion of the gastrointestinal tract, from mouth to anus. Although CD primarily localizes to the ileum and colon, increasing evidence indicates that the esophagus, stomach and duodenum are also greatly affected in a substantial percentage of patients. The prevalence of upper gastrointestinal CD, however, has not been firmly established and is dependent on the diagnostic tool used. Retrospective studies using symptoms and radiography place incidence below 5%, while use of upper endoscopy and histology approximate occurrence as high as 60%1,2. Regardless of prevalence, CD patients with upper gastrointestinal involvement, most commonly in the antrum and duodenum2, are generally asymptomatic, Helicobacter pylori (H. pylori)-negative, and display concomitant distal disease1. Involved areas present as diffuse, patchy redness or multiple, focal erythematous spots2,3. The most common cell types associated with focal lesions are lymphocytes (CD3+), granulocytes (neutrophils, eosinophils), and histiocytes (CD68+CD68R+)4, with a low percentage (<5%) of patients demonstrating granulomas or microgranulomas4,5.
Despite advances in screening and increased awareness of prevalence, the pathogenesis of Crohn’s gastritis remains poorly understood and therapeutic modalities limited. Treatment normally includes standard medical therapies for CD, with steroids and immunomodulators alone, or in combination with acid suppressors, being the most favorable and efficacious1. Additionally, only a limited number of experimental animal models exist to investigate gastritis, with Helicobacter infection or NSAID administration being the most commonly employed techniques; however, neither model has direct relevance to Crohn’s gastritis. Gastritis is also frequently noted in models of autoimmunity, including thymectomized mice, athymic (nu/nu) mice adoptively transferred with T cells, and mice possessing furin-deficient T cells6–8. Gastritis associated with these models results from self-reactive T cells and/or the inability of T regulatory cells to control effector cell function and expansion7,8.
Although autoimmunity has clear relevance, there remains a need to develop novel animal models highlighting the gastric manifestations of CD in order to provide more precise mechanistic insight into the etiology, pathogenesis, and treatment of Crohn’s gastritis. Herein, we demonstrate that SAMP1/YitFc (SAMP) mice, which represent a well-described model of spontaneous CD-like ileitis9, also display a progressive, chronic gastritis that is preceded by abnormally high, region-specific gastric epithelial permeability. SAMP gastritis occurs independent of Helicobacter infection and in fact, uniquely persists in the absence of commensal flora. Furthermore, gastritis is immunologically-mediated by transfer of pathogenic CD4+ T cells from donor SAMP and responds efficaciously to corticosteroids, but not PPI administration. These findings represent the first account of the gastric manifestations, and extend the utility, of the SAMP model to include Crohn’s gastritis.
Materials and Methods
Mice
SAMP and AKR/J mice were propagated at the University of Virginia (UVA) and Case Western Reserve University (CWRU), with founders provided by S. Matsumoto (Yakult Central Institute for Microbiological Research, Tokyo, Japan)10,11. Mice were maintained under SPF conditions, fed standard laboratory chow (Harlan Teklad, Indianapolis, IN), and kept on 12h light/dark cycles. C3Smn. CB17-Prkdcscid/J (SCID) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). GF SAMP were maintained at Taconic Farms (Albany, NY) and shipped in GF vessels for same-day experimentation. All procedures were approved by UVA’s and CWRU’s IACUC and AALAC guidelines.
Histology and Myeloperoxidase (MPO) Assays
Stomachs were opened along the greater curvature, immersed in Bouin’s fixative, rinsed in ethanol, and paraffin-embedded. Three μm sections were stained with H&E and gastric inflammation evaluated by a trained pathologist (JRM) in a blinded fashion using a modified scoring system previously described by Ismail et al.12. Briefly, each stomach region (forestomach, corpus and antrum) was assessed individually for 3 parameters: 1) thickening, 2) infiltration of polymorphonuclear (PMN) cells, and 3) infiltration of mononuclear cells (MNC). Severity was graded based on the absence (0) or presence (1) of each parameter, with PMN infiltration further examined for focal, diffuse or abscess involvement. Similarly, MNC infiltration was examined for focal, diffuse or aggregate involvement in the lamina propria. A total score was calculated by adding the values for each region of the stomach. Results are reported as total inflammatory scores (TIS).
Immunohistochemistry for MPO localization was performed on stomach tissue sections employing a rabbit anti-mouse MPO polyclonal detecting antibody (NB120-15484, Novus Biologicals, Littleton, CO), and visualized using the Rabbit IgG Vectastain® ABC kit according to manufacturer’s instructions (Vector Laboratories, Burlingame, CA). MPO activity was evaluated on gastric tissues as previously described13,14 and used as a quantitative measure of acute inflammation.
Helicobacter Spp. Testing
Testing of Helicobacter spp. was performed on total DNA isolated from fecal pellets using a modification of a previously described method15. Briefly, fecal pellets were collected from SAMP and AKR mice, resuspended in 1.7 ml PBS, pH 7.4, and centrifuged at 700 x g for 5 min. 100 μl volume of supernatant was diluted 1:2 with PBS and DNA isolated using the QIAamp Tissue Kit (QIAGEN, Inc., Valencia, CA). PCR was performed for 16S rRNA using degenerate primer sequences (5′-ATGAAKCTTYTAGCTTGCTA-3′; 5′-AGATACCGTCATWATCTTCTC-3′), recognizing all Helicobacter species. PCR reactions were heated to 94°C for 30s, followed by 45 cycles of denaturation at 94°C for 30s, primer annealing at 53°C for 30s, and extension at 72° for 60s in a Perkin-Elmer model 2400 thermocycler. Resulting PCR products were subjected to electrophoresis on an agarose gel containing 0.5 μg/ml ethidium bromide resolving sizes of 380 bp for H. hepaticus, H. rappini, H. muridarum, H. ganmani, H. flexispira, and H. felis, and of 560 bp for H. bilis and 460 bp for H. typhlonius, due to an intervening sequence. H. hepaticus and H. bilis were used as positive controls.
In Vivo Permeability
Following a previously described method16,17, gastric permeability was assessed in mice 3 to 20 weeks of age. Briefly, mice were fasted for 2h prior to the administration of a sugar probe (sucrose; 100 mg in 0.2 mL sterile H2O) via orogastric gavage, and placed individually in metabolic cages for a 22h urine collection. Urine volume was measured and sucrose concentration determined by high-performance liquid chromatography. Fractional excretion (FE) of sucrose was determined as the ratio of the amount of sucrose in the urine over the amount administered.
CXCL1 and Apical Junctional Complex (APC) Protein Expression
Gastric epithelial tight junction (TJ) proteins of SAMP and AKR mice were assessed by confocal microscopy and qRT-PCR. For confocal microscopy, OCT-embedded stomachs were sectioned at 3 μm and TJ proteins immunolocalized as previously described18. Quantitative RT-PCR was performed on full-thickness gastric tissue samples using primers for CXCL1, occludin (Occl), claudins (Cldns) 1–519,20, Cldn 15 (5′-ATGTCGGTAGCTGTGGAGAC-3′; 5′-CCCTGCAATGGCCAGCAGC-3′) and Cldn 18 (5′-GACCGTTCAGACCAGGTACA-3′; 5′-GCGATGCACATCATCACTC-3′), and mRNA transcripts measured relative to eukaryotic 18S rRNA (internal control). Analysis was performed using a Bio-Rad iCycler iQ Real Time Detection system, software, and reagents (Bio-Rad Laboratories, Hercules, CA). Reaction mixtures were comprised of primers (400 nM each), cDNA (5% by vol) and iQ SYBR Green Supermix (200 μM dNTPs, 3 mM MgCl2, 0.625 U iTaq DNA polymerase, SYBR Green I, 10 nM fluoresein) in a total volume of 25 μL. Thermal cycling conditions were 95°C for 3 min followed by 40 cycles of 95°C for 15s, 60°C for 15s and 72°C for 15s. Expression of target genes were normalized to 18S rRNA.
Adoptive Transfer and Cytokine Expression
Mesenteric lymph nodes (MLNs) were harvested from 10- to 12-week-old donor SAMP or AKR mice, rendered into single cell suspensions, and purified for CD4 using magnetic bead-activated cell sorting according to manufacturer’s instructions (MACS, Miltenyi Biotec, Auburn, CA). CD4+ T cells (400,000) were transferred i.p. into 6- to 8-week-old MHC-matched SCID mice. Six weeks post-transfer, recipient mice were euthanized and stomachs processed for histological evaluation of inflammation and MPO activity. Gastric cytokine mRNA expression was evaluated by qRT-PCR as described above, and using the following primer sequences: IFNγ (5′-GCCAAGTTTGAGGTCAACAAC-3′; 5′-CCGAATCAGCAGCGACTC-3′), TNF (5′-GCGGTGCCTATGTCTCAG-3′; 5′-GCCATTTGGGAACTTCTCATC-3′), IL-10 (5′-TTTAAGGGTTACTTGGGTTGC-3′; 5′-CCGCATCCTGAGGGTCTTC-3′), IL-4 (5′-GCTAGTTGTCATCCTGCTCTTC-3′; 5′-GGCGTCCCTTCTCCTGTG-3′), IL-5 (5′-GCTTCTGCACTTGAGTGTTCTG-3′; 5′-CCTCATCGTCTCATTGCTTGTC-3′), IL-13 (5′-TTGCTTGCCTTGGTGGTCTC-3′; 5′-GGGAGTCTGGTCTTGTGTGATG-3′), IL-17A (5′-TTTAACTCCCTTGGCGCAAAA-3′; 5′-CTTTCCCTCCGCATTGACAC-3′), IL-17F (5′-TGCTACTGTTGATGTTGGGAC-3′; 5′-AATGCCCTGGTTTTGGTTGAA-3′), and IL-22 (5′-TTCGAGGAGTCAGTGCTAAA-3′; 5′-AGAACGTCTTCCAGGGTGAA-3′).
Statistical Analysis
All results are expressed as mean ± SEM and contain a minimum of n=5/group. Data were analyzed using GraphPad Prism 5 (GraphPad Software Inc., San Diego, CA). Selection of appropriate statistical tests was based on the variance and underlying distribution of data. Global effects between groups were first assessed using a one-way analysis of variance (ANOVA) with Bonferroni correction for multiple comparisons. For correlation analysis, Pearson’s r was used as a measure of correlation. Differences between individual groups were then directly compared using two-sample unpaired Student’s t test. A probability of P<.05 was considered significant.
Results
SAMP Mice Display Gastric Inflammation that is Preceded by Altered Epithelial Paracellular Permeability
Histologic examination of stomachs from both SAMP and AKR mice displayed minimal to no gastritis at 3 weeks of age, with no difference in mean TIS (SAMP vs. AKR; 1.9±0.6 vs. 1.5±0.3, n.s.) (Figure 1A). However, as early as 4 weeks, SAMP mice demonstrated increased gastric inflammation compared to age-matched AKRs (3.3±0.4 vs. 1.0±0.1, P<.05) that was characterized by mononuclear cell aggregates and diffuse accumulations of neutrophils in all regions of the stomach (Figure 1B). SAMP mice showed disrupted epithelial architecture with marked dilation of gastric glands and evidence of metaplasia in some regions. Furthermore, prominent cellular infiltrates were commonly observed in the mucosa and submucosa of the corpus, and dilated gastric glands and focal crypt abscesses were also frequently present (Figure 1C, left & middle panels). Evaluation at higher magnification established that cellular infiltrates were mainly comprised of lymphocytes with interspersed neutrophils and macrophages. Inflammatory lesions were generally associated within the lower portion of the gastric gland residing in close proximity to the muscularis mucosae (Figure 1C, right panel); however, foci of active gastritis primarily consisting of neutrophils were also visible within the glandular epithelium.
Figure 1. Time course and histologic features of SAMP gastritis.

(A) Gastric TIS in SAMP (□) and AKR (■) mice, aged 3 to 20 weeks. n=5–12/group; *P<.05 vs. age-matched AKRs. (B) Representative photomicrographs demonstrate 3-week-old SAMP with normal gastric mucosa, while 12- and 20-week-olds show progression of pathologic features and severity of gastritis. Twelve-week-old SAMP demonstrate increased acute and chronic inflammation within the lamina propria with focal glandular abscesses (arrows), while 20-week-olds present with more diffuse, chronic transmural inflammatory infiltrates (corpus region depicted, left panels). AKR mice at 3 and 12 weeks have normal appearing, non-inflamed stomachs, while 20-week-olds show focal, minimal chronic inflammation along the base of the mucosa, consistent with normal, physiological inflammation (right panels). 200X magnification. (C) Representative photomicrographs of full-thickness corpus of 20-week-old SAMPs display a prominent inflammatory infiltrate within the mucosa and extending into the submucosa (left panel; 200X), with higher magnification of an inflammatory site consisting mostly of lymphocytes with occasional neutrophils and macrophages at the base of crypts (middle panel; 400X). Acute and chronic inflammation highlighting foci of active gastritis with neutrophils present within the glandular epithelium; focal crypt abscess (arrow) is depicted (right panel; 400X). (D) Prevalence and severity of SAMP gastritis in different age groups; n=16–78/group. (E) Severity of gastritis in male and female SAMP at different age groups. n=8–42/group; no significant differences in gastritis severity comparing males vs. females were detected. (F) Correlation analysis of gastric and ileal TIS showed a significant positive correlation between these two phenotypes. n=147; r=.412, P<.0001.
In regard to penetrance, the incidence of gastritis in SAMP mice increased with age, as did the percentages of mice developing more severe disease (Figure 1D). 19% of SAMP at weaning and up to 5 weeks of age displayed mild gastritis, while at 5–10 weeks, almost 60% of SAMP showed histological evidence of gastritis with about 2/3 of these mice having mild gastritis, and 1/3 moderate to severe gastritis. By 10–20 weeks, 76% of all SAMP developed gastritis with increasing incidence of disease severity. In addition and differently from SAMP ileitis, no gender differences were observed in either the onset or severity of gastritis in age-matched males vs. females (Figure 1E). However, the time course of SAMP gastritis was similar to ileitis and the severity of these two phenotypes was positively correlated (r=.412, P<.0001.; Figure 1F).
Interestingly, no positive correlation was observed between gastric inflammatory scores and MPO activity (SAMP, r=−.81; AKR, r=.39; n.s.). In fact, MPO activity in the stomach was greatest in young mice, with 3-week-old SAMP displaying higher MPO levels than age-matched AKR controls (33.4±8.8 vs. 17.7±1.1 U/mg tissue, P<.01). No differences in MPO activity were measured at older ages, with levels less than 5 U/mg tissue for both SAMP and AKR mice (Figure 2A). These results were confirmed by immunohistochemistry, with MPO immunostaining robustly expressed in 3-week-old SAMP (Figure 2B, upper right panel). However, despite obvious signs of inflammatory infiltrates in older SAMP, MPO immunoreactivity was relatively sparse (Figure 2B, lower right panel), supporting the histological findings that stomachs of older SAMP have limited foci of active gastritis.
Figure 2. Quantitative and qualitative assessment of MPO activity in SAMP gastritis.

(A) MPO activity was determined in gastric tissue samples from SAMP (□) and AKR (■) mice aged 3 to 20 weeks. MPO activity and (B) MPO immunoreactivity (corpus region depicted) was highest in 3-week-old SAMP and AKR mice; 200X magnification. (C) Assessment of gastric permeability (sucrose FE) in SAMP showed early and increased permeability compared to age-matched AKRs, while (D) young SAMP (3- to 4-week-old) demonstrated elevated levels of CXCL1 mRNA transcripts compared to age-matched AKRs. n=5–18/group for all experiments; *P<.05 vs. age-matched AKR.
In an effort to determine potential pathogenic mechanisms of gastritis associated with SAMP mice, epithelial barrier function was characterized in vivo by measuring gastric paracellular permeability to a disaccharide (i.e., sucrose) over the course of 17 weeks. Interestingly, as early as 3 weeks, immediately after weaning, and continuing for the duration of the study (20 weeks), SAMP mice exhibited greater than a 2.5-fold increase in gastric permeability over time versus age-matched AKRs (Figure 2C). Specifically, SAMP mice exhibited higher gastric permeability than AKRs as early as 3 weeks of age (0.0104±0.0011 vs. 0.0038±0.004, P<.05), and continued up to 20 weeks of age (0.0063±0.001 vs. 0.0014±0.001, P<.05). This difference in gastric barrier function was evident 1 to 2 weeks before the onset of histologically visible inflammation (Figure 1). In addition, gastric tissue samples from young SAMP mice (3- to 4-week-old) displayed increased mRNA expression of the chemokine CXCL1, known to be a potent neutrophil chemoattractant, versus age-matched AKR mice (5.3-fold, P<.05; Figure 2D). Similar results were noted for TNF mRNA expression (3.7-fold, P<.05; data not shown), a cytokine shown to play a critical role in other models of gastritis (i.e., NSAID-induced, H. felis-induced)21,22. These data suggest that the combination of epithelial barrier dysfunction and a luminal antigen may be the initiating factor in this model.
SAMP Gastritis and Associated Increase in Gastric Permeability Occur Independently of Helicobacter Infection and Persists in the Absence of Commensal Microflora
Commensal flora residing within the stomach represents a potential source for antigenic stimuli that may be responsible for inducing chronic gastritis characteristic of SAMP mice. Infection with Helicobacter spp. was ruled out as an etiologic factor as continuous surveillance of fecal pellets from SAMP and AKR, as well as co-housed sentinel mice, was found to be negative for H. hepaticus, H. bilis, H. rappini, H. muridarum, H. trogontum, H. ganmani, H. flexispira, H. typhlonius, H. mustelae and H. felis (data not shown). In addition, gastric inflammation and permeability were examined in <4-week-old and >20-week-old SAMP mice raised under GF conditions and compared to age-matched SAMPs raised under SPF conditions (Figure 3). Despite the absence of commensal flora, GF SAMP mice displayed overt gastric inflammation as early as 3–4 weeks of age compared to SPF-raised AKR controls (4.5±0.7 vs. 1.1±0.2, P<.05; Figure 3A). Examination of stomachs obtained from older GF SAMP mice (>20-weeks of age) showed that gastritis persisted and increased in severity (TIS=6.5±0.5, Figure 3A & D). Similar to inflammatory scores, gastric epithelial paracellular permeability was also found to be increased (P<.05 vs. SPF AKR) at 3–4 weeks of age (0.020±0.011 vs. 0.003±0.0003, Figure 3B), and remained elevated at >20 weeks of age. Interestingly, the occurrence of both histologically discernable gastritis and altered barrier function in <4-week-old SAMP GF mice was not accompanied by an increase in MPO activity (Figure 3C). MPO activity in young GF SAMP mice was potently attenuated compared to age-matched SPF SAMP (1.6±0.2 vs. 19.8±4.8 U/mg tissue; P<.05), while no difference was found between MPO activity levels from age-matched GF SAMP and SPF AKR (1.6±0.2 vs. 6.4±1.5 U/mg tissue; n.s.). Qualitatively, gastritis in GF SAMP presented with similar pathological features as those observed for SPF SAMP mice (Figure 3D).
Figure 3. Development of SAMP gastritis occurs in the absence of bacterial flora colonization and is likely mediated by an inherent epithelial barrier defect.

(A) Both SPF and GF SAMP displayed increased gastric permeability at 3–4 weeks, prior to and/or coincident with the onset of gastric inflammation, compared to age-matched SPF AKRs, as measured by (B) histological examination and (C) MPO activity. n=6–30/group for all experiments; *P<.05 vs. age-matched SPF AKR; ΨP<.05 vs. age-matched GF SAMP. (D) Severity and histologic features of gastritis in 3- to 4-week old and >20-week-old SAMP mice raised under GF conditions. GF SAMP at an early age demonstrate diffuse acute and chronic inflammation, present mostly within the lamina propria. Older GF SAMP display acute and chronic inflammation within the base of the lamina propria with superficial, mucosal clear cell metaplasia; 150X (upper panels) and 100X (lower panels) magnifications.
The findings in GF SAMP further promote that epithelial barrier dysfunction is the primary inherent defect for SAMP gastritis, and that commensal flora does not represent the source for antigenic stimuli. Therefore, expression levels of APC proteins were examined at the protein and mRNA levels. Unlike our previous findings of altered TJ protein expression in the ileum of SAMP mice20, no discernable changes in protein or mRNA expression for ZO-1, occludin and various claudins (Cldn-3, Cldn-5, Clnd-15 and Cldn-18) were seen (Figure 4). Both confocal microscopy and qRT-PCR failed to detect the expression of claudin-1, -2 and -4. Considering that the majority of claudins present in the epithelium are also present in the endothelium and that the gastrointestinal mucosa possesses an extensive microvascular bed, mRNA expression of various AJC proteins were examined in epithelial cell isolates collected from the upper and lower portions of the gastric glands by laser capture microdissection. Despite minimizing non-epithelial cell contaminates, no alterations in either occludin or claudins were found (data not shown).
Figure 4. Epithelial TJ protein expression in SAMP stomachs prior to the onset of gastritis.
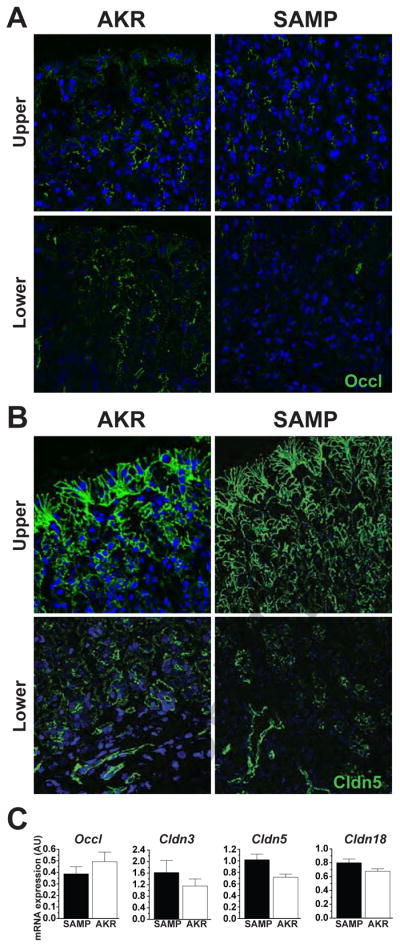
Representative photomicrographs of corpus tissue from 3- to 4-week-old SAMP and AKR mice immunostained for (A) occludin or (B) claudin-5 demonstrate that their distribution appears unaltered, both in the upper and lower portions of the gastric glands of SAMP compared to age-matched AKRs; 200X magnification. (C) Quantitative RT-PCR for Cldns-5, -3, -18 and Occl mRNA transcripts was performed on full-thickness corpus tissues obtained from young (3- to 4-week-old) AKR and SAMP mice. No significant alterations in gene expression for the TJ proteins measured were detected. Data are normalized to 18S rRNA and presented as mean ± SEM (n=7–8/group).
SAMP Gastritis Responds Therapeutically to Corticosteroid Administration, but not to an Acid Inhibitor
Acid suppressors and steroids represent two common therapeutic modalities used to treat Crohn’s gastritis2. As such, two independent groups of SAMP mice were treated either with a PPI (pantoprazole) or a corticosteroid (dexamethasone, DexM). Oral, daily treatment of 10-week-old SAMP with pantoprazole for 14 days had no effect on gastric TIS, but increased gastric permeability as measured by sucrose FE (P<.05) (Figure 5A). Conversely, daily treatment of SAMP with DexM for one week both significantly attenuated gastric TIS (P<.05) and improved epithelial barrier function (P<.05) (Figure 5B). DexM, therefore, has proven to be an efficacious treatment both for the gastritis and ileitis23 present in SAMP mice, and suggests common immune-mediated mechanism(s) in the pathogenesis of gastric and small intestinal inflammation characteristic of this unique mouse strain.
Figure 5. Decreased gastric inflammation in SAMP mice treated with corticosteroids, but not a PPI.

Ten-week-old SAMP with established gastritis were treated with either the PPI, pantoprazole, (50 mg/kg, p.o.) or DexM (5 mg/kg, i.p.). (A) Treatment with pantoprazole had no effect on gastric inflammation, but increased gastric paracellular permeability. Conversely, (B) treatment with DexM reduced both gastritis (left panel) as well as gastric paracellular permeability (right panel). n=6/group (histologic evaluation), *P<.05 vs. vehicle; n=6/group (permeability studies), *P<.05 vs. pre-treatment.
SAMP Gastritis can be Adoptively Transferred to Recipient Mice via Pathogenic CD4+ T Lymphocytes
In an effort to further delineate potential immunopathogenic mechanism(s) responsible for gastric inflammation in SAMP mice, CD4+ lymphocytes were purified from MLNs of 10- to 12-week-old SAMP and adoptively transferred into MHC-matched SCID mice. Similar to ileitis displayed by SAMP11,23, gastric inflammation was also found to be adoptively transferable (Figure 6). Mild to moderate gastric inflammation was observed in recipient SCIDs reconstituted with CD4+ T cells from donor SAMP 6 weeks post-transfer, while little to no gastritis was found in SCIDs receiving CD4+ T cells from AKR controls or untransferred SCIDs (P<.05 for SCIDs transferred with donor SAMP vs. either donor AKR or no cell transfer, Figure 6A). SCIDs receiving SAMP donor cells also displayed elevated gastric MPO activity vs. controls (P<.05) (Figure 6B & C), with pathological characteristics of gastritis consistent to those found in native SAMP mice (Figure 6C). Finally, gastric cytokine profiles from SCID recipients of SAMP donor cells were similar to that observed in SAMP ileitis, displaying a mixed Th1/Th2 cytokine profile24, as well as increased IL-17F levels (Garg RR et al., submitted paper, under review) (Figure 6D).
Figure 6. Pathogenic SAMP CD4+ T lymphocytes mediate gastritis when transferred into naïve SCID mice.

Six weeks post-transfer, SCIDs receiving SAMP CD4+ T cells developed mild to moderate gastritis as determined by (A) histological evaluation and (B) MPO activity. (C) Representative histology and immunohistochemical staining for MPO demonstrate the majority of inflammation and cellular infiltrates localized to the corpus region of the stomach in SCIDs receiving donor SAMP cells. Minimal to no inflammation was found in gastric tissues obtained from SCIDs adoptively transferred with AKR CD4+ T lymphocytes; 100X magnification. n=8/group; *P<.05 vs. control (SCIDs with no cell transfer) and AKR (SCIDs receiving donor AKR CD4+ T cells). (D) Full-thickness corpus tissues from SCIDs adoptively transferred with SAMP donor CD4+ T cells display increased Th1, Th2, and IL-17F cytokine mRNA transcripts compared to SCID recipients of AKR donor cells. Data are presented as fold-differences with AKR (SCIDs receiving donor AKR CD4+ T cells) average values set as 1. n=5–6/group; *P<.05 and ***P<.005.
Discussion
The results of the present study suggest common, immunopathogenic mechanism(s) mediate mucosal inflammation present in both the stomach and terminal ileum of SAMP mice, a unique mouse strain that represents a novel model of CD-like gastritis. These finding are of interest because, to date, no spontaneous models exist to study Crohn’s gastritis. The prevalence of Crohn’s gastritis has not been firmly established; however, recent studies have shown that at least 60% of histologically-diagnosed cases of chronic gastritis in the absence of H. pylori infection are associated with CD, making this condition one of the most common causes of Helicobacter-negative chronic gastritis, aside from NSAID-induced4,5,25,26.
Unfortunately, targeted treatment modalities for Crohn’s gastritis remain limited. Indeed, CD gastritis appear to respond to systemic treatment with either steroids or immunomodulators1. However, other drugs, such as mesalamine and low-bioavailability steroids, which are commonly used to treat IBD and act through direct contact with the diseased mucosa, appear to have little efficacy in treating gastric and gastroduodenal CD. This is likely due to the fact that these drugs are embedded in formulations targeting the terminal ileum or colon and are not released in the stomach27. Thus, current treatment options for mild to moderate gastric CD are largely unsatisfactory and the development of new compounds/formulations is greatly needed.
As such, development of animal models to examine the gastric manifestations and common pathogenic mechanisms with CD enteritis would also greatly improve our understanding and treatment of Crohn’s gastritis. The SAMP mouse represents an excellent model of CD as it shares several features in common with the clinical disease. That is, Crohn’s patients and SAMP mice develop disease that localizes to the upper (gastric) and lower (ileum) gastrointestinal tract. In addition, Crohn’s gastritis is often characterized or labeled as focally-enhanced gastritis4. Similarly, SAMP gastritis histologically presents primarily as focal inflammatory lesions that surround and/or infiltrate the gland(s) with inflammatory cell infiltrates comprised mainly of lymphocytes and granulocytes, and evidence of epithelial damage4,5. Although autoimmune mechanism(s) can not be excluded, the discontinuity of inflammation and the presence of abundant neutrophil infiltrates imply that SAMP gastritis closely resembles gastric CD rather than other idiopathic gastric inflammatory conditions, such as autoimmune gastritis, which is characterized by a diffuse lympho-monocytic infiltrate and diffuse parietal cell atrophy28. These findings appear to be specific for SAMP since parental AKR controls housed in the same animal facility did not show any sign of ileitis or gastritis. In addition to sharing pathologic characteristics of Crohn’s gastritis, SAMP mice respond therapeutically to corticosteroids, which represent one of the most common and efficacious drugs currently available to treat Crohn’s patients with gastric localization1.
Our results also showed high levels of MPO activity primarily in young compared to older SAMP mice. Specifically, gastric MPO activity was greatest in 3-week-old SAMP and somewhat decreased thereafter, indicating robust neutrophil infiltration early on during the acute inflammatory phase of gastritis, which also coincides with the early increased expression of CXCL1. In fact, although the incidence of gastritis severity continues to increase with age, the relative magnitude in disease severity after 5 weeks of age is not significantly different when compared to older (20-week-old) SAMP mice. These data are consistent with a recent study reporting the prevalence of Helicobacter-negative gastritis primarily in pediatric Crohn’s patients under 18 years of age29.
In addition, although it is not completely clear if the development of SAMP gastritis requires prior full expression of ileitis to occur, we did observe that the penetrance of gastritis is 76% at 20 weeks, while SAMP ileitis is at 100% at the same age. Therefore, the possibility exists that effector cells responsible for ileitis may also drive chronic gastritis similar to that observed for other mouse models of chronic intestinal inflammation with extraintestinal manifestations, including the upper GI involvement and liver inflammation described in the CD45RBhi adoptive transfer model30,31, or the arthritis demonstrated in the TNFΔARE mice32. This concept is supported by experiments performed in the present study wherein transfer of pathogenic CD4+ T cells from SAMP MLN have the ability to produce both the ileitis as well as the gastritis phenotype in naïve SCID recipient mice.
The results herein also suggest that similar to human CD, SAMP mice have abnormally high gastrointestinal epithelial permeability, and barrier dysfunction may be a predisposing factor to the spontaneous inflammation described in this model. Altered barrier function in the SAMP small intestine may stem from aberrant expression of occludin and claudin-220. Decreased occludin and increased claudin-2 expression in epithelial TJ fibrils have been shown to result in weaker anastomoses between neighboring cells, resulting in barrier dysfunction33–36. In fact, mice carrying a null mutation in the occludin gene also develop chronic gastritis and hyperplasia37. Further investigation into the precise mechanism(s) behind barrier dysfunction in the SAMP model may help to identify the basis of increased permeability as a primary initiating factor in the pathogenesis of IBD. In the present study, we have provided evidence supporting this concept and that the initial events leading to SAMP gastritis may also be due to inherent epithelial alterations and barrier dysfunction that occur even in the absence of enteric flora. However, definitive proof of a primary etiologic role for the permeability defect in SAMP mice, as well as in the pathogenesis of human CD, will come from the development of pharmacological agents and/or therapeutic interventions specifically targeted at correcting intestinal epithelial barrier dysfunction prior to or during the development of chronic intestinal inflammation.
In summary, we report herein a new model of immune-mediated CD-like gastritis, which appears to be mediated by a mechanism involving epithelial barrier dysfunction independent of bacterial colonization, including Helicobacter infection. This model has great potential to allow a better understanding of pathogenic mechanism(s) of CD gastritis and for the development of new therapeutic modalities for this chronic debilitating disease.
Acknowledgments
Grant Support: We acknowledge continued support from the National Institutes of Health: DK057880 (TTP & FC), DK056762 (TTP), and DK055812 (FC), and past support from the Crohn’s & Colitis Foundation of America (Career Development Award to BKR).
The authors acknowledge Muhammadreza Sachedina, Mary Riggins, Jason D’Antuono, Sharon Hoang, Brian Marks, and Jami Southard for outstanding technical support. Special appreciation is given to Kevin Scott for intellectual/technical contributions to gastric in vivo permeability studies, Asma Nusrat for expertise/technical guidance in characterizing epithelial TJ expression, and Peter Ernst for initial assistance/familiarization with the gastritis histologic scoring system.
Abbreviations used in this paper
- CD
Crohn’s disease
- SAMP
SAMP1/YitFc
- SPF
specific pathogen-free
- GF
germ-free
- TJ
tight junction
- PPI
proton pump inhibitor
- IBD
inflammatory bowel disease
- UC
ulcerative colitis
- PMNs
polymorphonuclear cells
- MNCs
mononuclear cells
- FE
fractional excretion
- MPO
myeloperoxidase
- TEER
transepithelial electrical resistance
- AJ
adherens junction
- AJC
apical junctional complex
- ZO-1
zonula occludens 1
- Occl
occludin
- Cldn
claudin
- MLN
mesenteric lymph node
- DexM
dexamethasone
- BM
bone marrow
- TIS
total inflammatory scores
Footnotes
Disclosures: Authors have no financial, professional or personal interest to disclose.
Author Contributions: BKR: study concept and design, acquisition, analysis and interpretation of data, drafting of manuscript, statistical analysis; LP, RRG: drafting and critical revision of manuscript for important intellectual content; MB, JAM, RMR, MCA, XMW, EJK: acquisition of data; SHF: acquisition, analysis and interpretation of data; JRM: analysis and interpretation of data; FC: material support, critical revision of the manuscript for important intellectual content, obtained funding; JBM: study concept and design, acquisition, analysis and interpretation of data, study supervision; TTP: study concept and design, analysis and interpretation of data, drafting and critical revision of manuscript for important intellectual content, obtained funding, study supervision
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mottet C, Juillerat P, Gonvers JJ, et al. Treatment of gastroduodenal Crohn’s disease. Digestion. 2005;71:37–40. doi: 10.1159/000083871. [DOI] [PubMed] [Google Scholar]
- 2.van Hogezand RA, Witte AM, Veenendaal RA, et al. Proximal Crohn’s disease: review of the clinicopathologic features and therapy. Inflamm Bowel Dis. 2001;7:328–37. doi: 10.1097/00054725-200111000-00010. [DOI] [PubMed] [Google Scholar]
- 3.D’Inca R, Sturniolo G, Cassaro M, et al. Prevalence of upper gastrointestinal lesions and Helicobacter pylori infection in Crohn’s disease. Dig Dis Sci. 1998;43:988–92. doi: 10.1023/a:1018870415898. [DOI] [PubMed] [Google Scholar]
- 4.Oberhuber G, Puspok A, Oesterreicher C, et al. Focally enhanced gastritis: a frequent type of gastritis in patients with Crohn’s disease. Gastroenterology. 1997;112:698–706. doi: 10.1053/gast.1997.v112.pm9041230. [DOI] [PubMed] [Google Scholar]
- 5.Halme L, Karkkainen P, Rautelin H, et al. High frequency of helicobacter negative gastritis in patients with Crohn’s disease. Gut. 1996;38:379–83. doi: 10.1136/gut.38.3.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith H, Lou YH, Lacy P, et al. Tolerance mechanism in experimental ovarian and gastric autoimmune diseases. J Immunol. 1992;149:2212–8. [PubMed] [Google Scholar]
- 7.Tung KS. Mechanism of self-tolerance and events leading to autoimmune disease and autoantibody response. Clin Immunol Immunopathol. 1994;73:275–82. doi: 10.1006/clin.1994.1199. [DOI] [PubMed] [Google Scholar]
- 8.Pesu M, Watford WT, Wei L, et al. T-cell-expressed proprotein convertase furin is essential for maintenance of peripheral immune tolerance. Nature. 2008;455:246–50. doi: 10.1038/nature07210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pizarro TT, Pastorelli L, Bamias G, et al. The SAMP1/YitFc mouse strain: A spontaneous model of Crohn’s Disease-like ileitis. Inflamm Bowel Dis. 2011 doi: 10.1002/ibd.21638. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsumoto S, Okabe Y, Setoyama H, et al. Inflammatory bowel disease-like enteritis and caecitis in a senescence accelerated mouse P1/Yit strain. Gut. 1998;43:71–8. doi: 10.1136/gut.43.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kosiewicz MM, Nast CC, Krishnan A, et al. Th1-type responses mediate spontaneous ileitis in a novel murine model of Crohn’s disease. J Clin Invest. 2001;107:695–702. doi: 10.1172/JCI10956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ismail HF, Zhang J, Lynch RG, et al. Role for complement in development of Helicobacter-induced gastritis in interleukin-10-deficient mice. Infect Immun. 2003;71:7140–8. doi: 10.1128/IAI.71.12.7140-7148.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bradley PP, Priebat DA, Christensen RD, et al. Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J Invest Dermatol. 1982;78:206–9. doi: 10.1111/1523-1747.ep12506462. [DOI] [PubMed] [Google Scholar]
- 14.Boughton-Smith NK, Wallace JL, Whittle BJ. Relationship between arachidonic acid metabolism, myeloperoxidase activity and leukocyte infiltration in a rat model of inflammatory bowel disease. Agents Actions. 1988;25:115–23. doi: 10.1007/BF01969102. [DOI] [PubMed] [Google Scholar]
- 15.Beckwith CS, Franklin CL, Hook RR, Jr, et al. Fecal PCR assay for diagnosis of Helicobacter infection in laboratory rodents. J Clin Microbiol. 1997;35:1620–3. doi: 10.1128/jcm.35.6.1620-1623.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scott KG, Meddings JB, Kirk DR, et al. Intestinal infection with Giardia spp. reduces epithelial barrier function in a myosin light chain kinase-dependent fashion. Gastroenterology. 2002;123:1179–90. doi: 10.1053/gast.2002.36002. [DOI] [PubMed] [Google Scholar]
- 17.Meddings JB, Gibbons I. Discrimination of site-specific alterations in gastrointestinal permeability in the rat. Gastroenterology. 1998;114:83–92. doi: 10.1016/s0016-5085(98)70636-5. [DOI] [PubMed] [Google Scholar]
- 18.Ivanov AI, Nusrat A, Parkos CA. Endocytosis of epithelial apical junctional proteins by a clathrin-mediated pathway into a unique storage compartment. Mol Biol Cell. 2004;15:176–88. doi: 10.1091/mbc.E03-05-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alam MS, Kurtz CC, Rowlett RM, et al. CD73 is expressed by human regulatory T helper cells and suppresses proinflammatory cytokine production and Helicobacter felis-induced gastritis in mice. J Infect Dis. 2009;199:494–504. doi: 10.1086/596205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olson TS, Reuter BK, Scott KG, et al. The primary defect in experimental ileitis originates from a nonhematopoietic source. J Exp Med. 2006;203:541–52. doi: 10.1084/jem.20050407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hasegawa S, Nishikawa S, Miura T, et al. Tumor necrosis factor-alpha is required for gastritis induced by Helicobacter felis infection in mice. Microb Pathog. 2004;37:119–24. doi: 10.1016/j.micpath.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 22.Appleyard CB, McCafferty DM, Tigley AW, et al. Tumor necrosis factor mediation of NSAID-induced gastric damage: role of leukocyte adherence. Am J Physiol. 1996;270:G42–8. doi: 10.1152/ajpgi.1996.270.1.G42. [DOI] [PubMed] [Google Scholar]
- 23.Burns RC, Rivera-Nieves J, Moskaluk CA, et al. Antibody blockade of ICAM-1 and VCAM-1 ameliorates inflammation in the SAMP-1/Yit adoptive transfer model of Crohn’s disease in mice. Gastroenterology. 2001;121:1428–36. doi: 10.1053/gast.2001.29568. [DOI] [PubMed] [Google Scholar]
- 24.Bamias G, Martin C, Mishina M, et al. Proinflammatory effects of TH2 cytokines in a murine model of chronic small intestinal inflammation. Gastroenterology. 2005;128:654–66. doi: 10.1053/j.gastro.2004.11.053. [DOI] [PubMed] [Google Scholar]
- 25.Matsumura M, Matsui T, Hatakeyama S, et al. Prevalence of Helicobacter pylori infection and correlation between severity of upper gastrointestinal lesions and H. pylori infection in Japanese patients with Crohn’s disease. J Gastroenterol. 2001;36:740–7. doi: 10.1007/s005350170015. [DOI] [PubMed] [Google Scholar]
- 26.Petrolla AA, Katz JA, Xin W. The clinical significance of focal enhanced gastritis in adults with isolated ileitis of the terminal ileum. J Gastroenterol. 2008;43:524–30. doi: 10.1007/s00535-008-2191-8. [DOI] [PubMed] [Google Scholar]
- 27.Kesisoglou F, Zimmermann EM. Novel drug delivery strategies for the treatment of inflammatory bowel disease. Expert Opin Drug Deliv. 2005;2:451–63. doi: 10.1517/17425247.2.3.451. [DOI] [PubMed] [Google Scholar]
- 28.Srivastava A, Lauwers GY. Pathology of non-infective gastritis. Histopathology. 2007;50:15–29. doi: 10.1111/j.1365-2559.2006.02553.x. [DOI] [PubMed] [Google Scholar]
- 29.Sonnenberg A, Melton SD, Genta RM. Frequent occurrence of gastritis and duodenitis in patients with inflammatory bowel disease. Inflammatory bowel diseases. 2011;17:39–44. doi: 10.1002/ibd.21356. [DOI] [PubMed] [Google Scholar]
- 30.Ostanin DV, Pavlick KP, Bharwani S, et al. T cell-induced inflammation of the small and large intestine in immunodeficient mice. American journal of physiology Gastrointestinal and liver physiology. 2006;290:G109–19. doi: 10.1152/ajpgi.00214.2005. [DOI] [PubMed] [Google Scholar]
- 31.Ostanin DV, Bao J, Koboziev I, et al. T cell transfer model of chronic colitis: concepts, considerations, and tricks of the trade. American journal of physiology Gastrointestinal and liver physiology. 2009;296:G135–46. doi: 10.1152/ajpgi.90462.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kontoyiannis D, Pasparakis M, Pizarro TT, et al. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10:387–98. doi: 10.1016/s1074-7613(00)80038-2. [DOI] [PubMed] [Google Scholar]
- 33.Balda MS, Whitney JA, Flores C, et al. Functional dissociation of paracellular permeability and transepithelial electrical resistance and disruption of the apical-basolateral intramembrane diffusion barrier by expression of a mutant tight junction membrane protein. J Cell Biol. 1996;134:1031–49. doi: 10.1083/jcb.134.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCarthy KM, Skare IB, Stankewich MC, et al. Occludin is a functional component of the tight junction. J Cell Sci. 1996;109 ( Pt 9):2287–98. doi: 10.1242/jcs.109.9.2287. [DOI] [PubMed] [Google Scholar]
- 35.Furuse M, Furuse K, Sasaki H, et al. Conversion of zonulae occludentes from tight to leaky strand type by introducing claudin-2 into Madin-Darby canine kidney I cells. J Cell Biol. 2001;153:263–72. doi: 10.1083/jcb.153.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harhaj NS, Antonetti DA. Regulation of tight junctions and loss of barrier function in pathophysiology. Int J Biochem Cell Biol. 2004;36:1206–37. doi: 10.1016/j.biocel.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 37.Saitou M, Furuse M, Sasaki H, et al. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell. 2000;11:4131–42. doi: 10.1091/mbc.11.12.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]