

Abstract
Background
Brugada Syndrome (BrS) is associated with mutations in the cardiac sodium channel (Nav1.5). We previously reported that the function of a trafficking-deficient BrS Nav1.5 mutation, R282H, could be restored by co-expression with the sodium channel polymorphism H558R. Here, we tested the hypothesis that peptide fragments from Nav1.5, spanning the H558R-polymorphism, can be used to restore trafficking of trafficking-deficient BrS sodium channel mutations.
Methods and Results
Whole-cell patch-clamping revealed that co-transfection in HEK293 cells of the R282H channel with either the 40 or 20 amino acid cDNA fragments of Nav1.5 containing the H558R polymorphism restored trafficking of this mutant channel. Fluorescence Resonance Energy Transfer (FRET) suggested that the trafficking-deficient R282H channel was misfolded and this was corrected upon co-expression with R558-containing peptides which restored trafficking of the R282H channel. Importantly, we also expressed the peptide spanning the H558R-polymorphism with 8 additional BrS Nav1.5 mutations with reduced currents, and demonstrated that the peptide was able to restore significant sodium currents in 4 of them.
Conclusions
In the present study, we demonstrated that small peptides, spanning the H558R-polymorphism, are sufficient to restore trafficking defect of BrS-associated Nav1.5 mutations. Our findings suggest that it might be possible to use short-cDNA constructs as a novel strategy that is tailored to specific disease-causing mutants of BrS.
Keywords: arrhythmia, electrophysiology, gene therapy, ion channels, SCN5A
Brugada Syndrome (BrS) is associated with an increased risk of sudden cardiac death resulting from episodes of polymorphic ventricular tachycardia.1,2 Mutations in the α-subunit of the cardiac sodium channel (Nav1.5), encoded by the SCN5A gene, have been found in 15 to 30% of BrS patients, currently representing the most common BrS genotype.3–5 To date, more than 100 SCN5A mutations have been linked to BrS.6 Most of those are missense mutations leading to loss-of-function phenotypes either by decreasing sarcolemmal expression of Nav1.5 channels7,8, producing non-functional channels9, or altered biophysical properties10–13. Current treatment options for BrS include implantable cardioverter-defibrillator (ICD) and drug therapy mostly with isoproterenol.14 However, none of these treatment options directly addresses the channel dysfunction that is the underlying source of BrS associated arrhythmias.
Failure of sarcolemmal expression has been identified as one important cellular mechanism of ion channel dysfunction in inherited arrhythmias, including BrS.7,8 For example, we have demonstrated that the BrS-causing mutation R282H, produces a trafficking-deficient sodium channel.7 Interestingly, we have also identified a healthy proband despite carrying R282H. We demonstrated that this individual did not develop BrS because R282H trafficking was restored in presence of the common sodium channel H558R-polymorphism as second allele.7 Here, we test a new strategy using peptide fragments of the cardiac sodium channel spanning the H588R-polymorphism, to restore trafficking and function of BrS mutations, by directly aiding in channel folding. This is not without precedent, since peptide fragments have already shown potential for treating channelopathies. For example, the most common mutation of the cystic fibrosis transmembrane conductance regulator, ΔF508-CFTR, which inhibits CFTR processing and trafficking, was rescued by co-expression of specific peptide or protein fragments derived from wild-type CFTR.15,16 In addition, it has been shown that trafficking of a hERG mutant channel could be restored by peptide fragments spanning an ER retention signals exposed on the C-terminus of the channel protein.17
We show that co-expression of BrS mutant channels with peptide fragments containing the H558R-polymorphism similarly rescues protein trafficking, and restores functional sodium channels to the cell surface. Finally, we employ FRET to analyze the mechanism of current restoration and show that peptide fragments containing the H558R-polymorphism initiate the productive folding of originally processing-defective BrS mutants in the ER.
Methods
Mutagenesis
Nine BrS trafficking-deficient mutations located in the pore of domain 1 of the cardiac sodium channel were studied. The R282H, G292S, V294M, K317N, L325R, G351V, D356N, R367H and R376H mutations and H558R-polymorphism were created on the Nav1.5 background (PubMed Accession No. NM198056) expressed in the GFP-IRES vector (BD Biosciences Clonetech, San Jose, CA) using the Stratagene QuikChange XL Site Directed Mutagenesis Kit.
Creation of Channel Fragments
For FRET experiments, channel fragments were created by amplifying small pieces of either the wild-type or the H558R-polymorphic channel (Figure 1). Primers were designed to amplify 40 and 20 amino acids fragments from Nav1.5. Forward primers contained a Kozak sequence (5’-GCC GCC ACC-3’) before the start codon (5’-ATG G-3’) to facilitate efficient translation initiation; reverse primers for the peptides expressed in pcDNA3 contained a stop codon. In addition, for the electrophysiological experiments, peptides fused to Enhanced Cyan Fluorescence Protein (ECFP) in pECFP-N3 were also generated. Sequences for peptide fragments are: wild-type 20 amino acid- NSTAGESESHHTSLLVPWPL, Polymorphic 40 amino acid- GSEADFADDENSTAGESESHRTSLLVPWPLRRTSAQGQPS and polymorphic 20 amino acid- NSTAGESESHRTSLLVPWPL.
Figure 1.
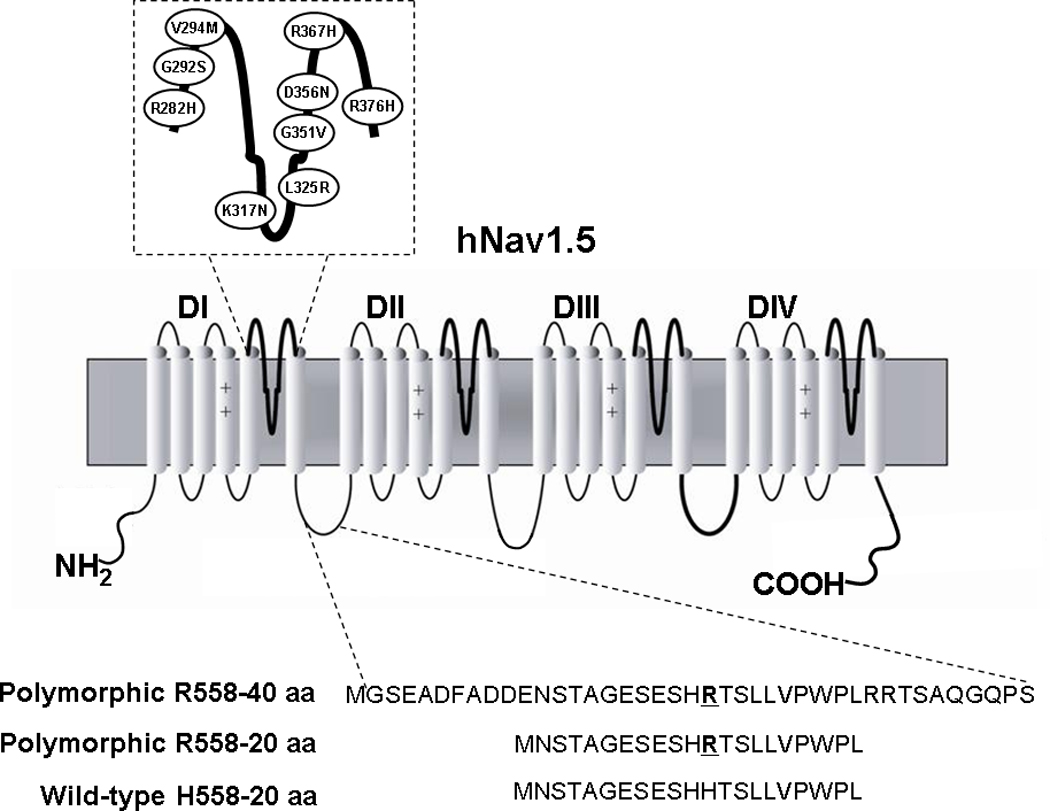
Schematic Representation of the α-Subunit of hNav1.5. Ovals represent the locations of the mutations characterized in this study. The lower panel shows the sequence of the fragments that were created by amplifying a piece of either the wild-type (H558) or the polymorphic channel (R558).
Expression of Nav1.5 and Fragments in HEK293 Cells
Transient transfection of full-length Nav1.5 cDNA and Nav1.5 fragments cDNA into human embryonic kidney (HEK293) cells was accomplished using the Polyfect transfection kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol. When channels were expressed alone, 1µg of full-length Nav1.5 cDNA was used. For co-transfections, 1µg of full-length Nav1.5 cDNA and 1µg of Nav1.5 fragment cDNA were combined leading to an excess of peptide in relation to full-length channel. Assuming similar transfection levels for both full-length channels and the fragments, this would result in an estimated 90:1 or 45:1 molar ratio (peptide fragment : full length sodium channel), due to their difference in sizes, for the 20 amino acid and 40 amino acid fragments, respectively. For direct application of the R558-containing peptide to the cell interior, a synthetic 20 amino acid peptide ([H]-NSTAGESESHRTSLLVPWPL-[OH]) was synthesized by Princeton BioMolecules which was added to the internal pipette solution at 10 µM final concentration. Purity of the peptide was ≥98%.
Biochemical Analysis of Nav1.5 Channels
HEK293 cells from similar confluent cultures were used to isolate proteins. Western blotting experiments were performed as previously described.18 To determine the expression level of the Nav1.5 protein in total protein lysates, proteins were extracted from HEK293 cells, transiently transfected with full-length Nav1.5 cDNA or co-transfections of full-length Nav1.5 cDNA with Nav1.5 fragment cDNA. Non-transfected cells were used as the negative control. Equal amounts of protein extracts were separated on 3–8% Tris-Acetate gel. Sodium channel proteins were detected using a rabbit polyclonal sodium channel antibody (Upstate).
Surface Biotinylation experiments were performed according to the manufacturer’s protocol. Briefly, before cell lysis, surface proteins were labeled at 4°C with 0.25mg/ml Sulfo-NHS-SS-Biotin (Pierce). After cell lysis, biotinylated proteins were isolated through a NeutrAvidin Agarose resin column (Pierce) and after elution, Western Blot experiments were performed.
Patch-Clamp Experiments
Macroscopic sodium currents from transfected cells were recorded using the whole-cell configuration of the patch-clamp technique as previously described.19 Cells that emitted green fluorescence (indicating the presence of the sodium channel) and cyan fluorescence (indicating the presence of the peptide) were considered successfully transfected. Low resistance electrodes (<2 MΩ) were used, and a routine series resistance compensation of an Axopatch 200A was preformed to values >80% to minimize voltage clamp errors. The uncompensated Rseries was therefore less than 2 MΩ. Voltage clamp command pulses were generated by a microcomputer using PCLAMP software version 9.02 (Axon Instruments, Foster City, CA). Membrane currents were filtered at 5 kHZ and digitized with 12-bit resolution. The internal solution contained (in mmol/L) NaCl 35, CsF 105, EGTA 10, and Cs-HEPES 10 adjusted to pH 7.4. The bath solution contained (in mmol/L) NaCl 140, KCl 5, MgCl2 1, CaCl2 2, HEPES 10, and glucose 10 adjusted to pH 7.4. Experiments were performed at room temperature (22°C to 23°C).
Whole-cell sodium current densities, cell capacitance, current-voltage relationship, conductance, time course of recovery from inactivation, steady-state inactivation and persistent current were measured as previously described.7,19
Constructs for FRET
The FRET construct was created using a transposon approach, which randomly inserts Enhanced Yellow Fluorescent Protein (EYFP) from the transposon construct into the Nav1.5 construct. The p(AmpR)R6Kyori transposon (Epicenter Technologies) was altered to contain EYFP upstream of the Ampicillin gene. Through mutagenesis, restriction sites were created on each end of the Amp gene to facilitate removal. The newly formed EYFP-transposon was then transposed into the pre-existing Nav1.5 vector with Kanamycin resistance and with ECFP fused to the C-terminal region of the sodium channel. The double antibiotic resistance, Amp/Kan, was used to select for transposed clones. Diagnostic digests followed by sequencing were used to ensure that the EYFP inserted correctly in the sodium channel gene. Removal of the Amp gene from correctly transposed clones was accomplished by digestion with engineered restriction sites. The construct containing EYFP inserted at position 1022 was used because it produced significant FRET measurement and functional sodium currents. Mutagenesis was done on this construct to insert the R282H BrS mutation.
As a positive control for the FRET experiments we used a CFP-YFP dimer, in which CFP and YFP were linked by a short amino acid chain. As a negative FRET control we used CFP and YFP co-expressed in HEK293 cells from separate vectors, pECFP-C1 and pEYFP-N1 (BD Biosciences Clonetech, San Jose, CA), respectively.
Image Analysis and Calculation of FRET Ratios
Images were acquired with an Olympus IX71 fluorescent microscope equipped with a Hamamatsu ORCA-ER charge coupled device (12 bit) and controlled by SLIDEBOOK software (Intelligent Imaging Innovations, Denver, CO). Filter cube specifications for the fluorescent channels were as follow for excitation and emission, respectively: ECFP, 430±25 and 470±30 nm; EYFP, 500±20 and 535±30 nm, and FRET, 430±25 and 535±30 nm.
Image analysis involved three basic operations: subtraction of background autofluorescence and blurred light, quantification of fluorescence intensity, and calculation of a corrected FRET (FRETc) using the following equation:
Where IDA is the fluorescence intensity of the cell measured from the FRET filter set and IDD and IAA are the fluorescent intensities of the same cell measured from the ECFP (donor) and EYFP (acceptor) filter sets, respectively. The cross-talk coefficients, a and d, are considered constant and a property of our microscope’s filter sets, with a=0.03 and d=0.63. The corrected FRET ratio (FRETc) includes normalization to IDD to correct for variances in protein expression from cell to cell. FRETc ratios reported, in the presence or absence of 5 µg/mL Brefeldin A, were measured from the ER in order to accurately compare between unrescued and rescued channels and avoid differences that could be due to channel processing.
Statistical Analysis
All data are reported as mean ± (standard error of the mean) SEM. The normality and equality of variance were tested using the Shapiro-Wilk test and Levene’s test, respectively. Two sets of data were compared using the unpaired Student’s t-test. One-way ANOVA with Dunnett’s multiple-comparison test using the statistical program SPSS16 was used for multiple sets of data. The level of significance for all statistical tests was P<0.05.
Results
Peptides Containing the H558R-Polymorphism Restore Trafficking of R282H Mutant Channels
To determine if channel fragments were capable to restore trafficking of the BrS mutant channel, whole-cell sodium currents were recorded from HEK293 cells co-expressing the R282H mutant channel and CFP-fused peptide fragments of either wild-type or polymorphic cardiac sodium channels (Figure 1).
Representative current traces elicited from transfected cells are shown in Figure 2A. Cells expressing R282H alone showed a severe reduction in sodium current density, in line with previous reports that this mutation produces a trafficking deficient channel.7,20 Interestingly, when the mutant channel was co-transfected with a 40 amino acid channel fragment containing the H558R-polymorphism, R282H+R558-40aa, whole-cell current was increased to 30% of wild-type (WT) level. Furthermore, co-expression of a 20 amino acid fragment containing the polymorphism, R282H+R558-20aa, further increased current density (Figure 2A–B) to 75% of the WT level. However, when WT channels were co-expressed with the 40 amino acid fragment containing the polymorphism, WT+R558-40-aa, the current density was not significantly different from WT channels alone (−474.1±125.9pA/pF vs −337.7±105.8pA/pF, P>0.05). These results indicate that a small R558-containing peptide is able to specifically restore trafficking of the mutant channel.
Figure 2.
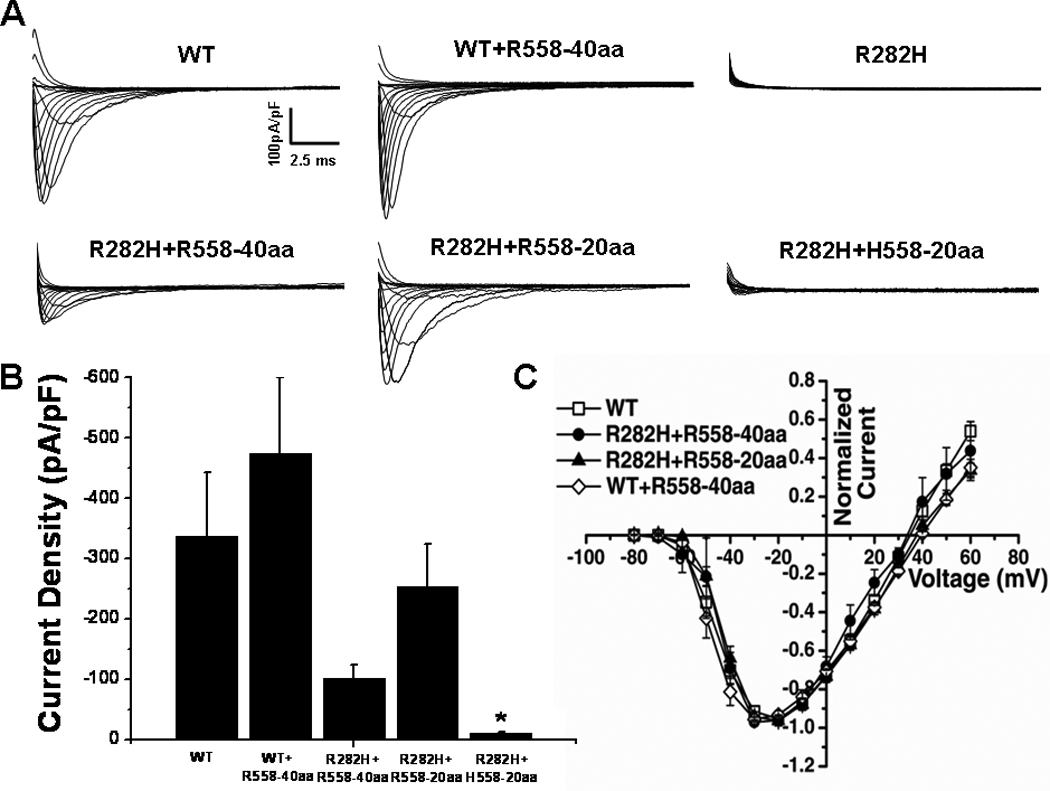
Peptides Containing the H558R-Polymorphism Restore Function of the R282H BrS Mutant Channel A. Whole-cell sodium current traces recorded from transfected HEK293 cells. The R282H mutation expressed alone does not generate functional sodium current. Current was restored when the mutant was co-expressed with R558-containing peptide fused with CFP (R282H+R558-40aa and R282H+R558-20aa), but not when co-expressed with the WT peptide (R282H+H558-20aa). B. Peak sodium current densities. Current density was similar for WT (n=6), R282H+R558-40aa (n=8), and R282H+R558-20aa (n=12) and significantly lower for R282H+H558-20aa (n=11). C. Current-Voltage Relationships were similar for all conditions. *P<0.05 compared to WT.
To ascertain that the polymorphism was necessary for rescue, the mutant channel was co-expressed with the corresponding 20 amino acid fragment of the WT channel, R282H+H558-20aa (Figure 2A–B). In this condition, restoration of current was not observed, indicating that the polymorphism is required to restore defective trafficking of the R282H mutant channel. To rule out any acute effects of the peptide, the 20 amino acid R558-containing peptide was synthesized and applied in the intracellular solution during whole-cell recordings. Acute expression of synthetic peptide did not alter peak current density of WT (−478.31±104.4pA/pF, n=6) and did not restore function of the R282H mutant channel (−23.8±4.2pA/pF, n=7).
Biophysical Properties of Rescued Currents
Having shown that R558-containing peptides fused to CFP were capable of restoring trafficking of the R282H mutant channel, we next compared the biophysical properties of rescued currents to WT currents. The current-voltage relationship, activation curve, voltage-dependence of steady-state inactivation, recovery from inactivation, and time constant of inactivation were measured for WT (open symbols) and rescued R282H currents (closed symbols). We found that normalized current-voltage relationships (Figure 2C) were not different between rescued and WT currents. The activation curve (Figure 3A) for rescued currents was also similar to WT. However, rescued R282H currents differed from WT currents in steady-state inactivation, which was shifted to more depolarized voltages, and recovery from inactivation was faster (Figures 3B–C and Table 1). The time course of inactivation (Figure 3D) was slower for channels rescued with the peptides, but only reached significance for channels treated with the smaller 20 amino acid peptide. Since the channel appeared to have impaired inactivation, we also measured the level of TTX-sensitive persistent current at the end of 300-ms depolarizing pulse for the R282H currents rescued by the R558-40aa peptide. These experiments demonstrated a significant increase in the level of persistent current (R282H:0.37±0.08% n=7 vs WT:0.15±0.07% n=5).
Figure 3.
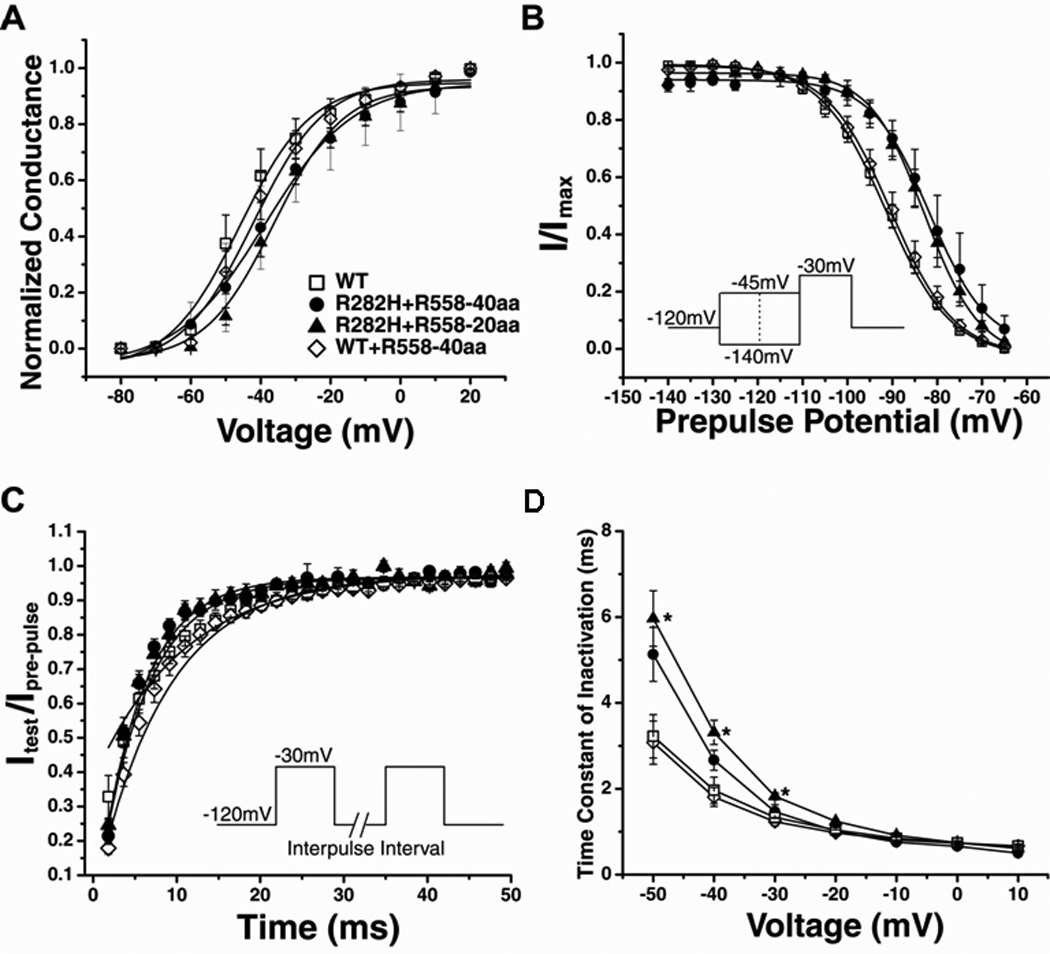
Biophysical Properties of Rescued Currents are Altered A. Conductance was similar for WT, rescued channels (R282H+R558-40aa and R282H+R558-20aa), and WT channels treated with the R558-containing peptide (WT+R558-40aa). B. Steady-State Inactivation of rescued currents (closed symbols) was significantly shifted toward depolarized potentials. C. Recovery from Inactivation. The time constants of recovery from inactivation were significantly faster for rescued channels. D. Time Constant of Inactivation. Inactivation of R282H+R558-20aa channels (n=9) was significantly slower than WT channels (n=7). R282H+R558-40aa channels (n=5) and WT+R558-40aa channels (n=9) behaved similarly to WT. *P<0.05 compared to WT.
Table 1.
Electrophysiological Properties for Wild-Type and BrS Mutant hNav1.5 Channels in the Presence or Absence of the Peptide Fragments.
| Na Channels hNav1.5 |
Peak Current Density (pA/pF) |
Steady-State Inactivation (V1/2), mV |
Recovery From Inactivation (τrec), ms |
|---|---|---|---|
| WT (n=7) | −337.7 ± 105.8 | −91.9 ± 1.7 | 8.4 ± 1.0 |
| WT+R558-40aa (n=9) | −474.1 ± 125.9 | −90.0 ± 1.6 | 7.0 ± 0.7 |
| R282H (n=10) | −9.7 ± 3.0a | n.a. | n.a. |
| R282H+H558-20aa (n=12) | −9.9 ± 2.8a | n.a. | n.a. |
| R282H+R558-40aa (n=9) | −101.9 ± 21.2b | −80.6 ± 2.7a | 4.5 ± 0.4a |
| R282H+R558-20aa (n=13) | −252.9 ± 71.8b | −82.8 ± 1.5a | 5.0 ± 0.5a |
P<0.01 compared with wild type.
P<0.01 compared with mutant alone.
n.a. = not available due to the small currents.
Importantly, none of these differences were observed when WT channels were treated with the R558-containing peptide, WT + R558-40aa (Figure 3). Taken together, the biophysical data suggests that the rescued R282H channels express a gain-of-function phenotype dominated by impaired inactivation, and that the fragments do not affect biophysical properties of WT channels.
Peptides Containing the H558R-Polymorphism Increase Nav1.5 protein Level in the Sarcolemma
We next sought to determine whether the restoration of the R282H mutation by the R558-containing peptide was due to alteration of Nav1.5 protein expression or correction of defective channel trafficking. To test this, we first transiently expressed the R282H mutation with or without the 20 amino acid fragment containing the polymorphism (R558-20aa). Then, we evaluated the expression level of the Nav1.5 proteins by Western blots. We first measured whole-cell lysate and noticed that the expression level of R282H was similar to that of WT Nav1.5 proteins (Figure 4A). Figure 4A shows that the presence of the fragment containing the H558R-polymorphism, R558-20aa, did not alter the total expression level of the R282H proteins in whole-cell lysate. In fact, sodium channel protein levels were similar for WT and R282H (with or without the R558-containing peptide). Since we observed an increase in peak current density for the R282H mutant in the presence of R558-containing peptides (Figure 2A–B), we performed surface biotinylation experiments to investigate the absence or presence of the R282H mutant in the cell membrane with or without the R558-containing peptide. Figure 4B and 4C confirm the absence of the R282H mutant in the cell membrane, as expected for a trafficking-deficient mutant. Importantly though, in presence of the R558-containing peptide, the R282H mutant sodium channel is inserted into the cell membrane, in line with current recordings.
Figure 4.
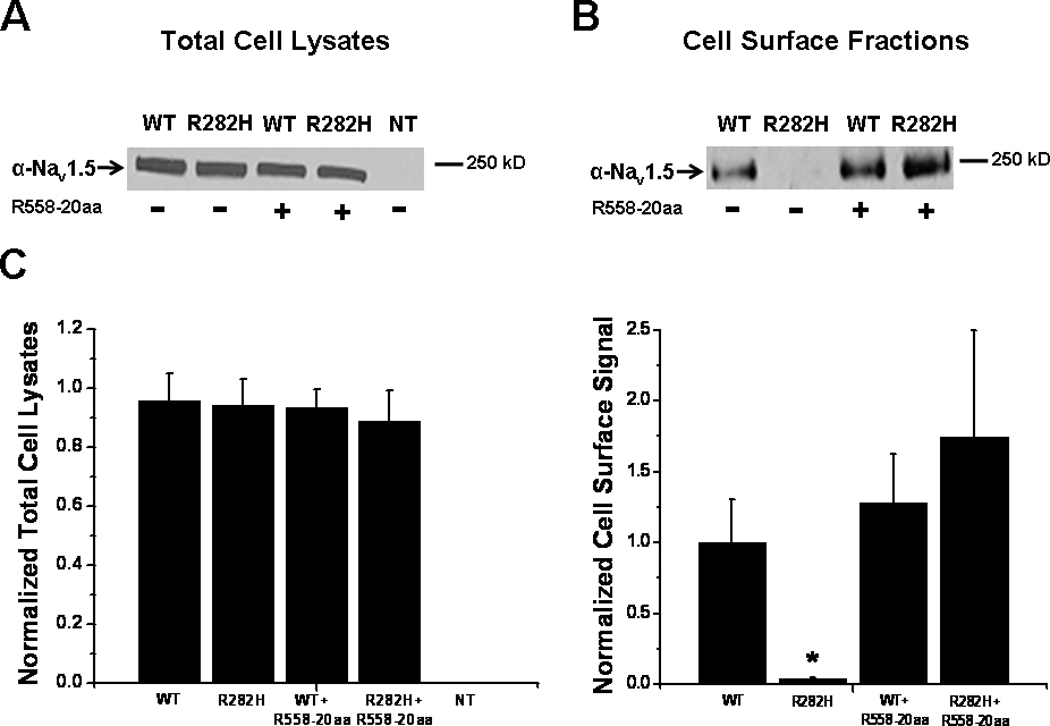
Biochemical Analysis A. Representative Western blot of three independent experiments showing WT and mutant proteins in total cell lysates in the presence or absence of the R558-containing peptide (R558-20aa). NT, non-transfected cells. B. Representative surface biotinylation experiments to investigate the absence or presence of the R282H mutant in the cell membrane with and without the peptide containing the polymorphism. Three independent biotinylation experiments were performed. C. Quantification normalized to WT. Data are presented as mean±SEM. *P<0.05 compared to WT.
R282H Affects Channel Conformation
In order to function properly, newly synthesized ion channels need to assemble and fold correctly in the ER, and traffic properly to their respective membrane subdomain.21 We have previously demonstrated that the absence of current observed with the R282H mutation is due to a trafficking defect.7 A common cause of trafficking deficient channels is misfolding and retention by the quality control machinery in the ER.22–24 Thus, one possible mechanism by which the R558-containing peptide may rescue the R282H mutant is by restoring proper folding of the mutated channel to an exportable conformation. FRET was used to examine folding of the trafficking-deficient mutant channel, R282H, in the presence and absence of the R558-containing peptide. To accomplish this, a sodium channel construct containing a pair of fluorescent proteins capable of transferring energy from one to the other was created.
The cardiac sodium channel used for these experiments, YFP-1022, had ECFP fused to the C-terminus of the channel and EYFP inserted into the Domain II-III linker at amino acid position 1022 (Figure 5A). This construct produced functional channels (Figure 5B). EYFP-1022 was associated with a corrected FRET (FRETc) value of 0.27±0.01 (Figure 5C). This value was significantly greater than the negative control (0.16±0.01, p<0.001). When the R282H mutation was created on EYFP-1022 (EYFP-1022/R282H), the FRETc value decreased significantly to 0.18±0.01 but was still significantly different from negative control (p=0.02). The FRET decrease indicated that the 3-D conformation of the channel has changed so that the EYFP and ECFP were now further apart. This is likely due to the fact that the R282H mutant channel is improperly folded. Interestingly, when fragments containing the polymorphism were co-expressed with the YFP-1022/R282H construct, FRETc was restored to values similar to non-mutated YFP-1022 p>0.05 (Figure 5C), which suggests that the mutated channel YFP-1022/R282H has now obtained a conformation similar to that of the functional YFP-1022 channel; this likely explains the ability of the mutant channel to traffic in presence of the R558-containing peptide. Similarly, when the YFP-1022/R282H construct was expressed in presence of the drug mexilitine which is known to restore trafficking of the R282H mutant, FRETc values were also restored to the level of the non-mutated YFP-1022 (data not shown). This supports the hypothesis that the mutant channel, either in presence of the drug or with the R558-containing peptide is now folding properly which allows the channel to traffic to the cell membrane forming functional channels.
Figure 5.
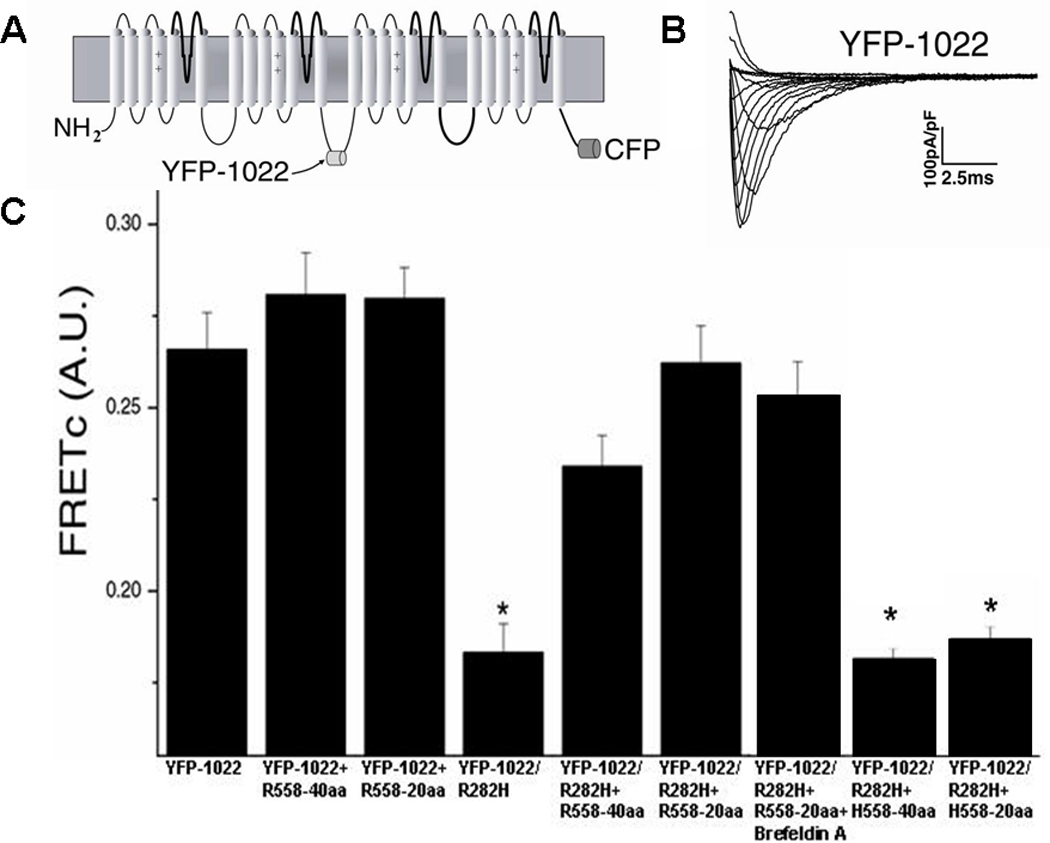
FRET in Normal and Mutant Channels A. Diagram of FRET construct containing CFP fused to the C-terminus of hNav1.5 and YFP inserted into the Domain II-III linker at amino acid position 1022. B. Currents elicited from the FRET construct are similar to WT currents. C. FRETc values for YFP-1022 (n=43) were similar to YFP-1022+R558-40aa (n=35), YFP-1022+R558-20aa (n=39), YFP-1022/R282H+R558-40aa (n=43), YFP-1022/R282H+R558-20aa (n=46), and YFP-1022/R282H+R558-20aa+Brefeldin A (n=34). FRETc of YFP-1022/R282H (n= 45) alone, YFP-1022/R282H+H558-40aa (n=36), and YFP-1022/R282H+H558-20aa (n=42) were significantly smaller than WT. *P<0.05 compared to WT.
To exclude non-specific effects of the peptide, non-mutated EYFP-1022 channels were co-expressed with R558-containing peptide. No FRETc increase was observed in these cells (Figure 5C), confirming that the increase in FRETc seen when EYFP-1022/R282H was co-expressed with the R558-containing peptide is specific. To further verify that the polymorphism was necessary for rescuing the improperly folded EYFP-1022/R282H channel, the EYFP-1022/R282H channel was co-expressed with the corresponding 40 or 20 amino acid fragment of the WT channel. In this experiment, restoration of FRETc values were not observed (Figure 5C), indicating that the polymorphism is required to promote the proper folding of the R282H channel.
Thus, these data suggest that restoration of folding in the ER will lead to protein trafficking to the sarcolemma which will result in the increase in peak current density observed (Figure 2A–B). To separate folding effects in the ER from forward trafficking to the cell surface membrane, we used the fungal metabolite Brefeldin A (BFA) which prevents anterograde transport from the Endoplasmic Reticulum (ER) to the Golgi apparatus. BFA treatment is thought to lead to a rapid accumulation of proteins within the ER following collapse of the Golgi apparatus.25,26 Consequently, incubation of cells co-expressing the R282H mutation and the R558-containing peptide (R282H+R558-20aa, n=6), together with BFA did not generate functional sodium current. In marked contrast, this blocking of forward trafficking with 5µg/mL BFA applied for 24 hours did not alter FRETc values p>0.05 (Figure 5C). These results indicate that conformational changes induced by EYFP-1022/R282H take place in the ER and not en route to the cell surface. Taken together, our FRET data suggest that the peptide containing the polymorphism rescues the R282H mutant by restoring proper folding of the mutated channel in the ER.
The Peptide Fragment Containing the H558R-Polymorphism Can Restore Currents for Multiple BrS Mutants
To expand on our observations with R282H, we tested 8 additional sodium channel BrS mutations located in the same region as R282H (pore domain I): G292S, V294M, K317N, L325R, G351V, D356N, R367H and R376H (Figure 1); mutations were taken from the Inherited Arrhythmia Database website (http://www.fsm.it/cardmoc/). Similarly to R282H, all mutants expressed alone resulted in significant decreased sodium currents compared to WT (Table 2). In fact, the K317N, L325R, G351V, D356N, R367H and R376H, resulted in non-measurable currents. For the G292S and V294M mutants, small and significantly reduced currents compared to WT were measured. Importantly, when these mutants were co-expressed with the R558-20aa peptide, the current density was increased for G292S, V294M, K317N and R376H (Table 2). The biophysical properties of these rescued currents were similar to WT channels, except for K317N which displayed a shift toward depolarized voltages for steady-state inactivation (Table 2). However, even in the presence of the R558-containing peptide, there were still no measurable currents for L325R, G351V, D356N and R367H (Table 2). Thus, we now show that out of 9 BrS-associated Nav1.5 mutations tested, the peptide was able to restore large sodium currents in 5 of them, confirming that this mechanism/strategy is not unique to the R282H mutation.
Table 2.
Electrophysiological Properties for BrS Mutant hNav1.5 Channels in the Presence or Absence of the R558-20aa Peptide.
| Na Channels hNav1.5 |
Peak Current Density (pA/pF) |
Steady-State Inactivation (V1/2), mV |
Recovery From Inactivationv (τrec), ms |
|---|---|---|---|
| G292S (n=10) | −129.2 ± 24.6a | −84.6 ± 1.3a | 5.9 ± 0.6 |
| G292S+R558-20aa (n=8) | −350.3 ± 25.5b | −89.8 ± 1.6 | 6.2 ± 0.3 |
| V294M (n=8) | −155.5 ± 24.8a | −87.8 ± 1.7 | 6.3 ± 0.7 |
| V294M+R558-20aa (n=10) | −339.7 ± 30.8b | −89.0 ± 0.8 | 5.9 ± 0.8 |
| K317N (n=7) | n.a. | n.a. | n.a. |
| K317N+R558-20aa (n=7) | −102.4 ± 14.8 | −83.8 ± 0.2a | 5.2 ± 0.3 |
| R376H (n=7) | n.a. | n.a. | n.a. |
| R376H+R558-20aa (n=7) | −110.3 ± 22.0 | −93.1 ± 3.8 | 5.9 ± 0.5 |
P<0.01 compared with wild type.
P<0.01 compared with mutant alone.
n.a. = not available due to the absence of currents.
The following mutants did not produce currents when co-expressed with the R558-20aa peptide: L325R, G351V, D356N and R367H.
Discussion
Brugada Syndrome is associated with mutations in the cardiac sodium channel that decrease whole-cell sodium currents, often by reducing channel expression at the cell surface. This lack of whole-cell sodium current is believed to cause an increased risk of sudden cardiac death in patients with BrS.27,28 In this study, we showed that small peptides, spanning the H558R-polymorphism, are sufficient to restore trafficking defect of a subset of BrS-associated Nav1.5 mutations. The central hypothesis of this work was first based on our previous study which provided us with enough supporting evidence of the rescue of the R282H mutant by the H558R-polymorphism (both clinical profile and cellular electrophysiology).7 However, we further hypothesized that this new strategy using peptide fragments of the cardiac sodium channel, spanning the H588R-polymorphism, could be used to restore trafficking and function of other BrS mutations in which reduced sodium current densities are also observed. Nine BrS trafficking-deficient mutations located in the pore of domain 1 of the cardiac sodium channel were studied. The peptide containing the H558R-polymorphism was able to increase current densities in five of these mutants. This is the first study showing that short fragments containing H558R-polymorphism can correct defective trafficking phenotype in BrS-associated Nav1.5 mutations. The FRET experiments suggest that the R558-containing peptides may act as “chemical chaperones” that promote proper protein folding and thereby facilitating the exit of rescued mutant ion channel from the ER.
It is commonly thought that channels which fail to traffic to the cellular membrane are misfolded, but very little work has been done to test this notion. Therefore, we employed a FRET based approach to study folding of the channel. To the best of our knowledge, this FRET approach has not been used to study folding of sodium channels. When R282H was introduced in a functional channel construct, containing CFP and YFP, the folding of the channel became altered, as shown by the decrease in FRETc signal and confirmed by the severe reduction of whole-cell sodium currents produced by the R282H mutation (Figure 5). Previous studies have shown that this mutant channel is retained in the ER7, and it appears that altered folding may be the cause of ER retention. In the presence of either of the peptides containing the H558R-polymorphism (40 aa and 20 aa), folding of the R282H mutant channel, as indicated by the restored FRETc value, was corrected. This data suggests that in the presence of the R558-containing peptide, the mutant channel is able to fold properly. Furthermore, blocking forward protein trafficking by BFA did not alter FRETc values. These data confirm that the R558-containing peptides specifically rescue the R282H mutant by restoring proper folding of the mutated channel in the ER. This indicates that the mechanism by which the polymorphism rescues the mutant channel involves either directly assisting in channel folding or allowing another protein to interact with the channel to correct folding.
A growing body of literature exists suggesting that trafficking defect of mutant channels can be rescued by fragments of polypeptides. Fragments of the HERG and CFTR channel have been shown to restore trafficking to mutant channels.15,16,29 Kupershmidt et al showed that a 100 amino acid peptide was able to restore trafficking to a HERG mutation in which the C-terminal region was truncated.17 When a peptide containing the Endoplasmic Retention signal, RXR, was co-expressed with the mutant channel, the peptide acted as a decoy, allowing the mutant channel protein to bypass ER quality control system. Moreover, Owsianik et al proposed a similar mechanism for the ability of peptides to rescue the ΔF508-CFTR mutation.16 However, after observing a similar rescue of the same channel, Cormet-Boyaka et al proposed that several different mechanisms may contribute to the fragments’ ability to help the mutant CFTR channel process, including that the fragments could be providing ER exit codes or burying areas on the CFTR channel that would lead to channel aggregation and destruction.15 However, none of these mechanisms seem to be occurring with our sodium channel fragments. Three RXR endoplasmic retention signals have been identified in the Domain I-II linker30 of Nav1.5, the region from which our fragments originate, but none are present in the fragments. Thus it seems unlikely that they are acting as decoys. The FRET data also suggest that the fragments are involved in folding, rather than providing or hiding ER signals.
The rescue of sodium channel mutations with mexilitine has been well documented.8,31,32 The theory behind these rescues is that the drug, which is a Nav1.5 blocker, stabilizes the pore of the channel allowing for trafficking. It is possible that the R558-containing peptides work similarly, in that they may be able to stabilize part of the channel for proper folding in the ER, and thus restore trafficking. However it appears unlikely that the fragments are acting as channel blockers. First, we did not observe a reduction of current density when WT channels were co-expressed with either the 40 amino acid fragment or in the presence of the synthesized 20 amino acid R558-containing peptide. If the fragment was acting as a blocker, we would expect to see some decrease in whole-cell current density in both transient transfection and acute effect experiments. Also, we know that the whole polymorphic channel can rescue the mutant channel7 and it is therefore unlikely that the polymorphic channel is blocking the mutant channel. Moreover, R558-containing peptide did not alter the expression level of the R282H proteins (Figure 4A). Thus we suspect that the mechanism of rescue with the R558-containing peptides differs from the one of sodium channel blockers. However, previous work in HERG has shown that channel blocking is not necessarily required for the rescue of trafficking-deficient channels to occur.33 Thus, it is possible for the mechanism of rescue of such drugs and of the peptide to be similar, and yet different than the blocking effect.
When trafficking is restored to the mutant R282H and K317N channels, changes in biophysical properties are uncovered suggesting the complexity of the disease. This finding is also consistent with a growing body of literature suggesting that some SCN5A mutations can cause both long QT and BrS phenotypes in members of one family, or LQT in one family and BrS in another.34–36 Pfahnl et al recently described a similar phenomenon, involving another trafficking-deficient BrS mutation, T353I, rescued by mexilitine.32 Interestingly, overnight exposure to 0.1 mM mexiletine increased T353I channel trafficking to the membrane to near normal levels. However, this mutant channels also showed a significant increased in late current by 1.6%, a finding seen with long QT mutations.
The destabilization of inactivation observed in rescued R282H mutant channels is consistent with the biophysical changes caused by mutations associated with Long QT Syndrome Type 3. It is plausible that the presence of a small late sodium current may be masked due to the inefficient channel trafficking, giving rise to the concern that protecting a patient from BrS may cause another type of arrhythmia. However, the initial patient, identified by Poelzing et al, who carries both the polymorphism and mutation, is healthy, displaying symptoms of neither BrS nor Long QT Syndrome.7 The possible explanation for the discrepancy in the alteration of inactivation kinetics following the R558-containing peptide treatment might be due to the difference in sodium channel population expressed in our heterologous expression system. In the present experiment, only the R282H channel is expressed, whereas in the previous report7, both H558R polymorphic channel and R282H channel were expressed. Therefore in the latter experiment, the whole-cell currents measured are the summation of the currents flowing through both H558R polymorphic channel and the rescued R282H channel. Thus, we speculate that the kinetic of the currents generated from the H558R polymorphic channel might conceal the alteration in fast inactivation of the rescued R282H mutant channel.
Conclusions and Study Limitation
We have thus shown that small peptides, spanning the H558R-polymorphism, are sufficient to restore trafficking defects of some BrS-associated Nav1.5 mutations. Due to their ability to rescue a mutant channel in vitro, we reasoned that it may be possible to use this R558-containing peptide as an alternative strategy aiming to rescue the trafficking defect of mutant cardiac sodium channels in vivo. We acknowledge the presence of limitations in our study. First, this rescuing strategy works on a subset of mutations and we are not aware yet of the exact mechanism which underlies this discrepancy between mutations. Of course, further studies are needed to test whether this rescue will occur in vivo. But importantly, although a future clinical application will require several further investigations, this report remains the first to present a new alternative strategy for treating the trafficking defects of BrS-associated sodium channels that for the first time addresses the underlying cause of the disease by restoring the function of the mutant channels. Additionally, this new approach has the advantage of using only a small fragment of the target gene which should reduce the problems usually seen with gene therapy when an entire gene is transferred. Therefore, polymorphic-containing peptides might represent a promising option to rescue trafficking-deficient channels.
Brugada Syndrome (BrS) is an inherited primary electrical cardiac disorder. It is characterized by ST-segment elevation in the right precordial leads (V1–V3) and increased susceptibility to sudden cardiac death due to episodes of polymorphic ventricular tachycardia. The genetic abnormalities that cause BrS have been linked to mutations in the SCN5A gene which encodes for the pore forming alpha-subunit of the cardiac sodium channel (Nav1.5). A trafficking defect of mutant cardiac sodium channels has been identified as one important cellular mechanism underlying a loss-of-function of cardiac sodium channels in BrS. Current treatment options for BrS include implantable cardioverter-defibrillator (ICD) and drug therapy mostly with isoproterenol. However, none of these treatment options directly addresses the channel dysfunction at the underlying source of BrS associated arrhythmias. We previously reported that the function of a trafficking-deficient BrS Nav1.5 mutation, R282H, could be restored by co-expression with the cardiac sodium channel containing the H558R-polymorphism. In the present study, we demonstrate that small peptides, spanning the H558R-polymorphism, are sufficient to restore trafficking defect of BrS-associated Nav1.5 mutations. Therefore, peptides containing the H558R-polymorphism might represent a promising option to rescue trafficking-deficient BrS mutations. In addition, our findings also suggest that it might be possible to use short-cDNA constructs as a novel strategy that is tailored to specific disease-causing mutations associated with BrS.
Acknowledgments
Funding Sources: This study was supported by an American Heart Association Scientist Development Grant (0635295N), R01HL094450 (ID), R21GM079467 (ID) and an AHA Pre-Doctoral Fellowship from the Great Rivers Affiliate (KS). SP was supported by the Michael Bilitch Fellowship in Cardiac Pacing and Electrophysiology from the Heart Rhythm Society.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Disclosures: None
References
- 1.Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20:1391–1396. doi: 10.1016/0735-1097(92)90253-j. [DOI] [PubMed] [Google Scholar]
- 2.Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, Gussak I, LeMarec H, Nademanee K, Perez Riera AR, Shimizu W, Schulze-Bahr E, Tan H, Wilde A. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111:659–670. doi: 10.1161/01.CIR.0000152479.54298.51. [DOI] [PubMed] [Google Scholar]
- 3.Priori SG, Napolitano C, Gasparini M, Pappone C, Della Bella P, Giordano U, Bloise R, Giustetto C, De Nardis R, Grillo M, Ronchetti E, Faggiano G, Nastoli J. Natural history of Brugada syndrome: insights for risk stratification and management. Circulation. 2002;105:1342–1347. doi: 10.1161/hc1102.105288. [DOI] [PubMed] [Google Scholar]
- 4.Schulze-Bahr E, Eckardt L, Breithardt G, Seidl K, Wichter T, Wolpert C, Borggrefe M, Haverkamp W. Sodium channel gene (SCN5A) mutations in 44 index patients with Brugada syndrome: different incidences in familial and sporadic disease. Hum Mutat. 2003;21:651–652. doi: 10.1002/humu.9144. [DOI] [PubMed] [Google Scholar]
- 5.Priori SG, Napolitano C, Gasparini M, Pappone C, Della Bella P, Brignole M, Giordano U, Giovannini T, Menozzi C, Bloise R, Crotti L, Terreni L, Schwartz PJ. Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome: A prospective evaluation of 52 families. Circulation. 2000;102:2509–2515. doi: 10.1161/01.cir.102.20.2509. [DOI] [PubMed] [Google Scholar]
- 6.Zimmer T, Surber R. SCN5A channelopathies-an update on mutations and mechanisms. Prog Biophys Mol Biol. 2008;98:120–136. doi: 10.1016/j.pbiomolbio.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 7.Poelzing S, Forleo C, Samodell M, Dudash L, Sorrentino S, Anaclerio M, Troccoli R, Iacoviello M, Romito R, Guida P, Chahine M, Pitzalis M, Deschenes I. SCN5A polymorphism restores trafficking of a Brugada syndrome mutation on a separate gene. Circulation. 2006;114:368–376. doi: 10.1161/CIRCULATIONAHA.105.601294. [DOI] [PubMed] [Google Scholar]
- 8.Valdivia CR, Tester DJ, Rok BA, Porter CB, Munger TM, Jahangir A, Makielski JC, Ackerman MJ. A trafficking defective, Brugada syndrome-causing SCN5A mutation rescued by drugs. Cardiovasc Res. 2004;62:53–62. doi: 10.1016/j.cardiores.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 9.Kyndt F, Probst V, Potet F, Demolombe S, Chevallier JC, Baro I, Moisan JP, Boisseau P, Schott JJ, Escande D, Le Marec H. Novel SCN5A mutation leading either to isolated cardiac conduction defect or Brugada syndrome in a large French family. Circulation. 2001;104:3081–3086. doi: 10.1161/hc5001.100834. [DOI] [PubMed] [Google Scholar]
- 10.Amin AS, Verkerk AO, Bhuiyan ZA, Wilde AA, Tan HL. Novel Brugada syndrome-causing mutation in ion-conducting pore of cardiac Na+ channel does not affect ion selectivity properties. Acta Physiol Scand. 2005;185:291–301. doi: 10.1111/j.1365-201X.2005.01496.x. [DOI] [PubMed] [Google Scholar]
- 11.Bezzina C, Veldkamp MW, van Den Berg MP, Postma AV, Rook MB, Viersma JW, van Langen IM, Tan-Sindhunata G, Bink-Boelkens MT, van Der Hout AH, Mannens MM, Wilde AA. A single Na(+) channel mutation causing both long-QT and Brugada syndromes. Circ Res. 1999;85:1206–1213. doi: 10.1161/01.res.85.12.1206. [DOI] [PubMed] [Google Scholar]
- 12.Dumaine R, Towbin JA, Brugada P, Vatta M, Nesterenko DV, Nesterenko VV, Brugada J, Brugada R, Antzelevitch C. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res. 1999;85:803–809. doi: 10.1161/01.res.85.9.803. [DOI] [PubMed] [Google Scholar]
- 13.Smits JP, Koopmann TT, Wilders R, Veldkamp MW, Opthof T, Bhuiyan ZA, Mannens MM, Balser JR, Tan HL, Bezzina CR, Wilde AA. A mutation in the human cardiac sodium channel (E161K) contributes to sick sinus syndrome, conduction disease and Brugada syndrome in two families. J Mol Cell Cardiol. 2005;38:969–981. doi: 10.1016/j.yjmcc.2005.02.024. [DOI] [PubMed] [Google Scholar]
- 14.Benito B, Brugada J, Brugada R, Brugada P. Brugada syndrome. Rev Esp Cardiol. 2009;62:1297–1315. doi: 10.1016/s1885-5857(09)73357-2. [DOI] [PubMed] [Google Scholar]
- 15.Cormet-Boyaka E, Jablonsky M, Naren AP, Jackson PL, Muccio DD, Kirk KL. Rescuing cystic fibrosis transmembrane conductance regulator (CFTR)-processing mutants by transcomplementation. Proc Natl Acad Sci U S A. 2004;101:8221–8226. doi: 10.1073/pnas.0400459101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Owsianik G, Cao L, Nilius B. Rescue of functional DeltaF508-CFTR channels by co-expression with truncated CFTR constructs in COS-1 cells. FEBS Lett. 2003;554:173–178. doi: 10.1016/s0014-5793(03)01162-1. [DOI] [PubMed] [Google Scholar]
- 17.Kupershmidt S, Yang T, Chanthaphaychith S, Wang Z, Towbin JA, Roden DM. Defective human Ether-a-go-go-related gene trafficking linked to an endoplasmic reticulum retention signal in the C terminus. J Biol Chem. 2002;277:27442–27448. doi: 10.1074/jbc.M112375200. [DOI] [PubMed] [Google Scholar]
- 18.Deschenes I, Armoundas AA, Jones SP, Tomaselli GF. Post-transcriptional gene silencing of KChIP2 and Navbeta1 in neonatal rat cardiac myocytes reveals a functional association between Na and Ito currents. J Mol Cell Cardiol. 2008;45:336–346. doi: 10.1016/j.yjmcc.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shinlapawittayatorn K, Du XX, Liu H, Ficker E, Kaufman ES, Deschenes I. A common SCN5A polymorphism modulates the biophysical defects of SCN5A mutations. Heart Rhythm. 2011;8:455–462. doi: 10.1016/j.hrthm.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Itoh H, Shimizu M, Mabuchi H, Imoto K. Clinical and electrophysiological characteristics of Brugada syndrome caused by a missense mutation in the S5-pore site of SCN5A. J Cardiovasc Electrophysiol. 2005;16:378–383. doi: 10.1046/j.1540-8167.2005.40606.x. [DOI] [PubMed] [Google Scholar]
- 21.Smyth JW, Shaw RM. Forward trafficking of ion channels: what the clinician needs to know. Heart Rhythm. 2010;7:1135–1140. doi: 10.1016/j.hrthm.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 23.Chevet E, Cameron PH, Pelletier MF, Thomas DY, Bergeron JJ. The endoplasmic reticulum: integration of protein folding, quality control, signaling and degradation. Curr Opin Struct Biol. 2001;11:120–124. doi: 10.1016/s0959-440x(00)00168-8. [DOI] [PubMed] [Google Scholar]
- 24.Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2003;4:181–191. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- 25.Donaldson JG, Kahn RA, Lippincott-Schwartz J, Klausner RD. Binding of ARF and beta-COP to Golgi membranes: possible regulation by a trimeric G protein. Science. 1991;254:1197–1199. doi: 10.1126/science.1957170. [DOI] [PubMed] [Google Scholar]
- 26.Lippincott-Schwartz J, Yuan LC, Bonifacino JS, Klausner RD. Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell. 1989;56:801–813. doi: 10.1016/0092-8674(89)90685-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coronel R, Casini S, Koopmann TT, Wilms-Schopman FJ, Verkerk AO, de Groot JR, Bhuiyan Z, Bezzina CR, Veldkamp MW, Linnenbank AC, van der Wal AC, Tan HL, Brugada P, Wilde AA, de Bakker JM. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: a combined electrophysiological, genetic, histopathologic, and computational study. Circulation. 2005;112:2769–2777. doi: 10.1161/CIRCULATIONAHA.105.532614. [DOI] [PubMed] [Google Scholar]
- 28.Papadatos GA, Wallerstein PM, Head CE, Ratcliff R, Brady PA, Benndorf K, Saumarez RC, Trezise AE, Huang CL, Vandenberg JI, Colledge WH, Grace AA. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene SCN5A. Proc Natl Acad Sci U S A. 2002;99:6210–6215. doi: 10.1073/pnas.082121299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kupershmidt S, Yang T, Chanthaphaychith S, Wang Z, Towbin JA, Roden DM. Defective human Ether-a-go-go-related gene trafficking linked to an endoplasmic reticulum retention signal in the C terminus. J Biol Chem. 2002;277:27442–27448. doi: 10.1074/jbc.M112375200. [DOI] [PubMed] [Google Scholar]
- 30.Zhou J, Shin HG, Yi J, Shen W, Williams CP, Murray KT. Phosphorylation and putative ER retention signals are required for protein kinase A-mediated potentiation of cardiac sodium current. Circ Res. 2002;91:540–546. doi: 10.1161/01.res.0000033598.00903.27. [DOI] [PubMed] [Google Scholar]
- 31.Valdivia CR, Ackerman MJ, Tester DJ, Wada T, McCormack J, Ye B, Makielski JC. A novel SCN5A arrhythmia mutation, M1766L, with expression defect rescued by mexiletine. Cardiovasc Res. 2002;55:279–289. doi: 10.1016/s0008-6363(02)00445-5. [DOI] [PubMed] [Google Scholar]
- 32.Pfahnl AE, Viswanathan PC, Weiss R, Shang LL, Sanyal S, Shusterman V, Kornblit C, London B, Dudley SC., Jr. A sodium channel pore mutation causing Brugada syndrome. Heart Rhythm. 2007;4:46–53. doi: 10.1016/j.hrthm.2006.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rajamani S, Anderson CL, Anson BD, January CT. Pharmacological rescue of human K(+) channel long-QT2 mutations: human ether-a-go-go-related gene rescue without block. Circulation. 2002;105:2830–2835. doi: 10.1161/01.cir.0000019513.50928.74. [DOI] [PubMed] [Google Scholar]
- 34.Grant AO, Carboni MP, Neplioueva V, Starmer CF, Memmi M, Napolitano C, Priori S. Long QT syndrome, Brugada syndrome, and conduction system disease are linked to a single sodium channel mutation. J Clin Invest. 2002;110:1201–1209. doi: 10.1172/JCI15570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Makita N. Phenotypic overlap of cardiac sodium channelopathies: individual-specific or mutation-specific? Circ J. 2009;73:810–817. doi: 10.1253/circj.cj-09-0014. [DOI] [PubMed] [Google Scholar]
- 36.Bezzina C, Veldkamp MW, van Den Berg MP, Postma AV, Rook MB, Viersma JW, van Langen IM, Tan-Sindhunata G, Bink-Boelkens MT, van Der Hout AH, Mannens MM, Wilde AA. A single Na(+) channel mutation causing both long-QT and Brugada syndromes. Circ Res. 1999;85:1206–1213. doi: 10.1161/01.res.85.12.1206. [DOI] [PubMed] [Google Scholar]