

Abstract
G-Protein coupled receptors (GPCRs) are intensely studied as drug targets and for their role in signaling. With the determination of the first crystal structures, interest in structure-based ligand discovery has increased. Unfortunately, most GPCRs lack experimental structures. The determination of the D3 receptor structure, and a community challenge to predict it, enabled a fully prospective comparison of ligand discovery from a modeled structure versus that of the subsequently released crystal structure. Over 3.3 million molecules were docked against a homology model, and 26 of the highest ranking were tested for binding. Six had affinities from 0.2 to 3.1μM. Subsequently, the crystal structure was released and the docking screen repeated. Of the 25 compounds selected, five had affinities from 0.3 to 3.0μM. One of the novel ligands from the homology model screen was optimized for affinity to 81nM. The feasibility of docking screens against modeled GPCRs more generally is considered.
GPCRs are a large family of membrane proteins that are critical for signal transduction. They have been a major focus of pharmaceutical research and are the primary targets of almost 30% of approved drugs1. All of these drugs were discovered without the aid of receptor structures by classical, ligand-based medicinal chemistry. Accordingly, many of these drugs reflect their origins as mimics of the natural signaling molecules. With the determination of the first drug-relevant GPCR structures in the last four years2-4, the opportunity for structure-based discovery of more novel scaffolds has arisen. Docking screens to these crystal structures have been unusually fruitful, with high hit-rates returning novel and potent ligands5-7. Still, the structures of most GPCRs remain undetermined. There are thought to be just over 360 pharmaceutically relevant GPCRs in the human genome8, and to date only five have had experimental structures determined, all by dint of extraordinary effort and innovation. For structure-based efforts to impact ligand discovery for most GPCRs, certainly in the near term, homology modeling of GPCR structures remains essential.
In the past, the structure of rhodopsin and, before that, bacteriorhodopsin9, were used to explore GPCR function and ligand recognition10-18. Several efforts to use homology models for ligand discovery, via docking, have also been undertaken19-25. With rare exceptions26,27, such docking screens use a hierarchy of pharmacophore filtering and ligand similarity to focus the molecules being docked. This will typically reduce an “unbiased” library by 10- to 100-fold to one more dominated by precedented chemotypes. Whereas this can be effective, such a combination of filtering and docking perforce removes unexpected chemotypes that a stand-alone, structure-based approach might otherwise find. Interestingly, two of these early studies included work on dopamine receptors, based on rhodopsin as a template20,21. Whereas both screens had high hit-rates, the pharmacophore filtering appears to bias the ligands discovered toward well-established chemotypes, a point to which we will return. More generally, the pharmacophore approach does not address those targets for which ligand information is weak, and does not illuminate how these models compare to what might be achieved with an experimental structure.
The opportunity to prospectively investigate how homology models compare to experimental structures for ligand discovery, and by extension to investigate what fraction of GPCRs might be exploitable for ligand discovery, emerged recently by way of a community challenge28. After the determination of the structure of the dopamine D329 and CXCR4 GPCRs in complex with antagonists (for D3, eticlopride, 1, Figure 1), the modeling community was asked to predict the structures of each complex before the coordinates were released. This provided an opportunity to not only predict the configuration of the single ligand bound to the complex, but also to use the homology model that emerged to discover new ligands, via structure-based docking screens, before the crystal structure was released. Once released, the same screen was prosecuted against the crystal structure. Since in each case the putative ligands would be tested for affinity, we could compare the two results to illuminate how successful the homology model was compared to the crystal structure in a situation where the predictions were truly blind.
Figure 1. Predicted Structure of the Dopamine D3 Receptor Binding Site.


(a) Comparison of the homology model of the dopamine D3 receptor in complex with eticlopride (light blue) to the crystal structure (yellow) visualized with PyMOL. The structures have been aligned using 15 binding site residues. Polar interactions for the crystal structure are shown in black dotted lines. (b) Chemical structure of eticlopride (compound 1).
We thus undertook the following calculations and experiments. Once we had submitted models of the D3/eticlopride complex, we turned to ligand discovery calculations. In these, over 3 million commercially available molecules were screened by docking to identify putative ligands that complemented the structure of the homology model. Before the crystal structure was released, 26 high scoring small molecules were purchased and tested for D3 receptor affinity (compounds 2-27). Finally, when the crystal structure was released several months later, a second docking campaign was prosecuted based on that structure; twenty-five more high scoring small molecules from this second screen were purchased and tested for D3 receptor affinity (compounds 28-52).
These calculations and experiments enabled the following investigation: Could a homology model—blinded to the (unknown) crystal structure—template the discovery of new ligands? How well would the homology model compare to the subsequent crystal structure, in terms of hit rate and affinity?
These questions thus directly addressed the possibility of using homology models for at least some of the vast majority of GPCRs whose structures will likely remain undetermined in the near term. More subtly, because the homology model was refined for its ability to enrich known D3 ligands, we wondered if ligands discovered against it would be biased toward known D3 chemotypes, and might therefore be less novel than those discovered using a crystal structure for docking-based discovery? From a chemical biology standpoint, we also wondered how ligand specificity would compare between the two screens. The template for the homology model was the β2 adrenergic receptor, and one might predict that the screened molecules might retain an activity for this target, or might be less specific than those screened against an experimental structure. Similarly, from a chemical probe standpoint, it is important to optimize for affinity, and we were unsure whether a homology model, selected for its ability to recognize general dopaminergic chemotypes, would be competent for such optimization. Here, we explored these questions by undertaking prospective docking screens against first a homology modeled structure of the D3 receptor and subsequently the crystal structure of the same receptor. Since the crystal structure was released after the screen was completed against the model structure, both screens were fully prospective. To our surprise, we found that the hit rates against both the modeled and experimental structures were not only high, but essentially equivalent. Notwithstanding the opportunities for bias toward known chemotypes in optimizing the homology model, both screens returned new scaffolds at a similar rate.
Results
Prediction of the Dopamine D3–Eticlopride Structure
The results of the D3/eticlopride structure prediction and docking challenge have been reported elsewhere 28 but will be briefly summarized here as they influence what follows. We were tasked with predicting the structure of the D3/eticlopride complex, without knowing either. We used the docking enrichment of known ligands among the top scoring molecules, from among a large number of decoys, as a criterion of model accuracy26,30,31. Almost 200,000 homology models were built using the program MODELLER-9v832 templated on the β13 and β22 adrenergic receptor crystal structures, and elastic network models calculated by the program 3K-ENM33 also based on these two structures. The top ranked 2964 of these 200,000 models, judged by MODELLER's internal DOPE score34, were advanced for docking. Up to 1,300 known dopaminergic ligands, along with up to 110,000 property-matched decoys, were docked35. Modeled receptor structures were prioritized for their ability to highly rank the known ligands compared to the decoys in the screens. Whereas this demanded a substantial amount of docking — 98,700,000 complexes calculated overall — it was largely automated. Our top model enriched the known ligands versus the decoys by 32-fold over what is expected at random among the 1% top ranking molecules. This enrichment was substantially higher than that found for docking of dopaminergic ligands against the β1 and β2 adrenergic templates used for the modeling, where the enrichments were 2 and 1, respectively.
Eticlopride was docked into each of the top models; five were selected for submission to the D3/eticlopride structure prediction and docking28. As was true of the predictions from several groups, our predicted structures showed overall fidelity to the subsequently released crystal complex. Our highest-ranked model had an overall Cα RMSD of 3.4Å, with an eticlopride RMSD of 1.65Å and an RMSD of the orthosteric site residues of 1.65Å (Figure 1). Most of the key ligand interactions14 observed in the crystal structure29 were also observed in this model, including the salt-bridge between the aminergic nitrogen of eticlopride and the recognition Asp1103.32 (Ballesteros-Weinstein numbering36). Similarly, two internal hydrogen bonds in eticlopride were captured by both the model and the crystal structure37. Intriguingly, all of the models that both enriched known ligands and docked eticlopride correctly were based on the templates from elastic network backbones; the higher range of motion explored by such models presumably contributed to the ultimate fidelity of the model to the experimental result. The 3K-ENM model captured backbone movements in several helices (III, IV, V and VI) that influence the shape of the binding site, e.g. a 1Å movement of transmembrane helix III in the region of Asp1103.32.
Docking for new D3 ligands
With this receptor model in hand, we next turned to ligand discovery. Over three million commercially available compounds were screened for complementarity to the receptor model, using DOCK3.638,39. Each molecule was fit into the site in an average of 1170 orientations, relative to the receptor, and for each orientation an average of 789 conformations, (thus over 900,000 configurations in total). Each configuration was scored for van der Waals and electrostatic complementarity, corrected for ligand desolvation39; thus for the 3.1 million compounds screened about 2 trillion complexes were evaluated. Prior to the release of the crystal coordinates, 26 top ranking compounds (2-27), all among the top 0.02% of docking-ranked molecules, were selected for experimental testing. The top scoring docking hits were dominated by mono-cationic molecules that all appeared to ion-pair with the key aminergic recognition residue Asp1103.32; most had overall good van der Waals complementarity to the orthosteric site. In the selecting the particular molecules for experimental testing, we corrected for energetic terms not included in the scoring function such as high ligand internal energy and receptor desolvation (detailed in Supplementary Methods). As far as we know, none had been previously tested for activity against dopaminergic receptors. Six of these, a hit-rate of 23%, bound to D3 measurably, with affinities ranging between 200 nM and 3.1 μM (Table 1, Figure 2, Figure 3 and Supplementary Results, Supplementary Figure 3). The similarity of the new compounds versus dopamine receptor ligands was assessed by calculating the Tanimoto coefficient (Tc) to the 10400 D3 annotations in the ChEMBL database (http://www.ebi.ac.uk/chembl). Four of the active compounds (2, 4, 5, and 7) resembled known ligands, with ECFP_4-based Tc values greater than 0.4. The two other active ligands, compounds 3 and 6, were topologically dissimilar to dopamine receptor ligands in the ChEMBL database (best Tc<0.35 by ECFP_4 fingerprints; novelty was also observed using Daylight fingerprints in Supplementary Table 2). These therefore appeared to be novel chemotypes for the D3 receptor.
Table 1. Discovered ligands from the docking screen against the dopamine D3 receptor homology model.
| # | Structure | TCa | Rankb (Model) |
Rankc (Crystal) |
Kid (μM) |
Closest Knownf |
|---|---|---|---|---|---|---|
| 2 |
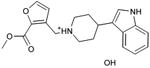
|
0.48 | 63 | 11,381 | 3.1 |
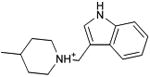
|
| 3 |
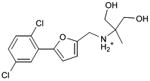
|
0.23 | 118 | 9,519 | 1.6 |
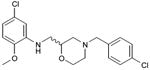
|
| 4 |
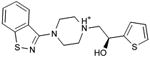
|
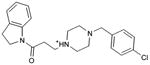
0.55 | 141 | 23,379 | 0.2 |
|
| 5 |
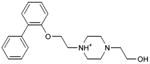
|
0.47 | 236 | 19,806 | 1.8 |
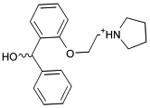
|
| 6 |
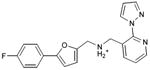
|
0.27 | 289 | 1,410 | 1.3 |
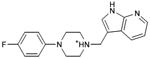
|
| 7 |
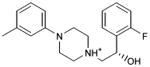
|
0.51 | 484 | 100,067e | 0.5 |
|
The ECFP_4 Tanimoto similarity (Tc) to the most similar dopamine receptor ligand in the ChEMBL database.
Rank of the compound in the screen against the homology model.
Rank of the compound in the screen against the crystal structure (PDB accession code: 3PBL).
Measured affinity for the dopamine D3 receptor. The uncertainty in each Ki is ±30%.
This compound was not in the ZINC lead like library at the time of the screen against the crystal structure. The rank has been calculated from a re-docking of the molecule.
The closest known dopamine receptor ligand from ChEMBL
Figure 2. Predicted binding modes of ligands found from the homology model screen.

Predicted binding poses for four ligands discovered in the docking screen against the dopamine D3 receptor homology model, visualized with UCSF Chimera: (a) 3 (b) 4 (c) 6 (d) 7 Predicted binding modes for the two analogs of compound 3 based on docking to the homology model (e) 56 and (f) 57.
Figure 3. Dose-response curves of discovered ligands.

Representative radioligand ([3H]N-methylspiperone) competition binding isotherms for compounds 3, 4, 7, 28, 30 and 31 (a-f). Data for a reference compound (chlorpromazine, black curve) are shown along with data for the test compound (red curve). Assays are performed using a final radioligand concentration between (0.5 × KD) and (1 × KD), where KD equals the radioligand dissociation constant, which is determined for each crude membrane preparation by radioligand saturation binding analysis. Data represent mean values ± standard error, performed on triplicate experiments.
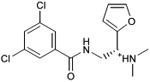
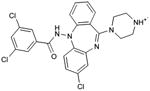
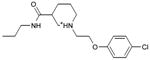
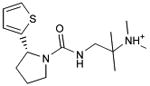
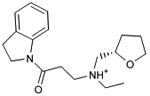
With access to the crystal structure (PDB code 3PBL)29, we then carried out a second docking screen of the 3.6 million lead-like molecules from ZINC40. Unlike the homology model, where side chain positions were optimized to enrich known ligands, the crystal structure heavy atom positions were unmodified. We selected 25 molecules (compounds 28-52) from among the top 0.02% of the crystal structure based screen for experimental testing. Five of these, molecules 28 to 32, were active, with Ki values between 300 nM and 3 μM, a hit-rate of 20% (Table 2, Figure 3, Figure 4 and Supplementary Figure 3). Whereas two of these, 28 and 30, resembled previously known scaffolds, and compound 32 was of intermediate similarity, two others, 29 and 31, represented novel scaffolds. Intriguingly, though 29 explored new substituents distal to the aminergic group, its aryl-amide core resembled that of eticlopride; such a chemotype was not observed among the actives from the homology-model screen. We note that compound 6, chosen from the homology model screen, also scored among the top 0.04% of the docking prioritized molecules from the crystal structure screen.
Table 2. Discovered ligands from the docking screen against the dopamine D3 receptor crystal structure.
| # | Structure | TCa | Rankc (Model) |
Rankb (Crystal) |
Kic (μM) |
Closest Knownf |
|---|---|---|---|---|---|---|
| 28 |
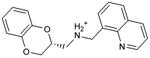
|
0.57 | 7,661 | 772 | 0.3 |
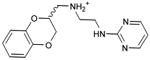
|
| 29 |
|
0.33 | 44,768e | 829 | 2.2 |
|
| 30 |
|
0.48 | 1,490e | 483 | 0.3 |
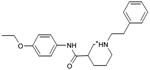
|
| 31 |
|
0.29 | 51,761e | 707 | 1.6 |
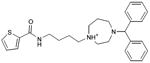
|
| 32 |
|
0.39 | 5,169e | 110 | 3.0 |
|
The ECFP_4 Tanimoto similarity (Tc) to the most similar dopamine receptor ligand in the ChEMBL database.
Rank of the compound in the screen against the homology model.
Rank of the compound in the screen against the crystal structure (PDB accession code: 3PBL.)
Measured affinity for the dopamine D3 receptor. The uncertainty in each Ki is ±30%.
This compound was not in the ZINC lead like library at the time of the screen against the homology model. The rank has been calculated from a re-docking of the molecule.
The closest known dopamine receptor ligand from ChEMBL
Figure 4. Predicted binding modes of ligands found from the crystal structure screen.

Predicted binding poses for the ligands discovered in the docking screen against the dopamine D3 receptor crystal structure, visualized with UCSF Chimera: (a) 28 (b) 29 (c) 31 (d) 32.
Ligand Bias in the Docking Screens
The homology model had been selected based on its ability to enrich known dopaminergic ligands and, in retrospective calculations it enriched known ligands substantially better than the crystal structure. Among those chemotypes most strongly enriched were phenyl-piperazines, which are characteristic for this target. The difference in enrichments between the screens was reflected in the compounds that were highly ranked in the prospective calculations. Overall, the overlap of the top 1000 docking hits from the two screens was only 90 molecules, two of which were selected for experimental evaluation; both were inactive. Eight of the compounds purchased for experimental testing from the homology model screen closely resembled known dopaminergic ligands, with ECFP_4 Tc values greater than 0.45 to annotated ligands in ChEMBL, but only four of the molecules purchased from the crystal structure screen had this level of similarity. The one instance where we observed a higher bias towards dopaminergic ligands from the crystal structure screen was in similarity to eticlopride itself. Indeed, nine of the molecules selected for testing had the aryl-amide-aminergic chemotype characteristic of eticlopride and its congeners, reflecting the many high-ranking molecules with this feature from the crystal structure screen. Conversely, only one compound from the homology model screen had this aryl-amide-aminergic chemotype. Notwithstanding these apparent biases going into experimental testing, novel chemotypes were ultimately confirmed for both screens.
The different ligands selected by the two screens reflect differences between the structures of the orthosteric sites in the homology model and the crystal structure. Whereas these two sites only differed by 1.65Å RMSD when superimposed, this was enough, when interrogated at a docking level, to change the identity, if not the nature, of the high-scoring docked molecules. The main difference between the two orthosteric sites is that the homology model is slightly more open and thus larger. For instance, the distance between the Cα atoms of Asp1893.32 and Ser1925.42 grew from 11.9Å in the crystal structure to 12.9Å in the model. More locally, Ile183 differs by 3.6Å between the two structures, while Val1895.39, Phe3456.51 and Phe3466.52 differ by 0.8 to 1.5Å. This overall opening reflects how the model was optimized: we docked ligands of all sizes to the model, and looked for enrichments. The model structure that was chosen could accommodate known ligands across a relatively wide size range, whereas the same site in the crystal structure more tightly encloses eticlopride, a relatively small ligand. Thus, many known phenyl-piperazine ligands that were enriched well by the model would clash with residues such as Val1895.39, Phe3456.51 and Phe3466.52 in the crystal structure.
Ligand Selectivity
An important challenge in dopaminergic receptor pharmacology is finding ligands that are specific for the D3 versus the D2 receptor. With few exceptions41, most D3 receptor ligands are also active on D2, making their use as chemical probes problematic. Methodologically, we were interested to learn if the new ligands derived from the homology-model docking retained activity for the β2 adrenergic receptor, from whose template the D3 model was derived. Active ligands were therefore counter-screened against the D2 and β2 receptors (Table 3). None had measurable activity against β2 AR at 10μM, suggesting that no significant template bias remained, and, also, that ligands specific for dopaminergic receptors had emerged. No effort was made to find D3 selective ligands, so achieving selectivity among the dopamine receptor subtypes would be fortuitous. Whereas most compounds showed little selectivity between D3 and D2, a few of the more novel scaffolds did, with affinities 6- and 20-fold better for the D3 over the D2 receptor for compounds 3 and 7, respectively (Table 3).
Table 3. Ligand selectivitiy for the dopamine D3, D2, and the β2 adrenergic receptor.
| Receptor Affinity (Ki, μM)a | |||
|---|---|---|---|
|
| |||
| Dopamine | Adrenergic | ||
|
| |||
| # | D3 | D2 | β2 |
| 2 | 3.1 | 1.3 | >10 |
| 3 | 1.6 | >10 | >10 |
| 4 | 0.2 | 0.4 | >10 |
| 5 | 1.8 | 0.4 | >10 |
| 6 | 1.3 | 4.5 | >10 |
| 7 | 0.5 | >10 | >10 |
| 28 | 0.3 | 0.9 | n.d. |
| 29 | 2.2 | 3.9 | n.d. |
| 30 | 0.3 | >10 | n.d. |
| 31 | 1.6 | >10 | n.d. |
| 32 | 3.0 | 3.8 | n.d. |
n.d.; Not determined
The uncertainty in each measured Ki is ±30%.
Optimization for Affinity
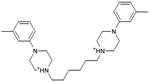
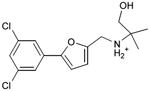
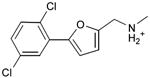
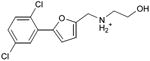
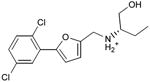
We were also interested in progressing a novel series for affinity, both as an end in itself and to explore whether model-based approaches could effectively guide this effort. Twenty analogs of compound 3, among the most dissimilar to known dopaminergic ligands, were found that had good complementarity to the D3 modeled structure (Table 4 and Supplementary Table 4, compounds 53 through 72). In its docked pose, the two hydroxyls and the aliphatic amine of compound 3 interacted with Asp1103.32 and Tyr3737.43. As it was difficult for these two residues to optimally interact with all the three ligand donors, we wanted to explore the possibility that the hydroxyls were not crucial for affinity. Commercially available molecules with this feature were extracted from the ZINC database40 and docked to the orthosteric site of the homology model. The docked poses were inspected and a set of analogs representing the diversity found in the database were selected for testing. All of these compounds retained the key Asp1103.32—cationic interaction, but explored variations in the hydroxyl groups and the substituents of the phenyl ring. In particular, we focused on compounds that preserved the meta-substituent on the phenyl ring, which fills a hydrophobic pocket formed by the side chains of residues Phe1643.28, Val1653.29, and Ser1684.57. Eleven analogs had substantially improved affinities, ranging from 4 to 20-fold better than the lead compound 3, with the most active reaching 81 nM (Table 4, Figure 2, Figure 3, and Supplementary Figure 3).
Table 4. Binding Affinities for eleven analogs of compound 3 against the dopamine D2 and D3 receptors.
|
|
||||
|---|---|---|---|---|
| # | Structure | TCa | Receptor Affinity (Ki, μM)b |
|
|
| ||||
| D3 | D2 | |||
| 53 |
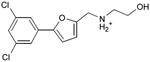
|
0.24 | 0.2 | 0.5 |
| 54 |
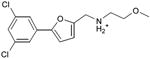
|
0.29 | 0.2 | 0.2 |
| 55 |
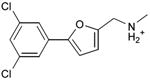
|
0.28 | 0.2 | 0.2 |
| 56 |
|
0.25 | 0.08 | 0.3 |
| 57 |
|
0.24 | 0.3 | 2.6 |
| 58 |
|
0.27 | 0.3 | 0.8 |
| 59 |
|
0.24 | 0.3 | 1.2 |
| 60 |
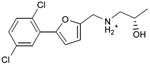
|
0.24 | 0.1 | 0.6 |
| 61 |
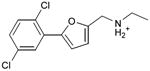
|
0.23 | 0.1 | 0.2 |
| 62 |
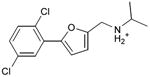
|
0.26 | 0.1 | 0.6 |
| 63 |
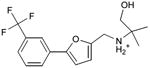
|
0.33 | 0.5 | 1.7 |
The Tanimoto similarity (Tc) to the most similar dopamine receptor ligand in the ChEMBL database.
The uncertainty in each measured Ki is ±30%.
Functional Activity of the Docking Hits
In previous docking screens against the GPCR crystal structures5-7, there has been a close correspondence between the function of the ligand co-crystalized with the receptor, either inverse agonist or neutral antagonist, and that of the docking hits. To explore whether this (presumably structural) bias was present in the D3 screens undertaken here, the docking hits (compounds 2-7 and 28-32) and several analogs of compound 3 (55-57 and 63) were investigated for agonism of the D3 and D2 receptors. With the possible exception of compound 28, which showed very weak partial agonism (Supplementary Figure 4 and Supplementary Table 3), none of these 15 compounds were agonists against either receptor and all appeared to function as antagonists. This corresponds with the known function of eticlopride, with which the receptor was co-crystalized and on whose binding mode the model was predicated.
Discussion
The determination of the structures of pharmacologically relevant GPCRs2-4 has sparked intense interest42. A crucial question is not only how these structures may themselves be exploited for ligand discovery, but what is the range of homologous targets that they illuminate. An astonishing result of this study was that the docking screen against the homology model was no less effective than that against the crystal structure; we would have been satisfied with the converse answer. The hit rates for both screens were high, at 23% and 20%, and their affinity ranges fully overlapped, with several molecules with 200 to 300 nM affinity from each screen. These predictions were likely right for the right reasons: the ligand poses overlapped with those adopted by eticlopride in its D3 complex, and this complex was itself well-predicted in the original blinded challenge. Homology models of proteins have been previously used for ligand discovery, including for GPCRs19-27. What was unusual, and perhaps unique to this study, was that a docking screen was prosecuted prospectively against a homology model and then, subsequently, the crystal structure. The results were thus doubly unbiased—the crystal structure was unknown at the time of the docking, and what we ultimately compared were new, experimentally-tested ligands.
A concern we harbored was whether the active molecules from the model-based screen—assuming any would be found—would be highly biased toward known dopaminergic ligands. A criterion for selecting effective models was their ability to enrich known ligands, and it seemed possible, even likely, that any actives that emerged from such a screen against it would simply recapitulate known D3 ligands. Indeed, the high-ranking molecules from the homology-model screen more closely resembled known dopaminergic ligands than did those from the crystal structure screen. Some of this bias can be seen among the experimentally tested molecules: for the model, three aryl-piperazines were confirmed as active, whereas for the crystal screen one eticlopride-like ligand was confirmed, the latter consistent with this structure's own conformational bias. In this sense the concern regarding bias was justified and may affect future studies. In the end, however, the experimentally active molecules were no more biased toward known ligands in one screen than the other. Meanwhile, from each screen emerged two novel scaffolds, four overall, and these not only differed from known ligands but also differed among themselves. Just as compelling, the active molecules from the homology model screen showed no measurable affinity for the β2-adrenergic receptor, which templated its modeling (Table 3). The model thus appeared to not only have captured the broad similarity that exists among aminergic GPCR targets, but also to have represented features specific to the D3 receptor. The lack of overlap among the hits from the two screens, and the observation that many ligands that dock well into the modeled structure did not fit into the corresponding crystallographic site, may reflect the many low energy conformations that GPCRs sample, both active and inactive43. The modeled and the experimental structures may thus represent different but viable low energy D3 receptor conformations, both likely inactive ones. This was also consistent with the satisfactory fidelity of the original D3/eticlopride structure prediction that was the point of departure for this study (Figure 1).
The relatively high affinities of the docking hits undoubtedly reflected the bias, among even large commercial libraries, toward molecules resembling known aminergic GPCR ligands5,7. It was encouraging that novel chemotypes could nevertheless be discovered. This is, after all, the promise of the structure-based enterprise: that based on complementarity to a protein structure novel ligands can be discovered and from these novel biologies might emerge. In this sense, it was instructive to compare the results of this study, which leveraged complementarity to a modeled structure alone in ligand selection, with an study that used a pipeline of pharmacophore filtering for dopaminergic chemotypes followed by docking20. Whereas this earlier study was in many ways path-breaking, and the hit rates and affinities achieved were high, the molecules discovered were typically much more similar to known dopaminergic ligands than those found here. This can be seen by inspection of the structures and comparison to the previously known ligands (Supplementary Table 5), or more quantitatively by considering the ECFP_4 Tanimoto coefficient values (Tc values). The Tc values averaged 0.55 to the most similar known dopaminergic, whereas, for the molecules discovered here, the average Tc value to the nearest known dopaminergic was 0.42, a large difference for this fingerprint that is born out by visual inspection (Table 1 and Supplementary Table 5).
Admittedly, the Ki of compound 3, which was among the most novel, was only 1.6 μM, probably too high (poor) to be useful as a probe or lead. As a new chemotype for this target, we wondered if its affinity could be improved. Structure-guided analog exploration led to derivatives of 3 with up to 20-fold improved affinity (Table 4), which may owe to elimination one of ethanolic group and exploration of small groups on the meta-position of the aryl ring. The most potent of these analogs, compound 56, had a Ki 81 nM, much more in the probe range and as potent as many approved dopaminergic drugs. With only 20 non-hydrogen atoms, 56 is only slightly larger than a fragment and is thus far from optimized; its ligand efficiency of 0.49 is promising for further elaboration44. This series may merit further consideration as D3 receptor probes.
Apart from bias in the ligand libraries, docking hits against GPCRs have previously recapitulated the functional properties of the ligand with which the receptor was co-crystalized, presumably reflecting a bias in the receptor structure used. Thus, earlier structure-based screens against inactive structures of GPCRs found only antagonists and inverse agonists5-7. This was true here too, both the ligands discovered against the homology model and those discovered against the crystal structure were essentially all antagonists. This likely reflected the inactive D3 conformation selected, from among those sampled in solution, by eticlopride in the experimental structure, and the bias toward such a structure from the modeling of a conformation competent to recognize the drug in the homology model. Additionally, most of the known ligands chosen for docking against the homology model were also antagonists.
GPCRs are central to cell signaling and are key targets for medicinal chemistry. The determination of the structures of pharmacologically-relevant GPCRs illuminates why they are so fruitful for drug discovery—their orthosteric sites are particularly well-suited to accommodate small organic molecules. This, and the substantial bias of chemical libraries towards the ligands of these targets, explains the high-potency of hits emerging from structure-based, and indeed high-throughput screens against them5-7. In this sense, the docking screens against the D3 receptor reprise what we learned from those against the β2-adrenergic and A2A adenosine receptors—hit rates are high, as are the affinities of the hits.
What was new to this study, in addition to the particular ligands discovered, was the direct, prospective comparison of the ability of homology models of GPCRs to template ligand discovery. The model used here was fully blinded from the crystal structure, but it was ultimately as effective in prioritizing active D3 ligands as judged by the hit-rate, the potency, and the novelty of the new ligands. Whereas we did not expect this result, it was encouraging for the structure-based enterprise against GPCRs. These receptors have advantages for homology modeling: the conservation of the seven trans-membrane helixes, and several strongly conserved residues, e.g. the DRY and NPXXY motifs, allow registry in sequence alignment to be determined with greater confidence than is typically possible. At a conservative cut-off of 35% transmembrane sequence identity, the five structures determined to date resemble 59 other GPCRs45(Supplementary Table 6). Whereas each new GPCR crystal structure will provide a rich vein for ligand discovery, their luster may reflect on a much larger number of exploitable targets.
Methods
Homology Models
The initial alignment was generated using PROMALS3D46 using a sequence profile that included all dopamine receptor sequences as well as the β1 and β2 adrenergic receptor sequences (PDB: 2VT4(chain B)3 and 2RH1(chain A)2). The initial alignment was manually refined to correctly align the residues forming the conserved disulfide bonds (C103-C181 and C355-C358). Alternative alignments of the extracellular loop 2 (EL2), which contacts the binding site14-16 were evaluated, resulting in the final alignment (Supplementary Figure 2). All homology models were built with MODELLER-9v832. The models were based on two types of templates: (1) the crystal structures of the β13 and β22 adrenergic receptors and (2) 710 elastic network models produced by 3K-ENM33, based on each of these two crystal structures. This led to almost 200,000 homology models. These were scored using DOPE34 resulting in 2964 models scoring well by modeling criteria, 4 from each 3K-ENM backbone, 64 from each crystal structure backbone.
The 2964 models were then evaluated for their ability to enrich known ligands among a large number of decoys. The models were ranked based on their adjusted logAUC and the enrichment factor at 1% (EF1) of the database39. Models had to score in the top quartile for logAUC and EF1, and more than 60% of the best scoring ligands had to form the conserved salt-bridge interaction with Asp1103.32, to be considered. The conformational sampling of eticlopride was restricted to conserve the internal hydrogen bonds observed in the Cambridge Structural Database47. Before the release of the crystallographic structure, five modeled eticlopride/D3 structures were submitted to the GPCRDOCK2010 competition28Of these, models #1 and #4 had ligand poses and orthosteric residue positions that resembled that of the crystal structure (to 1.65Å or better).
Molecular Docking Screens
A version of DOCK3.5.54 with an improved treatment of ligand solvation and with improved speed, DOCK3.638,39 (http://dock.compbio.ucsf.edu/), modified with scripting drawn from DOCK Blaster (http://blaster.docking.org) was used in docking calculations against the homology model and the crystallographic structure of the dopamine D3 receptor (PDB 3PBL29). The flexible-ligand sampling algorithm in DOCK3.6 superimposes atoms of the docked molecule onto binding site matching spheres, which represent favorable positions for individual ligand atoms. Forty-five matching spheres were used; for the crystal structure these were derived from the position of eticlopride while the spheres for the homology models were derived from overlaid docking poses of known ligands. The degree of ligand sampling is determined by the bin size, bin size overlap, and distance tolerance, set to 0.4Å, 0.1Å, and 1.5Å, respectively, for both the matching spheres and the docked molecules. Complementarity of each ligand pose is scored as the sum of the receptor–ligand electrostatic and van der Waals interaction energy, corrected for ligand desolvation39. The best scoring conformation of each docked molecule is then subject to 100 steps of rigid-body minimization. Partial charges from the united atom AMBER force field were used for all receptor atoms except for Ser1925.42, Ser1935.43, and Ser1965.46, for which the dipole moment was increased as previously described5. From the ZINC lead-like set of commercially available molecules, over 3 million compounds were docked. Prior to selecting compounds for experimental testing, the hit list was filtered to remove a previously known high internal energy motif that results in unreasonably favorable docking scores48, using automated scripts. The rankings reported here reflect this filtering (see Supplementary Methods for details).
Binding affinity and functional activity of the docking-predicted compounds
Affinities for D3, D2 dopaminergic and β2 adrenergic receptors were determined by radioligand competition binding at the NIMH Psychoactive Drug Screening Program49. Briefly, crude P2 (21,000 × g) membrane preparations were prepared from cell lines transiently expressing recombinant human GPCRs at about 50μg protein/microliter of 50mM Tris, 1% BSA, pH 7.4 (assayed by Bradford using a BSA standard). 50μL of membrane suspension were added to the wells of a 96-well plate containing 100μL of binding assay buffer, 50μL of radioligand present at five times its Kd, and 50μL of candidate ligand at a concentration five times that desired in the assay (Supplementary Table 1). Reactions were incubated for 60 to 90 min at room temperature in the dark and then harvested onto 0.3% PEI-treated GF/A filtermats (Wallac). After three washes with ice-cold wash buffer (50 mM Tris, pH 7.4), filter mats were dried in a microwave oven and impregnated with Meltilex scintillant (Wallac). Residual radioligand binding, measured by scintillation using a TriLux microbeta counter (Wallac), was plotted as a function of competitor and regressed using “one-site competition” in Prism4.0 (GraphPad) to obtain IC50 values. Ki values were calculated from the IC50 values using the Cheng-Prusoff approximation.
To investigate the functional activity of the new ligands (i.e., agonism or antagonism) at D2 and D3 receptors, we measured recruitment of β-arrestin2 to agonist-occupied receptors using the Tango assay50 (summarized here, see Supplementary Methods). HTLA cells were transfected with plasmid encoding either the hD2V2 Tango receptor or the hD3V2 Tango receptor. As a negative control, cells were transfected with pEYFP-N1 (Clontech). Subsequently, the cells were trypsinized, resuspended to 1 × 104 cells/50μl growth medium, and seeded in poly-D-lysine-coated glass-bottom 384-well plates (Costar). The next day, the medium was replaced with serum-free DMEM (Cellgro), and the cells were stimulated with reference agonist (Quinpirole), reference antagonist (Chlorpromazine), or test compounds. Assay concentrations of all compounds ranged from 3pM to 30μM. After an overnight incubation with reference or test compounds, the medium was removed and replaced with 1× BriteGlo (Promega). Luminescence was counted using a TriLux (PerkinElmer) plate reader. Quinpirole (Sigma-Aldrich), Chlorpromazine (Sigma-Aldrich), and the test compounds were all inactive on HTLA cells not expressing a Tango receptor. In additional control experiments, HTLA cells were transfected with a plasmid encoding the human V2 Tango receptor50; Quinpirole and Chlorpromazine had no effect on HTLA cells expressing this receptor.
Supplementary Material
Acknowledgments
We thank Drs. Qingyi Yang and Kim Sharp for help with 3K-ENM, Dr. Mark Burlingame with help with compound LC/MS, and Prof. Gilad Barnea (Brown University) for supplying beginning Tango constructs and a plasmid encoding the human V2 Tango receptor. Supported by NIH grants GM59957 and GM71630 (to BKS), by U54GM093342 (to AS and BKS), by GM71790 R01GM54762 (to AS), and the NIMH Psychoactive Drug Screening Program (to BLR), by postdoctoral fellowships from the Knut and Alice Wallenberg Foundation (to JC) and NRSA-Kirschstein F32GM096544 (to RGC) and F32GM088991 (to ASch)
Footnotes
Supplementary Information. Additional methods, both computational and experimental, are detailed as Supplementary Methods. Compound sourcing and purity are also described in Supplementary Methods. Supplementary Results include the sequence alignment used during modeling, the results of the functional assays, a table describing GPCRs with high sequence identity and known ligands, and all compounds tested for both screens and analogs are presented. The top lists produced by each docking screen are also presented in their entirety as Supplementary Dataset 1. Coordinates for the discovered ligands docked to the homology model and crystal structure are also supplied as Supplementary Dataset 2.
Author Contributions: Docking and homology modeling was conducted by JC and RGC, the latter with assistance from HF, ASch and AS. JJI and BKS assisted with compound selection and strategy, JJI lent expertise with the DOCK Blaster toolchain and fixed problems with ZINC. BLR and VS were responsible for the pharmacological guidance as to the appropriate specificity tests, while VS conducted all the experiments. All authors contributed to the writing of the manuscript.
The authors declare no competing financial interest.
References
- 1.Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 2.Cherezov V, et al. High-Resolution Crystal Structure of an Engineered Human β2-Adrenergic G Protein–Coupled Receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Warne T, et al. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaakola VP, et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carlsson J, et al. Structure-Based Discovery of A2A Adenosine Receptor Ligands. J Med Chem. 2010;53:3748–3755. doi: 10.1021/jm100240h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katrich V, et al. Structure-Based Discovery of Novel Chemotypes for Adenosine A2A Receptor Antagonists. J Med Chem. 2010;53:1799–2809. doi: 10.1021/jm901647p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kolb P, et al. Structure-based discovery of β2-adrenergic receptor ligands. Proc Natl Acad Sci USA. 2009;106:6843–6848. doi: 10.1073/pnas.0812657106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vassilatis DK, et al. The G protein-coupled receptor repertoires of human and mouse. Proc Natl Acad Sci USA. 2003;100:4903–4908. doi: 10.1073/pnas.0230374100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donnelly D, Findlay JBC. Seven-helix receptors: structure and modelling. Curr Opin Struct Biol. 1994;4:582–589. [Google Scholar]
- 10.Kurczab R, Nowak M, Chilmonczyk Z, Sylte I, Bojarski AJ. The development and validation of a novel virtual screening cascade protocol to identify potential serotonin 5-HT7R antagonists. Bioorg Med Chem Lett. 2010;20:2465–2468. doi: 10.1016/j.bmcl.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 11.Salo OMH, et al. Virtual Screening of Novel CB2 Ligands Using a Comparative Model of the Human Cannabinoid CB2 Receptor. J Med Chem. 2005;48:7166–7171. doi: 10.1021/jm050565b. [DOI] [PubMed] [Google Scholar]
- 12.Bissantz C, Schalon C, Guba W, Stahl M. Focused library design in GPCR projects on the example of 5-HT2c agonists: Comparison of structure-based virtual screening with ligand-based search methods. Proteins: Struct Funct Bioinf. 2005;61:938–952. doi: 10.1002/prot.20651. [DOI] [PubMed] [Google Scholar]
- 13.Kratochwil NA, et al. An Automated System for the Analysis of G Protein-Coupled Receptor Transmembrane Binding Pockets: Alignment, Receptor-Based Pharmacophores, and Their Application. J Chem Inf Model. 2005;45:1324–1336. doi: 10.1021/ci050221u. [DOI] [PubMed] [Google Scholar]
- 14.Shi L, Javitch JA. The Binding Site Of Aminergic G Protein-Coupled Receptors: The Transmembrane Segments and Second Extracellular Loop. Annual Review of Pharmacology and Toxicology. 2002;42:437–467. doi: 10.1146/annurev.pharmtox.42.091101.144224. [DOI] [PubMed] [Google Scholar]
- 15.Shi L, Javitch JA. The second extracellular loop of the dopamine D2 receptor lines the binding-site crevice. Proc Natl Acad Sci USA. 2004;101:440–445. doi: 10.1073/pnas.2237265100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Graaf C, Foata N, Engkvist O, Rognan D. Molecular modeling of the second extracellular loop of G-protein coupled receptors and its implication on structure-based virtual screening. Proteins: Struct Funct Bioinf. 2008;71:599–620. doi: 10.1002/prot.21724. [DOI] [PubMed] [Google Scholar]
- 17.de Graaf C, Rognan D. Customizing G Protein-Coupled Receptor Models for Structure-Based Virtual Screening. Curr Pharm Des. 2009;15:4026–4048. doi: 10.2174/138161209789824786. [DOI] [PubMed] [Google Scholar]
- 18.Michino M, et al. Community-wide assessment of GPCR structure modelling and ligand docking: GPCR Dock 2008. Nat Rev Drug Disco. 2009;8:455–463. doi: 10.1038/nrd2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tikhonova IG, et al. Discovery of novel agonists and antagonists of the free fatty acid receptor 1 (FFAR1) using virtual screening. J Med Chem. 2008;51:625–633. doi: 10.1021/jm7012425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Varady J, et al. Molecular Modeling of the Three-Dimensional Structure of Dopamine 3 (D3) Subtype Receptor: Discovery of Novel and Potent D3 Ligands through a Hybrid Pharmacophore- and Structure-Based Database Searching Approach. J Med Chem. 2003;46:4377–4392. doi: 10.1021/jm030085p. [DOI] [PubMed] [Google Scholar]
- 21.Becker OM, et al. G protein-coupled receptors: In silico drug discovery in 3D. Proc Natl Acad Sci USA. 2004;101:11304–11309. doi: 10.1073/pnas.0401862101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engel S, et al. A Virtual Screen for Diverse Ligands: Discovery of Selective G Protein-Coupled Receptor Antagonists. J Am Chem Soc. 2008;130:5115–5123. doi: 10.1021/ja077620l. [DOI] [PubMed] [Google Scholar]
- 23.Evers A, Klabunde T. Structure-based Drug Discovery Using GPCR Homology Modeling: Successful Virtual Screening for Antagonists of the Alpha1A Adrenergic Receptor. J Med Chem. 2005;48:1088–1097. doi: 10.1021/jm0491804. [DOI] [PubMed] [Google Scholar]
- 24.Evers A, Klebe G. Successful Virtual Screening for a Submicromolar Antagonist of the Neurokinin-1 Receptor Based on a Ligand-Supported Homology Model. J Med Chem. 2004;47:5381–5392. doi: 10.1021/jm0311487. [DOI] [PubMed] [Google Scholar]
- 25.Kellenberger E, et al. Identification of Nonpeptide CCR5 Receptor Agonists by Structure-based Virtual Screening. J Med Chem. 2007;50:1294–1303. doi: 10.1021/jm061389p. [DOI] [PubMed] [Google Scholar]
- 26.Cavasotto CN, et al. Discovery of Novel Chemotypes to a G-Protein-Coupled Receptor through Ligand-Steered Homology Modeling and Structure-Based Virtual Screening. J Med Chem. 2008;51:581–588. doi: 10.1021/jm070759m. [DOI] [PubMed] [Google Scholar]
- 27.Kiss R, et al. Discovery of novel human histamine H4 receptor ligands by large-scale structure-based virtual screening. J Med Chem. 2008;51:3145–3153. doi: 10.1021/jm7014777. [DOI] [PubMed] [Google Scholar]
- 28.Kufareva I, et al. Status of GPCR modeling and docking as reflected by community wide GPCR Dock 2010 assessment. Structure. 2011 doi: 10.1016/j.str.2011.05.012. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chien EYT, et al. Structure of the Human Dopamine D3 Receptor in Complex with a D2/D3 Selective Antagonist. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Evers A, Gohlke H, Klebe G. Ligand-supported Homology Modelling of Protein Binding-sites using Knowledge-based Potentials. J Mol Biol. 2003;334:327–345. doi: 10.1016/j.jmb.2003.09.032. [DOI] [PubMed] [Google Scholar]
- 31.Katritch V, Rueda M, Lam PCH, Yeager M, Abagyan R. GPCR 3D homology models for ligand screening: Lessons learned from blind predictions of adenosine A2a receptor complex. Proteins: Struct Funct Bioinf. 2010;78:197–211. doi: 10.1002/prot.22507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eswar N, et al. Comparative Protein Structure Modeling With MODELLER. Curr Prot in Bioinf. 2006;5:1–30. doi: 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang Q, Sharp KA. Building alternate protein structures using the elastic network model. Proteins: Struct Funct Bioinf. 2009;74:682–700. doi: 10.1002/prot.22184. [DOI] [PubMed] [Google Scholar]
- 34.Shen MY, Sali A. Statistical potential for assessment and prediction of protein structures. Prot Sci. 2006;15:2507–2524. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang N, Shoichet BK, Irwin JJ. Benchmarking Sets for Molecular Docking. J Med Chem. 2006;49:6789–6801. doi: 10.1021/jm0608356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 37.Kuhn B, Mohr P, Stahl M. Intramolecular Hydrogen Bonding in Medicinal Chemistry. J Med Chem. 2010;53:2601–2611. doi: 10.1021/jm100087s. [DOI] [PubMed] [Google Scholar]
- 38.Irwin JI, et al. Automated docking screens: a feasibility study. J Med Chem. 2009;52:5712–5720. doi: 10.1021/jm9006966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mysinger MM, Shoichet BK. Rapid Context-Dependent Ligand Desolvation in Molecular Docking. J Chem Inf Model. 2010;50:1561–1573. doi: 10.1021/ci100214a. [DOI] [PubMed] [Google Scholar]
- 40.Irwin JJ, Shoichet BK. ZINC - A free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Newman AH, et al. N-(4-(4-(2,3-Dichloro- or 2-methoxyphenyl)piperazin-1-yl)butyl)heterobiarylcarboxamides with Functionalized Linking Chains as High Affinity and Enantioselective D3 Receptor Antagonists. J Med Chem. 2009;52:2559–2570. doi: 10.1021/jm900095y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bucci M, Goodman C, Sheppard TL. A decade of chemical biology. Nat Chem Biol. 2010;6:847–854. doi: 10.1038/nchembio.489. [DOI] [PubMed] [Google Scholar]
- 43.Kobilka BK, Deupi X. Conformational complexity of G-protein-coupled receptors. Trends Pharmacol Sci. 2007;28:397–406. doi: 10.1016/j.tips.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 44.Hajduk PJ, Greer J. A decade of fragment-based drug design: strategic advances and lessons learned. Nat Rev Drug Disco. 2007;6:211–219. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]
- 45.Gloriam DE, Foord SM, Blaney FE, Garland SL. Definition of the G Protein-Coupled Receptor Transmembrane Bundle Binding Pocket and Calculation of Receptor Similarities for Drug Design. J Med Chem. 2009;52:4429–4442. doi: 10.1021/jm900319e. [DOI] [PubMed] [Google Scholar]
- 46.Pei J, Kim BH, Grishin NV. PROMALS3D: a tool for multiple protein sequence and structure alignments. Nuc Acid Res. 2008;36:2295–2300. doi: 10.1093/nar/gkn072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Allen FH. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Cryst B. 2002;58:380–388. doi: 10.1107/s0108768102003890. [DOI] [PubMed] [Google Scholar]
- 48.Ferreira RS, et al. Complementarity Between a Docking and a High-Throughput Screen in Discovering New Cruzain Inhibitors. J Med Chem. 2010;53:4891–4905. doi: 10.1021/jm100488w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jensen NH, et al. N-Desalkylquetiapine, a Potent Norepinephrine Reuptake Inhibitor and Partial 5-HT1A Agonist, as a Putative Mediator of Quetiapine's Antidepressant Activity. Neuropsychopharmacology. 2007;33:2303–2312. doi: 10.1038/sj.npp.1301646. [DOI] [PubMed] [Google Scholar]
- 50.Barnea G, et al. The genetic design of signaling cascades to record receptor activation. Proc Natl Acad Sci USA. 2008;105:64–69. doi: 10.1073/pnas.0710487105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.