

Abstract
We recently proposed a bias correction approach to evaluate accurate estimation of the odds ratio (OR) of genetic variants associated with a secondary phenotype, in which the secondary phenotype is associated with the primary disease, based on the original case-control data collected for the purpose of studying the primary disease. As reported in this communication, we further investigated the type I error probabilities and powers of the proposed approach, and compared the results to those obtained from logistic regression analysis (with or without adjustment for the primary disease status). We performed a simulation study based on a frequency-matching case-control study with respect to the secondary phenotype of interest. We examined the empirical distribution of the natural logarithm of the corrected OR obtained from the bias correction approach and found it to be normally distributed under the null hypothesis. On the basis of the simulation study results, we found that the logistic regression approaches that adjust or do not adjust for the primary disease status had low power for detecting secondary phenotype associated variants and highly inflated type I error probabilities, whereas our approach was more powerful for identifying the SNP-secondary phenotype associations and had better-controlled type I error probabilities.
Keywords: Odds ratio, bias, type I error, power, secondary phenotype, frequency-matched study, SNP, genome-wide association study
Introduction
Recently, we [Wang and Shete, 2011] proposed an approach to evaluate the accurate estimation of the odds ratio (OR) of genetic variants associated with a secondary phenotype, in which the secondary phenotype is associated with the primary disease, based on the original case-control data collected for the purpose of studying the primary disease. In that original study, we proposed bias correction approaches, which can provide more accurate OR estimates than those obtained using the standard logistic regression approaches (with or without adjustment for the primary disease status) or the extended inverse-probability-of-sampling-weighted (IPW) regression approach. Especially in the case-control study of primary disease frequency-matched with respect to the secondary phenotype, all the logistic and IPW regressions will be biased, but the proposed bias correction approach can estimate the true OR accurately. However, the original study only focused on the accurate estimation of ORs, it did not investigate the power and type I error rates of the different approaches to detect associated variants. Therefore in this communication, we would like to know whether the proposed approach can achieve a higher power for identifying associated variants than the standard approaches. As reported in this communication, we investigated the type I error probabilities and powers of our bias correction approach using simulation studies and compared the results to those obtained using logistic regression analysis (with or without adjustment for the primary disease status). We examined the empirical distribution of the natural logarithm of the corrected OR obtained using the bias correction approach and found it to be normally distributed under the null hypothesis, similar to the natural logarithms of ORs obtained using logistic regression approaches. Therefore, the p values associated with the corrected ORs can be evaluated assuming a normal distribution under the null hypothesis. The results from our simulation studies showed that the standard approaches had low power for detecting secondary phenotype associated variants and highly inflated type I error probabilities, whereas our bias correction approach is more powerful for identifying the single-nucleotide polymorphism (SNP)-secondary phenotype associations and has better-controlled type I error probabilities.
Methods
In this study, we considered scenarios with three variables: SNP (X), secondary phenotype (T), and primary disease (Y), where the secondary phenotype is always associated with the primary disease. We used directed acyclic graphs (DAGs) to represent the joint distributions of X, T, and Y, as shown in Figure 1. An arrow between two variables represents an association between them. We focused on a scenario in which both SNP and secondary phenotype are associated with primary disease, and we were interested in testing the association between the SNP and the secondary phenotype. Panel (A) in Figure 1 shows a model in which there is no SNP-secondary phenotype association (null hypothesis), and panel (B) shows a model in which there is a SNP-secondary phenotype association (alternative hypothesis). As in the original study, we assumed binary random variables for primary disease and secondary phenotype, denoted as Y = {0, 1} and T = {0, 1} , respectively, with 0 representing individuals without the primary disease/secondary phenotype and 1 representing individuals with the primary disease/secondary phenotype. Denote two alleles at a SNP locus by A and a. Let A be the deleterious allele and a be the normal allele. In this study, we will only investigate the scenario assuming the dominant genetic model, so the SNP variable is denoted as X = {0,1} , with 0 representing genotype(a,a) , and 1 representing genotypes (A,a) and (A,A) . However, our approach is not restricted to a dominant model, and other genetic models such as the additive or recessive model can also be applied (as shown in the original study).
Figure 1.
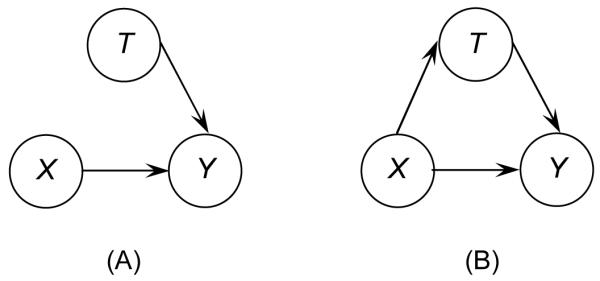
Directed acyclic graphs (DAGs) representing joint distribution of three variables: single-nucleotide polymorphism (X), secondary phenotype (T), and primary disease (Y).
According to the network structures illustrated in Figure 1, we can express the dependency of each random variable using conditional probabilities with logistic models, as below
| (1) |
and
| (2) |
where αi , i = 0, 1 and βj , j = 0,1, and 2 are regression coefficients. Note that the OR of interest representing SNP-secondary phenotype association is a function of the regression coefficient α1 : OR = exp(α1). Therefore, for testing the SNP-secondary phenotype association, the hypothesis of a two-sided test can be expressed as null hypothesis H0: α1 = 0 (Figure 1 (A)) versus alternative hypothesis H1:α1 ≠ 0 (Figure 1 (B)), given that the other regression coefficients, such as β1 and β2 , are free parameters under both the null and alternative hypotheses.
To evaluate the ORs for SNP-secondary phenotype association, we considered different approaches. We used the same notations as those in the original study: (1) was calculated from the logistic regression, without adjustment for the primary disease status; (2) was calculated from the logistic regression, with adjustment for the primary disease status; and (3) was calculated from the bias correction approach as proposed in the original study [Wang and Shete, 2011] for the frequency-matching design with respect to the secondary phenotype of interest. Briefly, our approach provides the accurate OR estimation by solving a system of non-linear equations iteratively, which account for the prevalence values of primary disease and secondary phenotype. Specifically, the biased OR from logistic regression and the prevalence values of primary disease and secondary phenotype can be expressed using non-linear functions of regression coefficients of α0 , β0 and α1 . The knowledge about the prevalence values is assumed to be known from literature. We evaluated the biased OR and the estimated values of other parameters necessary for the non-linear equations from data. The solution of the system of non-linear equations gives us the corrected OR for the SNP-secondary phenotype association.
Simulation Approach
We performed a simulation study to investigate the type I error probabilities and the observed powers of different approaches. For both models in Figure 1, we first generated genotypes of the SNP X with the use of the genotype frequencies, assuming that the SNP is in Hardy-Weinberg proportion. Given the dataset of realizations of SNP X, secondary phenotype T was generated using Equation (1) and then conditioned on the values of X and T, disease outcome Y was generated using Equation (2). We set β1 = 0.365 and β2 = 1.0986 , which correspond to ORs of 1.4 and 3, respectively. For null model, we set α1 = 0 , corresponding to an OR of 1 (null hypothesis of no association between the SNP and the secondary phenotype); for alternate model, we set α1 = 0.365 , corresponding to an OR of 1.4 (alternative hypothesis of association between the SNP and the secondary phenotype). We used the prevalence values 10% for both the primary disease and the secondary phenotype. On the basis of these pre-defined parameters, we obtained the values for the intercept regression coefficients α0 and β0 . For null model, α0 = −2.1972 and β0 = −2.5812 ; for alternate model, α0 = −2.4228 and β0 = −2.5844 .
Using all of the pre-defined parameters, we simulated a large amount of data on the population of interest and then randomly sampled 5,000 cases (individuals with the primary disease). We considered the frequency-matching design based on secondary phenotype in this communication; therefore, the 5,000 controls were sampled so that the proportion of the presence of the secondary trait in the controls was approximately equal to that in the cases [Rothman and Greenland, 1998]. We assumed that the difference between the proportions of the presence of the secondary trait in cases and controls was ±2% with equal probability. In this way, we simulated 100,000,000 replicates of data, each with values for the SNP, the secondary phenotype, and the primary disease. For each replicate, we evaluated the , , and for SNP-secondary phenotype association, and obtained 100,000,000 observations of , , and .
Empirical Distributions
In general, when a logistic regression is performed, a single logistic regression coefficient (i.e., natural logarithm of OR) can be tested for statistical significance by using the Wald test, where the regression coefficient is assumed to follow a normal distribution, with a mean of 0 and a standard error of the coefficient. For the purpose of comparison, we examined the empirical distributions of the random variables corresponding to the natural logarithms of ORs, using the simulated values under the null hypothesis that there is no association between the SNP and the secondary phenotype (Figure 1 (A)). We denoted the random variables of interest as , , and for , , and , respectively. Based on 100,000,000 observations from simulations, which can be considered as random samples drawn from the underlying populations for the random variables, we plotted the histograms for the random variables , , and . We also plotted the corresponding curves of normal distributions under the null hypotheses, with a mean of 0 and the estimated standard errors of , , and , respectively (Figure 2). We observed that the histograms of random variables and were shifted to the left (Figure 2 (A) and (B)) compared to the theoretical normal distribution curves, that is, the corresponding and obtained using logistic regression approaches were underestimated. However, the histogram of random variable (Figure 2 (C)) agreed well with the theoretical normal distribution curve. This observation confirmed the conclusion that bias is present when the OR is estimated using logistic regression approaches and also confirmed that the bias correction approach proposed in the original study provided a very accurate estimate of OR. On the basis of the simulation results, the asymptotic normality of under the null hypothesis was found to be a good fit. Therefore, we evaluated p values associated with , assuming a normal distribution under the null hypothesis.
Figure 2.

Histograms of random variables , , and based on 100,000,000 replicates under the null hypothesis that there is no association between the SNP and the secondary phenotype (Figure 1 (A)). The theoretical curves were plotted following normal distributions, with a mean of 0 and the estimated standard errors of , , and , respectively.
RESULTS
Table 1 lists the summary statistics of the obtained ORs (, , and ) using 100,000,000 replicates, for the null or alternative hypothesis. When both the SNP and the secondary phenotype are associated with the primary disease, the logistic regression approaches provided biased estimates of ORs under both the null and alternative hypotheses. Under the null hypothesis, the median values for and were 0.9579 and 0.9574, respectively. Under the alternative hypothesis, the median values for and were 1.3381 and 1.3403, respectively. Both logistic regression approaches underestimated the true ORs, which was consistent with the results for frequency-matching study design in the original study. On the other hand, the median values of the corrected had good agreement with the ORs used to generate the datasets for both hypotheses (1.0003 versus 1 for the null hypothesis and 1.4001 verses 1.4 for the alternative hypothesis, respectively).
Table 1.
Summary statistics of odds ratios obtained using different approaches
| Null hypothesis (OR = 1) |
Alternative hypothesis (OR = 1.4) |
|||||
|---|---|---|---|---|---|---|
| Median | 0.9579 | 0.9574 | 1.0003 | 1.3381 | 1.3403 | 1.4001 |
| Q1-Q3 * | 0.9253-0.9921 | 0.9252-0.9911 | 0.9658-1.0364 | 1.2907-1.3879 | 1.2932-1.3897 | 1.3499-1.4529 |
Q1: 1st quartile, Q3: 3rd quartile
Type I Error Probabilities
We investigated the type I error probabilities of the logistic regression approaches (with and without adjustment for the primary disease status) and our bias correction approach. Table 2 reports the observed type I error probabilities of the different approaches, at different pre-specified significance levels (from 0.05 to 10−5), based on 100,000,000 replicates under the null hypothesis that there is not an association between the SNP and the secondary phenotype. It is not surprising to see that the type I error rates of both logistic regression approaches were inflated dramatically, because all the estimated and values were biased as illustrated in Figure 2 (A) and (B). For example, at a nominal significance level of 0.01, the type I error rates were 0.0456 and 0.0443 for the logistic regression approaches without and with adjustment, respectively, which are about 4.5 times the nominal significance level. In contrast, we observed that the proposed bias correction approach can control the type I error probability much better than the biased logistic regression approaches. For example, at a nominal significance level of 0.01, the type I error rate for the bias correction approach was 0.0121, which closely agrees with the nominal significance level.
Table 2.
Estimated type I error probabilities at different significance levels in simulation studies based on 100,000,000 replicates under the null hypothesis, each replicate with 5,000 cases and 5,000 frequency-matched controls with respect to the secondary phenotype.
| Type I error probabilities |
|||
|---|---|---|---|
| Significance | Logistic (without adjustment) |
Logistic (with adjustment) |
Our approach |
| 5.00E-02 | 1.39E-01 | 1.36E-01 | 5.65E-02 |
| 1.00E-02 | 4.56E-02 | 4.43E-02 | 1.21E-02 |
| 5.00E-03 | 2.80E-02 | 2.63E-02 | 6.09E-03 |
| 1.00E-03 | 8.38E-03 | 7.59E-03 | 1.26E-03 |
| 5.00E-04 | 4.97E-03 | 4.49E-03 | 7.42E-04 |
| 1.00E-04 | 1.38E-03 | 1.24E-03 | 1.72E-04 |
| 5.00E-05 | 8.77E-04 | 6.77E-04 | 1.01E-04 |
| 1.00E-05 | 2.52E-04 | 2.09E-04 | 1.03E-05 |
Power Comparison
We also compared the observed power of the bias correction approach to those of the logistic regression approaches for evaluating the OR of the SNP-secondary phenotype association (Table 3). Based on our simulation of 100,000,000 replicates under the alternative hypothesis that the OR of the SNP associated with the secondary phenotype is 1.4, we reported the observed powers at different nominal significance levels (from 10−4 to 10−8), including 5×10−8, which is the commonly used genome-wide association significance threshold [Altshuler et al., 2008]. For all the significance levels tested, the bias correction approach was more powerful for identifying the SNP-secondary phenotype association than both logistic regression approaches. As in the type I error probabilities estimation, the observed powers of the two logistic regression approaches were very similar owing to the frequency-matching design. For example, at a nominal significance level of 10−6, the observed powers for declaring the significance of the SNP-secondary phenotype association were 73.04% and 73.48% for the logistic regression approaches without and with adjustment for the primary disease status, respectively. When the bias correction approach was used, the observed power was 93.23% at a 10−6 significance level. When the nominal significance level was 10−8, the observed powers were only about 40% for both logistic regression approaches, while the bias correction approach still had 73.42% power to identify the SNP-secondary phenotype association.
Table 3.
Observed powers at different significance levels in simulation studies based on 100,000,000 replicates under the alternative hypothesis, each replicate with 5,000 cases and 5,000 frequency-matched controls with respect to the secondary phenotype.
| Observed powers |
|||
|---|---|---|---|
| Significance | Logistic (without adjustment) |
Logistic (with adjustment) |
Our approach |
| 1.00E-04 | 95.12% | 95.19% | 99.41% |
| 5.00E-05 | 93.12% | 93.24% | 99.03% |
| 1.00E-05 | 86.58% | 86.77% | 97.60% |
| 5.00E-06 | 83.10% | 83.20% | 96.58% |
| 1.00E-06 | 73.04% | 73.48% | 93.23% |
| 5.00E-07 | 68.31% | 68.77% | 91.23% |
| 1.00E-07 | 56.50% | 57.03% | 85.19% |
| 5.00E-08 | 51.38% | 51.91% | 82.08% |
| 1.00E-08 | 39.88% | 40.46% | 73.42% |
DISCUSSION
In this communication, we report the results of our investigation of the type I error rates and observed powers of the bias correction approach for estimating the OR of association between the SNP and the secondary phenotype proposed in the original study [Wang and Shete, 2011]. We focused on the case-control study design frequency-matched on the secondary phenotype. We performed a simulation study to compare the observed type I errors and powers obtained using the bias correction approach with those obtained using logistic regression approaches with and without adjustment for the primary disease. Our results show that the standard approaches have low power for identifying the SNP-secondary phenotype association and highly inflated type I error rates; the bias correction approach is a more powerful approach for identifying this association and has a better-controlled type I error probability. Furthermore, the misspecification of primary disease and secondary phenotype prevalence does not have any major impact on the power and type 1 error of the bias correction approach.
In this study, we focused on a scenario in which both the SNP and the secondary phenotype are associated with the primary disease. Other scenarios, with different associations among the primary disease, the secondary phenotype, and the SNP, have been studied by [Monsees et al., 2009]. In general, when there is no association between the secondary phenotype and the primary disease, there is no bias arising for measures of marker-secondary trait association under both null and alternative hypotheses. When the secondary phenotype is associated with the primary disease, but the marker is not associated with the primary disease, there are very interesting observations. Monsees et al. [2009] showed that, in this scenario, there is no bias for estimating measures of marker-secondary trait association under the null hypothesis, but there is a bias under the alternative hypothesis. They showed a biased estimate of the regression coefficient of the marker with a continuous secondary trait by using simulation studies. Interestingly, in this situation, Kraft [2007] derived a simple proof about the bias in estimating ORs relating a binary secondary phenotype and a binary marker. He showed that if the marker is not associated with the primary disease conditional on the secondary phenotype, the naïve estimate using logistic regression is not biased. We also investigated this particular scenario where only the secondary phenotype is associated with the primary disease and that the study is frequency-matched case-control study with respect to the secondary phenotype. We found that under both null and alternative hypotheses, the bias correction approach, as well as the logistic regression approaches, can provide unbiased estimates of the OR of association between the SNP and the secondary phenotype. All the observed type I error probabilities were well controlled for all the different approaches. Also, all the approaches had very similar powers to identify the SNP-secondary phenotype associations. Therefore, we can conclude that the bias correction approach is also robust in this scenario. A proof that shows the bias in OR estimation for the frequency-matching study with respect to the secondary phenotype is given in the Supplementary Materials.
In summary, we further confirm that the bias correction approach proposed in our original study can provide very accurate OR estimates of SNP-secondary phenotype association. Moreover, we conclude that the proposed bias correction approach has more power to detect the SNP-secondary phenotype association and better control of the type I error.
Supplementary Material
ACKNOWLEDGMENTS
This research is supported by the National Institutes of Health grant 1R01CA131324.
References
- Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science. 2008;322:881–8. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft P. Analyses of genome-wide association scans for additional outcomes. Epidemiology. 2007;18:838. doi: 10.1097/EDE.0b013e318154c7e2. [DOI] [PubMed] [Google Scholar]
- Monsees GM, Tamimi RM, Kraft P. Genome-wide association scans for secondary traits using case-control samples. Genet Epidemiol. 2009;33:717–28. doi: 10.1002/gepi.20424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman KJ, Greenland S. Modern epidemiology. Lippincott Williams & Wilkins; Philadelphia, PA: 1998. [Google Scholar]
- Wang J, Shete S. Estimation of odds ratios of genetic variants for the secondary phenotypes associated with primary diseases. Genet Epidemiol. 2011;35:190–200. doi: 10.1002/gepi.20568. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.