

A 12-year-old boy currently is followed by multiple sub-specialists for problems caused by the chromosome 22q11.2 deletion syndrome (22q11DS) (Figure). He was born via spontaneous vaginal delivery, weighing 3033 g, to a 31-year-old G3P3 mother after a full-term pregnancy complicated only by mild polyhydramnios. Family history was non-contributory. Apgar scores were 8 at 1 minute and 9 at 5 minutes. With the exception of a weak cry, the results of the infant’s initial examination were unremarkable, and he was moved to the well-baby nursery. Shortly thereafter, a cardiac murmur was noted, the cardiology department was consulted, and the child was transferred to a local tertiary care facility with a diagnosis of tetralogy of Fallot. Stable, he was discharged home at 3 days of life.
Figure.
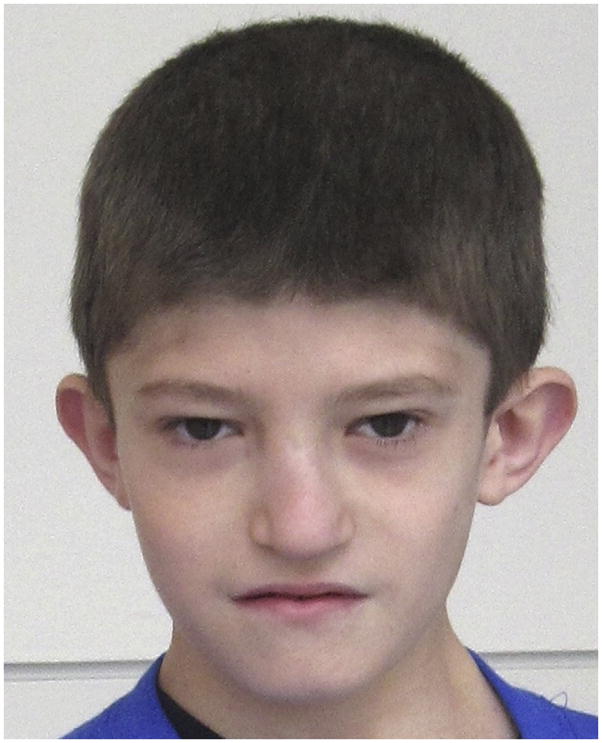
Mild dysmorphic facial features of a boy aged 11 years with 22q11.2DS, including a short forehead, hooded eyelids with upslanting palpebral fissures, malar flatness, bulbous nasal tip with hypoplastic alae nasi, and protuberant ears.
At 5 days of life, he had jerky movements. On presentation to the local emergency department, his total calcium level was 4.7 mg/dL, and later partial hypoparathyroidism was diagnosed. At that time, a consulting geneticist suggested the diagnosis of chromosome 22q11DS. Weeks later, the family received a telephone call confirming the diagnosis with fluorescence in situ hybridization (FISH). No additional information about the diagnosis, prognosis, etiology, or recurrence risk was provided until the child was 5 months of age, when he underwent cardiac repair at a third hospital, where a comprehensive 22q11DS program was in operation. In the interim, the child had feeding difficulties requiring supplemental nasogastric tube feeds, nasal regurgitation, and gastroesophageal reflux, while the parents searched the internet for reliable information about their son’s diagnosis.
Subsequent notable abnormalities and interventions included: recurrent otitis media with bilateral myringotomy tube placement at 6 months; angioplasty with left pulmonary artery stent placement after the identification of pulmonary artery stenosis with bilateral pleural effusions at age 6 years; chronic upper respiratory infections with significant T cell dysfunction requiring live viral vaccines to be held until age 7 years; velopharyngeal incompetence necessitating posterior pharyngeal flap surgery at 7 years; enamel hypoplasia and numerous caries resulting in 3 separate dental procedures under general cardiac anesthesia beginning at age 7 years; multiple cervical and thoracic vertebral anomalies with thoracic levoconvex scoliosis and upper lumbar dextroscoliosis requiring growing rod placement at age 11 years with subsequent rod extension at ages 11.5 and 12 years; postoperative hypocalcemia; short stature; constipation; and persistent idiopathic thrombocytopenia. Pertinent negative test results included normal renal ultrasound scanning and parental 22q11.2 deletion studies.
On physical examination, the boy’s height and weight have consistently tracked just below the fifth percentile, with no evidence of growth hormone deficiency. His head circumference is within reference range at the 25th percentile. Dysmorphic features include: a low anterior hairline; hooded eyelids; malar flatness; normally formed but protuberant ears with attached lobes; a mildly deviated nose with a bulbous nasal tip and hypoplastic alae nasi; asymmetric crying facies with a thin upper lip; mild micrognathia; a sacral dimple; and soft tissue syndactyly of the second and third toes.
Developmentally, the boy had mild delays in achieving motor milestones, sitting at 11 months and walking at 18 months. However, he exhibited significant delays in the emergence of language: he never babbled, spoke his first words at age 3 years, and only achieved full conversational speech at 7 years. However, he had relative strengths in receptive language and communicated appropriately by the use of sign language. Now quite conversant, he is mainstreamed in the seventh grade with resource room supports. Moreover, he is affable, but exhibits anxiety and perseverations. Lastly, despite numerous medical, academic, and social challenges, he participates in assisted athletics, is an avid wrestling fan, and enjoys travel. However, his exceptionally supportive parents, siblings, and extended family continue to worry about his long-term outcome and transition of care as he approaches adulthood.
As demonstrated by this boy’s complicated course, practical multi-system guidelines are needed to assist the general practitioner and specialists in caring for patients with 22q11DS. Although still under-recognized, detection, including in the prenatal setting, is increasing. Moreover, the phenotypic spectrum is highly variable, and patients may present at any age. Thus, initial guidelines developed by an international panel of experts present the best practice recommendations currently available across the lifespan, with a major focus on the changing issues through childhood development.
Background
Although clinically under-recognized, 22q11DS is the most common microdeletion syndrome (MIM #188400/#192430), with an estimated prevalence of 1 in 4000 live births.1–3 However, the actual occurrence may be higher because of variable expressivity.4 In comparison, Down syndrome is seen in 1 in 1200 newborns.5 The 22q11.2 deletion is the second most common cause of developmental delay and major congenital heart disease after Down syndrome, accounting for approximately 2.4% of individuals with developmental disabilities6 and approximately 10% to 15% of patients with tetralogy of Fallot.7,8 22q11.2 deletions have been identified in most patients with DiGeorge syndrome, velocardiofacial syndrome, and conotruncal anomaly face syndrome9–14 and in a subset with autosomal dominant Opitz G/BBB syndrome and Cayler cardiofacial syndrome.15,16 Although this list of associated disorders may appear quite perplexing, it is understandable because the diagnoses were originally described by clinicians concentrating on their particular areas of interest. After the widespread use of FISH, however, patients with a deletion became collectively referred to by their chromosomal etiology: the 22q11.2DS.
Clinical features prompting a clinician to perform 22q11.2 deletion studies may vary depending on the age of the patient. However, they commonly include two or more of these classic findings: developmental disabilities, learning disabilities, or both17–19; conotruncal cardiac anomalies, palatal defects, nasal regurgitation, and/or hypernasal speech; behavioral problems, psychiatric illness, or both20,21; immunodeficiency22; hypocalcemia; and characteristic facial features (Figure).23–26 However, because of the significant variability of expression, especially in the absence of classic findings, the diagnosis may be missed.34,35 This variable expression also means that 22q11.2 deletions may be detected in patients in whom other clinical syndromes were previously diagnosed, such as Goldenhar for example.4 Identification of 22q11DS, especially in adolescents and adults, often requires an enhanced index of suspicion.23–25 Male and female children are equally affected.26
The hemizygous 22q11.2 deletion (ie, on only one of the chromosome pair) is almost always too small to be identified with cytogenetic studies using standard chromosome banding techniques alone. Since 1992, FISH studies, with probes such as N25 or TUPLE1 within the most commonly deleted region, have allowed clinical laboratories to identify patients with submicroscopic 22q11.2 deletions. Most patients (approximately 85%) have a large (approximately 3 Mb) deletion, encompassing approximately 45 functional genes, whereas the remaining patients have smaller atypical or “nested” deletions, usually within the 3 Mb deletion region.27,28 FISH is limited to one single target sequence within the proximal 22q11.2 deletion region. Some “atypical” deletions do not include the region containing FISH probes generally used for clinical testing,29 thus patients studied only with these methods would remain undetected. More sophisticated techniques that can detect 22q11.2 deletions of any size, such as array comparative genomic hybridization (aCGH), genome-wide microarrays and multiplex ligation-dependent probe amplification (MLPA), will eventually replace FISH studies in most laboratories.30
The occurrence of 22q11.2 deletions is related to the genomic architecture of the chromosome 22q11.2 region. Low copy repeat (LCR) sequences with high homology to each other make this region especially susceptible to rearrangements because of unequal meiotic crossovers and thus aberrant interchromosomal exchanges (non-allelic homologous recombination).31 These LCR sequences flank the common 22q11.2 deletions and define the common breakpoints. Breakpoints that are not flanked by LCRs, however, may involve other repeat elements and mechanisms that are yet to be defined.32,33
Most 22q11.2 deletions (>90%) are found to have arisen as de novo (spontaneous) events, with both parents unaffected.4,28 However, in as many as 10% of individuals, a 22q11.2 deletion is identified in a parent, approximately equally in mothers and fathers.28,34 Therefore, on the basis of the significant variability of expression and somatic mosaicism (the deletion is present only in a subset of tissues; eg, lymphocytes),39 parental testing is recommended for all, with appropriate follow-up and genetic counseling when a deletion is identified.4,35
Considering mortality, unlike the early reports of patients with DiGeorge syndrome, with improved palliative cardiac repair and medical management of immunodeficiency, infant mortality in 22q11DS is now relatively low (approximately 4%).34 However, compared with population-based expectations, the overall mortality rate is elevated, especially in adults.42
22q11DS is quintessentially a multi-system syndrome with a remarkable variability in the severity and extent of expression in individuals,35 even in affected members of the same family.34 Moreover, the presence of one feature does not predict the presence of any other feature. Also, to date there are no convincing data indicating major differences in clinical expression related to the variable size and extent of the 22q11.2 deletions.4,28 Thus, although there are some recommendations that are relevant for all patients, treatment must be targeted to best suit the individual, their age or developmental stage, and their particular constellation of associated features, severity, and need for treatment. For example, as seen in our illustrative case report, in infancy and preschool classic features such as any combination of feeding problems, infection, hypocalcemia, and structural cardiac and palatal anomalies may be accompanied by speech, learning, and/or developmental difficulties. When the child is of school age, parental concerns often shift to a focus on finding appropriate educational support, helping foster peer relationships, and coping with a variety of medical issues such as non-specific but activity-limiting leg pains, scoliosis, autoimmune diseases, and short stature, at times caused by growth hormone deficiency. Recurrent infections may affect school attendance; secondary cardiac procedures may be needed as the child grows. Adolescents and young adults may have new onset or recurrence of seizures, treatable psychiatric illness, or both. In adulthood, a noteworthy proportion of individuals find employment and normal social relationships difficult to establish or sustain. Moreover, throughout the lifespan, new syndrome-related local and systemic conditions may present, which can be especially stressful when the underlying link to 22q11DS is not recognized.36
Clearly, diagnosis at any age significantly changes genetic counseling and patient treatment.4,25 Early diagnosis provides the best opportunity for affecting the course of illness and optimizing outcomes. Anticipatory care includes screening for and coordinated management of associated conditions.4,24,25 Available evidence indicates that standard treatments are effective for related problems, from congenital cardiac anomalies to thyroid disease to psychiatric illness.37 All management strategies should be pursued, however, in the context of the multi-system nature of 22q11DS. Specialty clinics, or so-called “clinical centers of excellence,” can, as seen in this case, provide support for both the parents and treating clinicians while facilitating access to peer-support networks.23–25 Such clinics also can provide careful monitoring of the possibilities and challenges faced by the patient, allowing for timely interventions as needed. Because of the complexity of 22q11DS in many cases, when geographically and economically feasible, we recommend that all affected individuals be evaluated periodically at a comprehensive care center. However, the availability of 22q11DS specialty clinics is limited. Thus, these guidelines are designed to assist the primary care physician in caring for the patient with a 22q11.2 deletion.
Methods
The guidelines were developed in two steps. First, there were two international 22q11DS consensus meetings, in Marseilles, France, in 2006 and in Utrecht, the Netherlands, in 2008, at which clinicians and researchers with broad expertise (18 subspecialties representing >15 countries) met in focus groups to discuss best practice recommendations on the basis of experiences and data.
Second, a systematic literature review of 239 clinically relevant publications was performed in an effort to support consensus recommendations with scientific evidence when possible,38 recognizing there is a relatively limited literature for this complex condition, particularly for management issues. Consequently, at this relatively early stage in our knowledge, virtually all the evidence for 22q11DS would be levels III or IV (descriptive studies, expert opinions, or both).43 Thus we have not formally graded the individual recommendations presented.
A draft consensus document stemming from these two steps was refined at the international 22q11DS meeting in Coventry, England, in 2010 with a goal of transcending nationalities, health care system differences, and subspecialty biases. Like all clinical practice guidelines, these initial recommendations will change with time as more data become available. Furthermore, it is likely that some or many clinicians will be unable to perform all the studies or evaluations because of costs, varying patterns of practice, and other reasons. As for most guidelines, there are no data available on cost effectiveness of anticipatory care (eg, identifying and treating hypocalcemia to prevent seizures) in 22q11DS. However, these guidelines, while tending to err on the side of being overly complete, strive to embrace what is collectively considered current best practice, allowing the caregiver to be aware of potential associations that may be clinically significant.
Guidelines Summarized
Table I presents the multi-system features, including both those that are common and those that are rarer but may be significant for diagnosis, follow-up, or both.25,35 Table I also provides an overview of management and the specialties commonly involved. Table II is organized with recommendations for the “at diagnosis” stage and later developmental stages. Table III presents important cautions and considerations that may be encountered by any clinician involved in the patient’s care. These are overarching general principles. Practical recommendations for an international audience were prioritized.
Table I.
Multisystem features of 22q11.2 deletion syndrome
| Common features* | Relevant age groups
|
Selected rarer features† | Management
|
Specialties commonly involved (in addition to family medicine, pediatrics, general internal medicine, radiology) |
|||
|---|---|---|---|---|---|---|---|
| Prenatal | Infant to child |
Teen to adult |
Standard‡ | Special considerations or attention | |||
General genetics
|
✓ | ✓ | ✓ |
|
✓ |
|
|
Cardiovascular (conotruncal/other)
|
✓ | ✓ | ✓ |
|
✓ |
|
|
Palatal and related (75%)
|
✓ | ✓ | ✓ | ✓ |
|
|
|
Immune-related¶
|
✓ | ✓ |
|
✓ |
|
|
|
Endocrine
|
✓ | ✓ |
|
✓ |
|
|
|
Gastroenterological
|
✓ | ✓ | ✓ |
|
✓ |
|
|
Genitourinary
|
✓ | ✓ | ✓ |
|
✓ |
|
|
Ophthalmology
|
✓ |
|
✓ |
|
|
||
Skeletal
|
✓ | ✓ | ✓ |
|
✓ |
|
|
Hematology/Oncology
|
✓ | ✓ |
|
✓ |
|
||
Neurologic
|
✓ | ✓ |
|
✓ |
|
|
|
Growth and development
|
✓ | ✓ | ✓ | ✓ |
|
|
|
Neuropsychiatric disorders
|
✓ | ✓ | ✓ |
|
|
||
Other
|
✓ | ✓ | ✓ |
|
|||
Rates are estimates only of lifetime prevalence of features for 22q11DS and will vary depending on how cases are ascertained and age of the patient. Features included have prevalence >1% in 22q11DS and significantly higher than general population estimates.
A selected (and to some extent arbitrary) set of rarer features of note in 22q11DS, emphasizing patients needing active treatment.
Standard surveillance, investigations, and management according to involved condition(s).
Characteristic facial features include long narrow face, malar flatness, hooded eyelids, tubular nose with bulbous tip, hypoplastic alae nasae, nasal dimple or crease, small mouth, small protuberant ears with thick overfolded/crumpled helices, and asymmetric crying facies.
Infants only: minimize infectious exposures; initially withhold live vaccines; cytomegalovirus-negative irradiated blood products; influenza vaccinations; respiratory syncytial virus prophylaxis.
All patients should have vitamin D supplementation; patients with documented hypocalcemia, relative or absolute hypoparathyroidism, or both may have to have prescribed hormonal forms (eg, calcitriol) supervised by endocrinologist.
May be important for diagnostic purposes.
Table II.
Recommended assessments for 22q11.2 deletion syndrome*
| Assessment | At diagnosis |
Infancy (0–12 months) |
Preschool age (1–5 years) |
School age (6–11 years) |
Adolescence (12–18 years) |
Adulthood (>18 years) |
|---|---|---|---|---|---|---|
| Ionized calcium, parathyroid hormone† | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Thyrotropin (thyroid-stimulating hormone)† | ✓ | ✓ | ✓ | ✓ | ✓ | |
| Complete blood cell count and differential (annual) | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Immunologic evaluation‡ | ✓ | ✓§ | ✓§ | |||
| Ophthalmology | ✓ | ✓ | ||||
| Evaluate palate¶ | ✓ | ✓ | ✓ | |||
| Audiology | ✓ | ✓ | ✓ | ✓ | ||
| Cervical spine (>age 4 years) | ✓|| | |||||
| Scoliosis examination | ✓ | ✓ | ✓ | |||
| Dental evaluation | ✓ | ✓ | ✓ | ✓ | ||
| Renal ultrasound | ✓ | |||||
| Electrocardiogram | ✓ | ✓ | ||||
| Echocardiogram | ✓ | |||||
| Development** | ✓ | ✓ | ✓ | |||
| School performance | ✓ | ✓ | ||||
| Socialization/functioning | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Psychiatric/emotional/behavioral†† | ✓ | ✓ | ✓ | ✓ | ✓ | |
| Systems review | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Deletion studies of parents | ✓ | |||||
| Genetic counseling‡‡ | ✓ | ✓ | ✓ | |||
| Gynecologic and contraceptive services | ✓ | ✓ |
These recommendations are proposed as at year end 2010. Each ✓ refers to a single assessment except as stated above and below. We have tended to err on the side of overinclusiveness. Local patterns of practice may vary.
In infancy, test calcium levels every 3 to 6 months, then every 5 years through childhood, and every 1 to 2 years thereafter; thyroid studies annually. Check calcium preoperatively and postoperatively and regularly in pregnancy.
In addition to complete blood cell count with differential, in newborns: flow cytometry; and at age 9 to 12 months (before live vaccines): flow cytometry, immunoglobulins, T-cell function. Expert opinion is divided about the extent of needed immune work-up in the absence of clinical features.
Evaluate immune function before administering live vaccines (see ‡).
In infancy, visualize palate and evaluate for feeding problems, nasal regurgitation, or both; in toddlers to adults, evaluate nasal speech quality.
Cervical spine films to detect anomalies: anterior/posterior, lateral, extension, open mouth, skull base views. Expert opinion is divided about the advisability of routine radiography. Symptoms of cord compression are an indication for urgent neurological referral.
Motor and speech/language delays are common; rapid referral to early intervention for any delays can help to optimize outcomes.
Vigilance for changes in behavior, emotional state, and thinking, including hallucinations and delusions; in teens and adults, assessment would include at-risk behaviors (sexual activity, alcohol/drug use, etc).
See text for details.
Table III.
Important cautions and considerations for patients with 22q11DS
| Feature | Management suggestions |
|---|---|
| Aspiration pneumonia | Suctioning and chest physiotherapy may be necessary as preventions; small food portions may help; tube feeding frequently necessary |
| Autonomic dysfunction | Careful monitoring perioperatively and postoperatively and at times of major biological stress (eg, infections, major medical crises); provision of necessary support |
| Surgical complications of all types at a somewhat elevated likelihood compared to other patients (bleeding, atelectasis, seizures, difficult intubation) | Careful monitoring perioperatively and postoperatively, including ionized calcium, oxygen levels; availability of small intubation equipment |
| Narrow lumens (eg, airway, spinal canal, ear canals) | May need smaller sized intubation equipment |
| Often need regular ear syringing to maximize hearing | |
| Aberrant anatomy (anywhere) | Preparatory investigations and consideration before surgery |
| Aberrant vascular anatomy | Consider magnetic resonance angiography before pharyngoplasty |
| Adenoidectomy may worsen velopharyngeal insufficiency | Consider risk/benefit |
| Posterior pharyngeal flap intervention may cause sleep apnea | Consider risk/benefit |
| Hypocalcemia risk elevated at times of biological stress (eg, surgery, infection, burn, peripartum) | Monitoring of ionized calcium levels and consideration of increased dose of vitamin D, calcium treatment, or both |
| Hypocalcemia worsening factors (eg, alcohol, fizzy drinks, pancreatitis) | Minimize alcohol and pop intake; extra caution with pancreatitis; monitor calcium levels more closely |
| Hypocalcemia treatments may cause nephrocalcinosis | Carefully monitor therapy |
| Seizure diathesis | Consider myoclonic, absence or generalized seizures with apparent clumsiness/tripping, poor concentration or falls, respectively; investigate low calcium and magnesium levels and ensure adequate treatment; consider anticonvulsant medications as adjunctive medications for other medications that often lower the seizure threshold (eg. clozapine, other antipsychotic medications) |
| Sensitivity to caffeine | Reduce caffeine intake, especially cola, “energy” drinks, and coffee; consider as a contributory factor to anxiety and/or agitation and/or tremor |
| Developmental delays common in all aspects of development, structural and functional | Anticipating a slower trajectory and changing capabilities over time, with necessary supports provided, can help reduce frustrations and maximize function; a good match between the expectations and demands of the environment and the social and cognitive capabilities of the individual will minimize the risk of chronic stress and of exploitation |
| Increased need for sleep | Regular, early bedtime and more hours of sleep than other same-aged individuals can help reduce irritability and improve learning and functioning |
| Increased need for structure, routine, certainty, sameness | Environmental adjustments to improve stability and limit changes can help reduce anxiety and frustration |
| Constipation | Consider with verbal and especially non-verbal patients as a cause of agitation, pain, or both; routine measures, including hydration, exercise, fiber, bowel routine |
| Tendency to form cysts of all types | Routine |
| Pregnancy complications | Consider as a biological stressor for the individual in the context of their associated features and risks (eg, hypocalcemia, adult congenital heart disease, psychiatric diseases, seizure diatheses, and social situation) |
Genetic Counseling
Genetic counseling for 22q11DS includes a discussion on prevalence, etiology, detection, variability, interventions, and prenatal/preconception options.4 Some affected adults, especially those detected through their more severely affected child, have been found to have the deletion despite minimal clinical findings.34 Moreover, somatic mosaicism has been reported.39 Thus, despite the absence of obvious clinical features, parental studies are always warranted to provide appropriate recurrence risk counseling. Likewise, the occurrence of germ line mosaicism results in a small recurrence risk for parents of children with de novo deletions.40,41 Rarely, patients have both a 22q11.2 deletion and another confounding diagnosis, such as a familial single gene disorder or other sporadic cytogenetic abnormality. This can complicate the assessment of features attributable to the 22q11.2 deletion and genetic counseling. Even more rarely the deletion occurs as a result of an unbalanced chromosome translocation,10 and this too influences the recurrence risk counseling. Thus, the provider must exclude the possibility of a rearrangement before providing counseling.
Affected individuals, regardless of sex and similar to patients with an autosomal dominant condition, have a 50% chance of having an affected child in each pregnancy. In light of the variability of the syndrome, however, it is impossible to predict the range and severity of manifestations in the offspring. Prenatal diagnostic options for such patients include: ultrasound scanning and fetal echocardiography, which is non-invasive but only detect some of the congenital anomalies associated with 22q11DS, prenatal deletion testing, such as via chorionic villus sampling or amniocentesis, which is highly accurate, or both. Preconception options include donor gametes with or without confirmatory prenatal testing or preimplantation genetic diagnosis utilizing in vitro fertilization.4
Counseling includes up-to-date information on the associated conditions commonly found, conditions likely to develop at the different developmental stages in 22q11DS, or both (Table I). In addition, information about management strategies, local resources, and supports should be provided to the patients, their families, and their clinicians.
Ideally, genetic counseling would be repeated at each life stage, with updated information about 22q11DS provided and questions answered. This is particularly important during transition to adolescence and adulthood when reproductive issues and treatable late onset conditions, such as psychiatric illness, are prominent features.25
Conclusion
In summary, these guidelines present the best practice recommendations currently available across the lifespan of a patient, with a major focus on the changing issues through childhood development. These guidelines will require updating as new information becomes available.
Glossary
- 22q11DS
22q11.2 deletion syndrome
- aCGH
Array comparative genomic hybridization
- FISH
Fluorescence in situ hybridization
- LCR
Low copy repeat
- MLPA
Multiplex ligation-dependent probe amplification
Appendix
Members of The International 22q11.2 Deletion Syndrome Consortium include: Véronique Abadie, Jeremy Allgrove, Francesca Amati, Kate Baker, Adriane Baylis, Marie-Paule Beaujard, Frits Beemer, Maria Boers, Patrick Bolton, Erik Boot, Sophie Brigstocke, Stephane Burtey, Linda Campbell, Melanie Chabloz, Eva Chow, Jill Clayton-Smith, Joseph Cubells, Martin Debbané, Marie-Ange Delrue, Bert De Smedt, Sasja Duijff, Peggy Eicher, Beverly Emanuel, Laurens Evers, Astrid Flahault, Alex Forsythe, Thierry Frebourg, Andy Gennery, Elizabeth Goldmuntz, Anne Gosling, Steven Handler, Damian Heine-Suñer, Aaron Hilmarsson, Annique Hogan, Roel Hordijk, Sarah Howley, Elizabeth Illingworth, Oksana Jackson, Hillary Joyce, Hiroshi Kawame, Robert Kelly, Alexandra Kemp, Lucas Kempf, J.L.L. Kimpen, Richard Kirschner, Petra Klaassen, Dinakantha Kumararatne, Michelle Lambert, Kari Lima, Elizabeth Lindsay, Silvia Macerola, Merav Burg Malki, Sandrine Marlin, Maria Mascarenhas, Stephen Monks, Veronica Moran, Bernice Morrow, Ed Moss, Clodagh Murphy, Nitha Naqvi, Bent Windelborg Nielsen, Lena Niklasson, Hilde Nordgarden, C.E. Oenema-Mostert, Marie-Christine Ottet, Catherine Pasca, Patrick Pasquariello, Christina Persson, Marie-France Portnoi, Sarah Prasad, Kimberly Rockers, Sulagna Saitta, Peter Scambler, Marie Schaer, Maude Schneider, Debbie Sell, Cindy Solot, Brian Sommerlad, Nancy Unanue, Frederick Sundram, Katrijn Van Aken, Therese van Amelsvoort, Aebele Mink van der Molen, Josine Widdershoven, and Elaine H. Zackai.
Footnotes
The authors declare no conflicts of interest.
References
- 1.Goodship J, Cross I, LiLing J, Wren C. A population study of chromosome 22q11 deletions in infancy. Arch Dis Child. 1998;79:348–51. doi: 10.1136/adc.79.4.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tezenas Du Montcel ST, Mendizabal H, Ayme S, Levy A, Philip N. Prevalence of 22q11 microdeletion. J Med Genet. 1996;33:719. doi: 10.1136/jmg.33.8.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oskarsdottir S, Vujic M, Fasth A. Incidence and prevalence of the 22q11 deletion syndrome: a population-based study in Western Sweden. Arch Dis Child. 2004;89:148–51. doi: 10.1136/adc.2003.026880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McDonald-McGinn DM, Zackai EH. Genetic counseling for the 22q11.2 deletion. Dev Disabil Res Rev. 2008;14:69–74. doi: 10.1002/ddrr.10. [DOI] [PubMed] [Google Scholar]
- 5.Cocchi G, Gualdi S, Bower C, Halliday J, Jonsson B, Myrelid A, et al. International trends of Down syndrome 1993–2004: births in relation to maternal age and terminations of pregnancies. Birth Defects Res A Clin Mol Teratol. 2010;88:474–9. doi: 10.1002/bdra.20666. [DOI] [PubMed] [Google Scholar]
- 6.Rauch A, Hoyer J, Guth S, Zweier C, Kraus C, Becker C, et al. Diagnostic yield of various genetic approaches in patients with unexplained developmental delay or mental retardation. Am J Med Genet A. 2006;140A:2063–74. doi: 10.1002/ajmg.a.31416. [DOI] [PubMed] [Google Scholar]
- 7.Goldmuntz E, Driscoll DA, Budarf ML, Zackai EH, McDonald-McGinn DM, Biegel JA, et al. Microdeletions of chromosomal region 22q11 in patients with congenital conotruncal cardiac defects. J Med Genet. 1993;30:807–12. doi: 10.1136/jmg.30.10.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carotti A, Digilio MC, Piacentini G, Saffirio C, Di Donato RM, Marino B. Cardiac defects and results of cardiac surgery in 22q11.2 deletion syndrome. Dev Disabil Res Rev. 2008;14:35–42. doi: 10.1002/ddrr.6. [DOI] [PubMed] [Google Scholar]
- 9.de la Chapelle A, Herva R, Koivisto M, Aula P. A deletion in chromosome 22 can cause DiGeorge Syndrome. Hum Genet. 1981;57:253–6. doi: 10.1007/BF00278938. [DOI] [PubMed] [Google Scholar]
- 10.Kelley RI, Zackai EH, Emanuel BS, Kistenmacher M, Greenberg F, Punnett HH. The association of the DiGeorge anomalad with partial monosomy of chromosome 22. J Pediatrics. 1982;101:197–200. doi: 10.1016/s0022-3476(82)80116-9. [DOI] [PubMed] [Google Scholar]
- 11.Scambler PJ, Carey AH, Wyse RKH, Roach S, Dumanski JP, Nordenskjold M, et al. Microdeletions within 22q11 associated with sporadic and familial DiGeorge syndrome. Genomics. 1991;10:201–6. doi: 10.1016/0888-7543(91)90501-5. [DOI] [PubMed] [Google Scholar]
- 12.Driscoll DA, Salvin J, Sellinger B, Budarf ML, McDonald-McGinn DM, Zackai EH, et al. Prevalence of 22q11 microdeletions in DiGeorge and velocardiofacial syndromes: implications for genetic counselling and prenatal diagnosis. J Med Genet. 1993;30:813–7. doi: 10.1136/jmg.30.10.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burn J, Takao A, Wilson D, Cross I, Momma K, Wadey R, et al. Conotruncal anomaly face syndrome is associated with a deletion within chromosome 22q11. J Med Genet. 1993;30:822–4. doi: 10.1136/jmg.30.10.822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsuoka R, Takao A, Kimura M, Imamura S, Kondo C, Joh-o K, et al. Confirmation that the conotruncal anomaly face syndrome is associated with a deletion within 22q11.2. Am J Med Genet. 1994;53:285–9. doi: 10.1002/ajmg.1320530314. [DOI] [PubMed] [Google Scholar]
- 15.McDonald-McGinn DM, Driscoll DA, Bason L, Christensen K, Lynch D, Sullivan K, et al. Autosomal dominant “Opitz” GBBB syndrome due to a 22q11.2 deletion. Am J Med Genet. 1995;59:103–13. doi: 10.1002/ajmg.1320590122. [DOI] [PubMed] [Google Scholar]
- 16.Giannotti A, Digilio MC, Marino B, Mingarelli R, Dallapiccola B. Cayler cardiofacial syndrome and del 22q11: part of CATCH22 phenotype. 1994;53:303–4. doi: 10.1002/ajmg.1320530320. [DOI] [PubMed] [Google Scholar]
- 17.Chow EWC, Watson M, Young DA, Bassett AS. Neurocognitive profile in 22q11 deletion syndrome and schizophrenia. Schizophr Res. 2006;87:270–8. doi: 10.1016/j.schres.2006.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Antshel KM, Fremont W, Kates WR. The neurocognitive phenotype in velo-cardio-facial syndrome: a developmental perspective. Dev Disabil Res Rev. 2008;14:43–51. doi: 10.1002/ddrr.7. [DOI] [PubMed] [Google Scholar]
- 19.De Smedt B, Devriendt K, Fryns JP, Vogels A, Gewillig M, Swillen A. Intellectual abilities in a large sample of children with velo-cardio-facial syndrome: an update. J Intellect Disabil Res. 2007;51:666–70. doi: 10.1111/j.1365-2788.2007.00955.x. [DOI] [PubMed] [Google Scholar]
- 20.Green T, Gothelf D, Glaser B, Debbane M, Frisch A, Kotler M, et al. Psychiatric disorders and intellectual functioning throughout development in velocardiofacial (22q11.2 deletion) syndrome. J Am Acad Child Adolesc Psychiatry. 2009;48:1060–8. doi: 10.1097/CHI.0b013e3181b76683. [DOI] [PubMed] [Google Scholar]
- 21.Fung WLA, McEvilly R, Fong J, Silversides C, Chow E, Bassett AS. Elevated prevalence of generalized anxiety disorder in adults with 22q11.2 deletion syndrome. Am J Psychiatry. 2010;167:998. doi: 10.1176/appi.ajp.2010.09101463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sullivan KE. Chromosome 22q11.2 deletion syndrome: DiGeorge syndrome/velocardiofacial Syndrome. Immunol Allergy Clin North Am. 2008;28:353–66. doi: 10.1016/j.iac.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 23.Oskarsdottir S, Persson C, Eriksson BO, Fasth A. Presenting phenotype in 100 children with the 22q11 deletion syndrome. Eur J Pediatr. 2005;164:146–53. doi: 10.1007/s00431-004-1577-8. [DOI] [PubMed] [Google Scholar]
- 24.Greenhalgh KL, Aligiania IA, Bromilow G, Cox H, Stait Y, Leech BJ, et al. 22q11 deletion: a multisystem disorder requiring multidisciplinary input. Arch Dis Child. 2003;88:523–4. doi: 10.1136/adc.88.6.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kapadia R, Bassett AS. Recognizing a common genetic syndrome: 22q11.2 deletion syndrome. Can Med Assoc J. 2008;178:391–3. doi: 10.1503/cmaj.071300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McDonald-McGinn DM, Kirschner RE, Goldmuntz E, Sullivan K, Eicher P, Gerdes M, et al. The Philadelphia story: the 22q11.2 deletion: report on 250 patients. Genet Couns. 1999;10:11–24. [PubMed] [Google Scholar]
- 27.Emanuel BS. Molecular mechanisms and diagnosis of chromosome 22q11.2 rearrangements. Dev Disabil Res Rev. 2008;14:11–8. doi: 10.1002/ddrr.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bassett AS, Marshall CR, Lionel AC, Chow EWC, Scherer SW. Copy number variations and risk for schizophrenia in 22q11.2 deletion syndrome. Hum Mol Genet. 2008;17:4045–53. doi: 10.1093/hmg/ddn307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amati F, Conti E, Novelli A, Bengala M, Digilio MC, Marino B, et al. Atypical deletions suggest five 22q11. 2 critical regions related to the DiGeorge/velo-cardio-facial syndrome. Eur J Hum Genet. 1999;7:903–9. doi: 10.1038/sj.ejhg.5200399. [DOI] [PubMed] [Google Scholar]
- 30.Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–64. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edelmann L, Pandita RK, Morrow BE. Low-copy repeats mediate the common 3-Mb deletion in patients with velo-cardio-facial syndrome. Am J Hum Genet. 1999;64:1076–86. doi: 10.1086/302343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shaikh T, Kurahashi H, Saitta SC, O’Hare AM, Hu P, Roe BA, et al. Chromosome 22-specific low copy repeats and the 22q11. 2 deletion syndrome: genomic organization and deletion endpoint analysis. Hum Mol Genet. 2000;9:489–501. doi: 10.1093/hmg/9.4.489. [DOI] [PubMed] [Google Scholar]
- 33.Baumer A, Riegel M, Schinzel A. Non-random asynchronous replication at 22q11.2 favours unequal meiotic crossovers leading to the human 22q11.2 deletion. J Med Genet. 2004;41:413–20. doi: 10.1136/jmg.2003.016352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McDonald-McGinn DM, Tonnesen MK, Laufer-Cahana A, Finucane B, Driscoll DA, Emanuel BS, et al. Phenotype of the 22q11.2 deletion in individuals identified through an affected relative: cast a wide FISHing net! Genet Med. 2001;3:23–9. doi: 10.1097/00125817-200101000-00006. [DOI] [PubMed] [Google Scholar]
- 35.Bassett AS, Chow EWC, Husted J, Weksberg R, Caluseriu O, Webb GD, et al. Clinical features of 78 adults with 22q11 deletion syndrome. Am J Med Genet A. 2005;138:307–13. doi: 10.1002/ajmg.a.30984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cohen E, Chow EWC, Weksberg R, Bassett AS. The phenotype of adults with the 22q11 deletion syndrome: a review. Am J Med Genet. 1999;86:359–65. doi: 10.1002/(sici)1096-8628(19991008)86:4<359::aid-ajmg10>3.0.co;2-v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bassett AS, Chow EWC. Schizophrenia and 22q11.2 deletion syndrome. Curr Psychiatry Rep. 2008;10:148–57. doi: 10.1007/s11920-008-0026-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Toriello HV, Goldenberg P. Evidence-based medicine and practice guidelines: application to genetics. Am J Med Genet C. 2009;151C:235–40. doi: 10.1002/ajmg.c.30222. [DOI] [PubMed] [Google Scholar]
- 39.Consevage MW, Seip JR, Belchis DA, Davis AT, Baylen BG, Rogan PK. Association of a mosaic chromosomal 22q11 deletion with hypoplastic left heart syndrome. Am J Cardiol. 1996;77:1023–5. doi: 10.1016/s0002-9149(97)89165-5. [DOI] [PubMed] [Google Scholar]
- 40.Hatchwell E, Long F, Wilde J, Crolla J, Temple K. Molecular confirmation of germ line mosaicism for a submicroscopic deletion of chromosome 22q11. Am J Med Genet. 1998;73:103–6. doi: 10.1002/(sici)1096-8628(19980630)78:2<103::aid-ajmg1>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 41.Sandrin-Garcia P, Macedo C, Martelli LR, Ramos ES, Guion-Almeida ML, Richieri-Costa A, et al. Recurrent 22q11.2 deletion in a sibship suggestive of parental germline mosaicism in velocardiofacial syndrome. Clin Genet. 2002;61:380–3. doi: 10.1034/j.1399-0004.2002.610511.x. [DOI] [PubMed] [Google Scholar]
- 42.Bassett AS, Chow EWC, Husted J, Hodgkinson KA, Oechslin E, Harris L. Premature death in adults with 22q11.2 deletion syndrome. J Med Genet. 2009;46:324–30. doi: 10.1136/jmg.2008.063800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shekelle P, Woolf SH, Eccles M, Grimshaw J. Clinical guidelines: Developing guidelines. Brit Med J. 1999;318:593–6. doi: 10.1136/bmj.318.7183.593. [DOI] [PMC free article] [PubMed] [Google Scholar]