

Abstract
Hemorrhagic shock (HS) due to major trauma and surgery predisposes the host to the development of systemic inflammatory response syndrome (SIRS) including acute lung injury (ALI) through activating and exaggerating the innate immune response. IL-1β is a crucial pro-inflammatory cytokine that contributes to the development of SIRS and ALI. Lung endothelial cells (EC) are one important source of IL-1β, and the production of active IL-1β is controlled by the inflammasome. In this study, we addressed the mechanism underlying HS activation of the inflammasome in lung EC. We show that high mobility group box 1 (HMGB1) acting through TLR4, and a synergistic collaboration with TLR2 and RAGE signaling, mediates HS-induced activation of EC NAD(P)H oxidase. In turn, reactive oxygen species (ROS) derived from NAD(P)H oxidase promote the association of thioredoxin-interacting protein (TXNIP) with the Nod-like receptor protein NLRP3 and subsequently induce inflammasome activation and IL-1β secretion from the EC. We also show that neutrophil-derived ROS play a role in enhancing EC NAD(P)H oxidase activation, and therefore an amplified inflammasome activation in response to HS. The present study explores a novel mechanism underlying HS activation of EC inflammasome and, thus, presents a potential therapeutic target for SIRS and ALI induced after HS.
Keywords: IL-1β, NAD(P)H oxidase, Reactive oxygen species, Toll-like receptors
Introduction
Systemic inflammatory response syndrome (SIRS) and multi-organ failure (MOF) are common complication following trauma, severe surgery, or hemorrhagic shock (HS), and result in high mortality and morbidity (1, 2). Acute lung injury (ALI) is an important component of MOF and often serves as a direct cause of death in these patients (3). HS promotes the development of ALI by inducing the immune system to produce an exaggerated inflammatory response to endogenous and exogenous danger signals (4–6). The lung vascular endothelium is an active organ that critically contributes to the pathogenesis of ALI following trauma, sepsis, and shock by affecting pulmonary and systemic homeostasis, including secretion of cytokines, chemokines, and adhesion molecules (7, 8). There is a significant gap in our knowledge concerning the mechanisms of HS regulation of lung endothelial cell (EC) activation, and subsequent promotion of lung inflammation.
The cytokine IL-1β is a key pro-inflammatory mediator involved in the development of post-trauma SIRS (9–14). EC are one important source of IL-1β and conversely, are also a target of IL-1β, which causes them to release a range of inflammatory molecules in response to IL-1β stimulation (7, 13, 15). Thus, EC are postulated to be an amplifier of inflammation through the sensing and release of IL-1β. The production of active IL-1β is tightly controlled by the formation and activation of the inflammasome, which is comprised of NOD-like receptors (NLRs), caspase-1 (or caspase-5) and apoptosis-associated speck-like protein containing a CARD domain (ASC) (16, 17). IL-1β is synthesized initially as an inactive precursor molecule (pro- IL-1β p35), which must be cleaved by caspase-1 at amino acid position 116 to produce the actively mature IL-1β (p17) that is then secreted in response to stimulating signals. Caspase-1 is also synthesized as an inactive 45-kDa protein (pro-caspase-1) that undergoes autocatalytic processing after assembly of the inflammasome in response to an appropriate stimulus (18). However, it is not clear how HS induces inflammasome activation and IL-1β maturation in the lung.
Reactive oxygen species (ROS) have been implicated as regulator of inflammasome activation (19). The major source of ROS within pulmonary EC is the non-phagocytic NAD(P)H oxidase (20), which is composed of membrane-bound gp91phox and p22phox, as well as cytosolic subunits such as p47phox, p67phox and the small GTPase Rac. Endothelial NAD(P)H oxidase can be activated in many ways including by growth factors, cytokines, shear stress, and hypoxia (21). We have recently reported that HS activates lung endothelial NAD(P)H oxidase through high-mobility group box 1 (HMGB1)-TLR4 signaling (22).
HMGB1 can be secreted by innate immune cells in response to microbial products or other inflammatory stimuli (23, 24), as well as be released by injured cells and is a prototypical damage-associated molecular pattern molecule (DAMP) (25–27). HMGB1 has been identified as an inflammatory cytokine that mediates lethality in sepsis (23, 24), development of ALI after trauma/hemorrhage (28–32), and hepatic injury after liver ischemia-reperfusion (33). We have shown that HMGB1 acting as an early mediator stimulates the activation of macrophages, polymorphonuclear neutrophils (PMN), and EC in a setting of HS/resuscitation (HS/R) (4, 22, 31, 32, 34).
In the present study, we investigated the mechanism of HS regulation of the inflammasome in lung EC. We demonstrate that HS, through HMGB1, activates endothelial NAD(P)H oxidase and the created oxidants cause thioredoxin (TRX)-interacting protein (TXNIP) to associate with NLRP3 leading to inflammasome activation and IL-1β secretion. We also show that HS-activated PMN and PMN NAD(P)H oxidase are required for augmented activation of the endothelial inflammasome through enhanced ROS production in lung EC. These findings reveal a pathway that links the global insult of HS to activation of the inflammasome molecules in lung EC.
Materials and Methods
Materials
Recombinant HMGB1 was purchased from R&D systems (Minneapolis, MN). Stimulating activity of recombinant HMGB1 was confirmed in mouse macrophages by assay of TNF release, with an ED50 of 3 ~ 12 μg/ml. Polyclonal neutralizing antibody against HMGB1 prepared as described previously (24) was provided by Dr. K. J. Tracey (Feinstein Institute for Medical Research, North Shore-LIJ Health System, Manhasset, NY). Nonimmune rabbit IgG (Catalog #: I5006) and all other chemicals were obtained from Sigma-Aldrich, except where noted.
Mouse model of Hemorrhagic shock and resuscitation
Male C57BL/6 WT mice and gp91phox knockout (gp91 phox−/−) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). TLR4 knockout (TLR4−/−) mice, TLR2 knockout (TLR2−/−) mice, and receptor for advanced glycation endproducts (RAGE) knockout (RAGE−/−) mice were bred in Dr. Billiar’s lab at the University of Pittsburgh; caspase-1 knockout (caspase-1−/−) mice were originally provided by Dr. Richard Flavell (Yale University) and bred in Dr. Billiar’s lab; NLRP3 knockout (NLRP3−/−) mice were obtained from Millennium Pharmaceuticals (Cambridge, MA). All mice used are on a C57BL/6 background. All experimental protocols involving animals were approved by Institutional Animal Care and Use Committee of VA Pittsburgh Healthcare System and University of Pittsburgh. Mice were 12–14 weeks of age at the time of experiments and were maintained on standard rodent chow and water ad libitum. The mice were not fasted before experiments. Animals were anesthetized with 50 mg/kg of ketamine and 5 mg/kg of xylazine via intraperitoneal (i.p.) administration. Femoral arteries were cannulated for monitoring of mean arterial pressure (MAP), blood withdrawal and resuscitation. HS was initiated by blood withdrawal and reduction of the MAP to 30 mmHg within 20 min. Blood was collected into a 1 ml syringe and heparinized to prevent clotting. In order to exclude the effect of heparin on immune processes, equal amounts of heparin (10 U) were injected into sham animals through the cannulated femoral artery during the sham operation. After a hypotensive period of 2 h, animals were resuscitated by transfusion of the shed blood and Ringer’s Lactate (RL) in a volume equal to that of shed blood, over a period of 60 min. The catheters were then removed, the femoral artery was ligated, and the incisions were closed. Sham animals underwent the same surgical procedures without hemorrhage and resuscitation. In some experiments, neutralizing antibodies against HMGB1 (600 μg per mouse) or nonimmune control IgG was injected intraperitoneally into the mice 10 min before hemorrhage. The animals were kept anesthetized during the whole experiment period by Xylazine and Ketamine. At various time points after resuscitation (0 to 8 h), bronchoalveolar lavage (BAL) was performed and BAL fluid collected. Lung vasculature was flushed by injection of phosphate buffered saline (PBS) through the right ventricle followed by collection of lung tissue for experimental analysis.
In vivo neutrophil depletion and repletion
PMN depletion was induced using RB6-8C5 monoclonal antibody (Ly-6G/Gr-1 specific) (eBioscience, San Diego, CA) (35). At approximately 16 h before performing shock or sham operation, 10 μg of the anti-mouse Ly-6G/Gr1 antibody or control antibody (rabbit anti-mouse IgG; Sigma-Aldrich) was administered i.p. to mice in 100 μl saline. Our previous studies have shown that during the period of 16 to 24 h after injection of anti-mouse Ly-6G/Gr1 antibody the circulating PMN count in the antibody-treated group was decreased to 0.08 ± 0.02% of total white blood cells vs. 22.2 ± 1.9% in the control group (36). There were no statistically significant differences in the number of peripheral lymphocytes, atypical lymphocytes, monocytes, or eosinophils between the antibody-treated and control groups (36). In order to determine the role of PMN NAD(P)H oxidase in the mechanism of HS-induced activation of lung EC inflammasome, PMN repletion in neutropenic mice was performed by tail vein injection of PMN (~2 × 106 cells) isolated from WT or gp91phox−/− mice that were subjected to either HS or sham operation. An immunomagnetic separation system (BD Biosciences Pharmingen, San Diego, CA) (37) was used to isolate PMN. Viability of the isolated PMN was > 95%, and PMN purity was >95% as assessed by trypan blue exclusion and Wright-Giemsa staining, respectively.
Mouse Lung Vascular Endothelial Cell (MLVEC) Isolation and Characterization
MLVEC were isolated using a previously described method (38) that was modified in our laboratory as follows: Briefly, mice were anesthetized with 50 mg/kg of ketamine and 5 mg/kg of xylazine i.p. The chest cavity was opened and the right ventricle was cannulated. PBS was infused to remove blood from lungs. Peripheral lung tissue dices in a size about 1 mm3 were prepared and cultured in a 60-mm culture dish in growth medium (MEM D-Val medium containing 2 mM glutamine, 10% FBS, 5% human serum, 50 μg/ml penicillin/streptomycin, 5 μg/ml heparin, 1 μg/ml hydrocortisone, 80 μg/ml endothelial cell growth supplement from bovine brain, 5 μg/ml amphotericin, and 5 μg/ml mycoplasma removal agent) at 37°C with 5% CO2 for 60 h. The adherent cells were continued in culture for 3 days after removal of the tissue dices, followed by purification using biotin-conjugated rat anti-mouse CD31 (PECAM-1) monoclonal antibody and BD IMag streptavidin particles plus-DM, and the immunomagnetic separation system (BD Biosciences Pharmingen, San Diego, CA) following the manufacturer’s instructions. The cells were allowed to grow for 3 to 4 days after purification. The cells were characterized by their cobblestone morphology, uptake of Dil-Ac-LDL (Biomedical Technologies Inc., Stoughton, MA), and staining for factor VIII-related antigen (Sigma Chemical Co., St. Louis, MO). MLVEC passaged between 3 and 5 times were used in experiments, in which cells were treated with HMGB1 (0.5μg/ml) for 0–6 h, washed with HBSS three times, and harvested for further analysis.
PMN-MLVEC co-incubation
PMN-MLVEC co-incubation was performed using Transwell™ plates (Corning Incorporated Life Sciences, Acton, MA). The preparation of MLVEC and isolation of peripheral PMN are as described above. The co-cultures were then stimulated with HMGB1 (0.5μg/ml) for up to 6 h in DMEM containing10% FCS at a PMN concentration of 1 × 106 cells/ml medium.
Coimmunoprecipitation and immunoblot analysis
Mouse lung tissue or MLVEC were homogenized or lysed (~1 × 106 cells/ml) in lysis buffer (10 mM Tris, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 10 mM NaF, 1 mM Na3VO4, 10 μg/ml leupeptin, 10 μg/ml aprotinin and 20 mM PMSF). The supernatants were quantified, and 600 μg of total protein for each sample was then immunoprecipitated with anti-ASC antibody (Santa Cruz Biotechnologies, CA) or anti-TXNIP antibody (MBL International Co., MA). The immunoprecipitated proteins were separated on a 10% SDS-PAGE gel, and were then electroblotted onto PVDF membrane and blocked for 1 h at room temperature with Tris-buffered saline containing 3% non-fat dried milk. NLRP3 was detected by probing the membranes with anti-NLRP3 antibody (Santa Cruz Biotechnologies, CA) at 1:500 dilution and detected with Clean-Blot IP Detection Reagent (Thermo Scientific, Rockford, IL) following the manufacturer’s instructions. Blots were then stripped and reprobed with anti-ASC antibody or anti-TXNIP antibody and again detected with Clean-Blot IP Detection Reagent. Caspase-1 cleavage in the lung tissue and MLVEC was measured by detecting its p10 fragment in Western blot using rabbit polyclonal anti-mouse caspase-1 p10 (Santa Cruz Biotechnologies, CA).
Caspase-1 detection in MLVEC
MLVEC (104~105 cells) were seeded onto the chamber slide in a 35 mm petri dish and cultured in the EC culture media for 12 h at 37°C. MLVEC were then washed twice with serum-free DMEM and incubated in serum-free DMEM for 30 min. The cells were stimulated with 0.5 μg/ml of HMGB1 for 0–6 h followed by staining of the cells with 1 × Caspase-1 FLICA Regent – cell permeable carboxyfluorescein-labeled fluorochrome inhibitor of caspase-1 -(ImmunoChemistry Technologies, Bloomington, MN), which binds to activated caspase-1, in serum-free DMEM for 1 h. The cells were then washed three times with PBS, fixed with 4% paraformaldehyde in PBS for 15 min at room temperature, and further washed with ice cold PBS for three times. The nucleus of the cells were stained with 1 μg/ml of Hoechst for 15 minutes at room temperature in the dark. The green fluorescence of caspase-1 positive cells was imaged by confocal microscope with excitation at 480 nm, and Hoechst-stained nuclei were observed with excitation at 365 nm.
Measurement of IL-1β
IL-1β in BAL fluid and cell culture media was measured using ELISA Ready-Set-Go kit for mouse IL-1β (eBioscience, San Diego, CA) following the manufacturer’s instructions.
Measurement of superoxide generation in live MLVEC
Live MLVEC cultured in 12-well cell culture plates were stained with the cell permeable ROS detection reagent H2DFFDA (Invitrogen Molecular Probes, Carlsbad, CA) at the concentration of 10 μM for 10 min. Cells were then washed with HBSS three times followed by incubation in the growth medium in the presence or absence of HMGB1 (0.5 μg/ml) for 0.5–2 h. ROS production was then detected by fluorescence microscopy at different time points.
Transfection of siRNA in MLVEC
TXNIP siRNA, control siRNA, and transfection kit were purchased from Santa Cruz Biotechnologies. MLVEC (2 × 105 cells) were seeded in a six well tissue culture plate, and incubated at 37°C in a CO2 incubator until the cells were 80% confluent. The cells were then transfected with TXNIP siRNA or control siRNA using the siRNA transfection kit following the manufacturer’s instructions. At 24 – 72 h after the transfection, TXNIP protein expression in the transfected cells was analyzed by Western blot. Since we observed a confirmed knockdown of TXNIP in the EC at 48 h after siRNA transfection, we set this time point as time 0 for the experiments using HMGB1 treatment.
Data Presentation and Statistical Analysis
The data are presented as mean ± SEM of the indicated number of experiments. Statistical significance among group means was assessed by ANOVA. Student Neuman- Keuls post-hoc test was performed. Differences were considered significant at p<0.05.
Results
HS activates NLRP3 inflammasome in the lung
The assembly of inflammasome components including NLRP3, ASC and pro-caspase-1 is an initial step of inflammasome activation, and subsequent pro-caspase-1 cleavage to an active form, leading to pro-IL-1β cleavage to produce mature IL-1β (39). Thus, the association of NLRP3 and ASC, as well as caspase-1 cleavage, represent intracellular activation of inflammasome. We first determined the ability of HS to induce inflammasome activation in the lung using the mouse model of HS/R as described in the Methods. Lung tissue and BAL fluid were collected up to 8 h after HS/R. As shown in Figure 1A, the association of NLRP3 and ASC in WT lung tissue was induced at 1 h after HS/R, and further increased between 2 and 8 h. Caspase-1 cleavage was detected starting at 2 h, reached a peak at 4h, and was maintained up to 8 h after HS/R. IL-1β concentration in BAL fluid, which represents the released IL-1β from pulmonary cells, was elevated at 2h after HS/R and sustained at a high level for at least 8 h. However, the HS-induced release of IL-1β was absent in NLRP3−/− mice and caspase-1−/− mice (Figure 1B). Additionally, lack of caspase-1 also prevented the association of NLRP3 and ASC (Figure 1A). Altogether these data demonstrate NLRP3 inflammasome-dependent and caspase-1-dependent mechanisms of IL-1β release at an early stage following HS/R.
Figure 1. HS induces NLRP3 inflammasome activation and IL-1β secretion in the lungs.
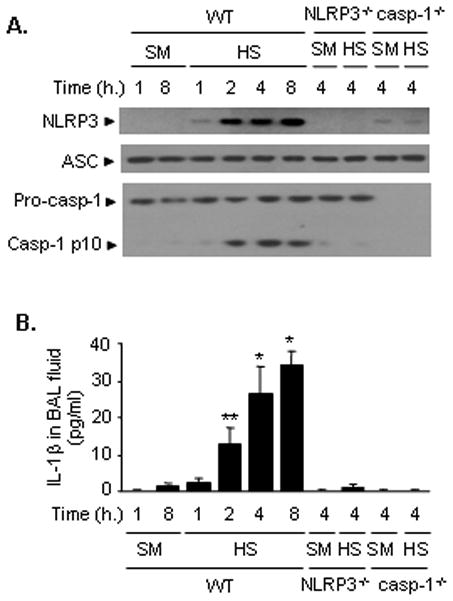
WT (C57BL/6) mice, NLRP3−/− mice, and caspases-1−/− mice were subjected to HS/R (HS) or sham operation (SM). Lung tissue and BAL fluid were recovered 1, 2, 4, and 8 h after HS/R or 1 and 8 h after sham operation. A. The association of NLRP3 and ASC was detected using immunoprecipitation with anti-ASC antibody followed by immunoblotting for NLRP3 and ASC, as well as for caspase-1 cleavage product p10 fragments. Western blotting as described in Material and Methods. The images are representatives of five independent experiments. B. IL-1β in BAL fluid was measured with ELISA. The graph shows the mean and SEM from five mice. ** p < 0.01 compared with all other groups; *p < 0.01 compared with the groups labeled with no asterisk.
HMGB1 is responsible for HS-induced lung NLRP3 inflammasome activation
We have previously reported that HS/R causes a significant increase of HMGB1 in the serum, lung, and liver at 2 h after HS/R (31). To determine if endogenous HMGB1 contributes to HS/R-induced inflammasome activation in the lung, neutralizing antibody to HMGB1 was administered to mice 15 min before HS/R. Treatment with anti-HMGB1 antibody resulted in a significant decrease in the HS/R-induced NLRP3-ASC association in the lung and IL-1β concentration in BAL fluid at 4 h after HS/R as compared to non-specific IgG-treated animals (Figure 2A and 2B). Known HMGB1 receptors include TLR4, TLR2, and RAGE. We thus further defined the role of HMGB1 and its receptors by using TLR4-, TLR2- and RAGE-deficient mice. The results show that knockout of TLR4 prevented the HS/R-induced inflammasome activation and IL-1β release in the lung (Figure 2A and 2B), whereas, either TLR2 knockout or RAGE knockout mice exhibited only a partial reduction of the association of NLRP3-ASC and caspase-1 cleavage (Figure 2A) and IL-1β in BAL fluid (Figure 2B) in response to HS/R.
Figure 2. HMGB1 acting through TLR4 mediates HS/R-induced NLRP3 inflammasome activation and IL-1β secretion.
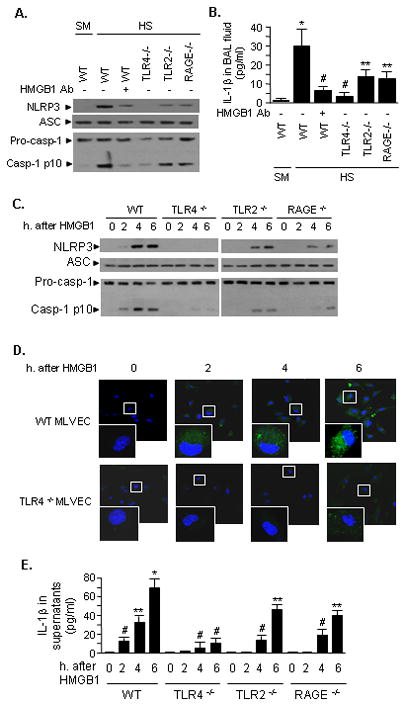
A and B. WT (C57BL/6) mice, TLR4−/− mice, TLR2−/− mice and RAGE−/− mice were subjected to HS/R (HS) or sham operation (SM). Some WT mice received anti-HMGB1 antibody (600 μg per mouse) by i.p. injection 10 min before HS or sham operation. Lung tissue and BAL fluid were then recovered at 4 h after HS/R or sham operation. In the lung tissue, the association of NLRP3 and ASC was detected using immunoprecipitation with anti-ASC antibody and immunoblotting with anti-ASC and NLRP3, as well as caspase-1 p10 fragments - all detected by Western blotting (A), and IL-1β in BAL fluid was measured with ELISA (B). The images are representatives of five independent experiments. The graph shows the mean and SEM from five mice. ** p < 0.01 compared with all other groups; *p < 0.01 compared with all other groups; # p < 0.05 compared with the groups labeled with no asterisk. C, D, and E. In vitro stimulation of MLVEC with HMGB1. MLVEC isolated from WT, TLR4−/−, TLR2−/− and RAGE−/− mice were treated with HMGB1 (0.5 μg/ml) for the time as indicated, and the association of NLRP3 and ASC and production of caspase-1 p10 fragments were detected (C); the activated caspase-1 in WT and TLR4−/− was visualized with caspase-1 FLICA reagent and observed under confocal microscope (D); IL-1β in the cell culture media was measured with ELISA (E). The images are representatives of three independent experiments. The graph shows the mean and SEM from five independent experiments. ** p < 0.01 compared with all other groups; *p < 0.01 compared with all other groups; # p < 0.05 compared with the groups labeled with no asterisk.
Lung vascular endothelial cells are one of the major pulmonary cell populations and the important source of inflammatory cytokines. We therefore directly stimulated MLVEC isolated from WT, TLR4−/−, TLR2−/−, or RAGE−/− mice with HMGB1 for up to 6 h, and then assessed NLRP3-ASC association, caspase-1 cleavage, and IL-1β release. In WT MLVEC, we observed a significant association of NLRP3-ASC and caspase-1 cleavage after HMGB1 treatment for 4 h and 6 h (Figure 2C). HMGB1-treatment of TLR4−/− MLVEC failed to induce NLRP3-ASC association and HMGB1-treatment of TLR2−/− and RAGE−/− MLVEC showed only a partial inhibition of expected NLRP3-ASC association (Figure 2C).
To verify that caspase-1 cleavage detected in the Western blot reflects actual caspase-1 activation, the active caspase-1 was visualized in MLVEC by staining the cells with capase-1 FLICA, which binds to active caspase-1. Confocal microscopy showed that HMGB1 induced increases in caspase-1 activity in WT, but not in TLR4−/− MLVEC (Figure 2D), which is consistent with the observations shown in Figure 2C. The changes in medium IL-1β concentration, shown in Figure 2E, further confirm our findings and demonstrate a critical role of HMGB1 and TLR4 in inducing IL-1β release from MLVEC.
HS-activated PMN augment HS-induced activation of inflammasome in lung EC
We have previously shown that PMN and PMN-derived oxidants play an important role in mediating an augmented activation of lung EC following HS/R (22, 36). Accordingly, we hypothesized that HS-activated PMN might also contribute to an enhanced activation of the inflammasome in lung EC through PMN-EC interactions. To test this hypothesis we depleted circulating PMN prior to HS/R, and in some cases, we replenished the neutropenic mice during the resuscitation phase with PMN that were isolated from either sham operated or HS mice at 2 h after HS/R. As shown in Figure 3A, at 4 h after HS/R, neutropenia induced in mice subjected to HS was associated with a significant reduction in the association of NLRP3-ASC and cleavage of caspase-1 in the lung, as well as a lower concentration of IL-1β in the BAL fluid, as compared to the mice subjected to HS/R with no PMN depletion. Repletion with WT PMN that were isolated from HS animals at 2 h after resuscitation, but not from sham animals, restored the NLRP3-ASC association in the lung and IL-1β level in the BAL fluid in response to HS/R (Figure 3A). These in vivo observations were further recapitulated in vitro using PMN-MLVEC co-culture system. MLVEC that were stimulated with HMGB1 and co-cultured with HS-activated PMN demonstrated an enhanced intracellular NLRP3-ASC association and caspases-1 cleavage, as well as a higher concentration of IL-1β in the medium as compared to those co-cultured with no PMN or with the PMN isolated from sham animals (Figure 3B). Taken together, these data indicate an important role of HS-activated PMN in augmenting HS/R-induced endothelial inflammasome.
Figure 3. HS-activated PMN are required for augmented activation of inflammasome.
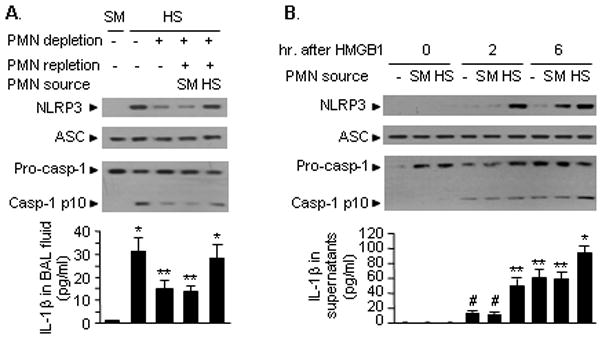
A. In vivo studies show that HS-activated PMN enhance HS/R-induced coupling of NLRP3 and ASC as well as caspase-1 cleavage in the lungs and IL-1β release in BAL fluid. PMN depletion was performed as described in Material and Methods 16 h before HS/R. PMN repletion was performed in neutropenic mice during resuscitation using tail vein injection of PMN that were isolated from blood of WT mice subjected to sham (SM) or HS at 2 h after resuscitation. The lung tissue and BAL fluid were collected at 4 h after resuscitation. The images are representatives of five independent experiments. The graph shows the mean and SE from five mice. ** p < 0.01 compared with all other groups; *p < 0.01 compared with the groups labeled with no asterisk. B. In vitro studies show that HS-activated PMN augment HMGB1-induced binding association of NLRP3 and ASC and caspase-1 cleavage in MLVEC with IL-1β release in the cell culture media. PMN (5 × 105 cells) were isolated from blood of WT mice subjected to sham (SM) or HS at 2 h after resuscitation, and co-cultured with MLVEC in the presence or absence of HMGB1 (0.5 μg/ml) for 0, 2, and 6 h. The images are representatives of five independent experiments. The graph shows the mean and SEM from five independent studies. ** p < 0.01 compared with all other groups; *p < 0.01 compared with all other groups; # p < 0.01 compared with the groups labeled with no asterisk.
NAD(P)H oxidase is required for HS-induced activation of inflammasome
To further define the role of PMN NAD(P)H oxidase in the activation of EC inflammasome, we replenished neutropenic WT HS mice with PMN isolated from gp91phox−/−mice, in which the gp91 subunit of NAD(P)H oxidase was genetically deleted and therefore a dysfunction of NAD(P)H oxidase. As shown in Figure 4A, PMN collected from gp91phox−/− mice, which were subjected to either sham or shock, failed to restore HS/R-induced NLRP3-ASC association and caspases-1 cleavage in the lung and IL-1β release into the BAL fluid (Figure 4A, lane 3 & 4). In order to test the role of EC endogenous NAD(P)H oxidase in mediating HS/R-induced inflammasome activation, PMN in gp91phox −/− mice were depleted, and following HS procedure, the gp91phox −/− mice were replenished with WT PMN during resuscitation phase. Figure 4A shows that in NAD(P)H oxidase-deficient mice the HS/R-induced inflammasome activation in the lung and IL-1β release were significantly diminished, even though the mice were replenished with WT PMN (Figure 4A, lane 7 & 8). This result suggested a critical role of endogenous NAD(P)H oxidase in mediating HS/R-induced inflammasome activation in EC.
Figure 4. NAD(P)H oxidase is required for HS-induced activation of inflammasome.
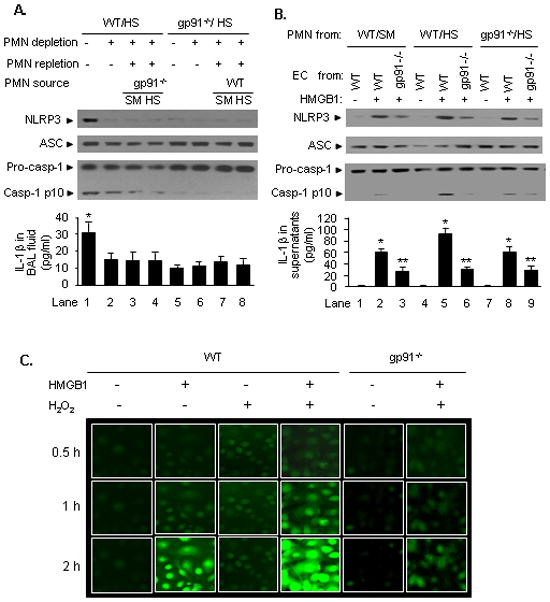
A. Effect of deficiency of NAD(P)H oxidase on HS/R-induced inflammasome activation in the lung. In WT or gp91phox−/− mice, PMN depletion and repletion were performed as described in the Materials and Methods. Particularly, PMN repletion was performed during resuscitation phase after HS, and in WT mice, PMN were replaced with gp91phox−/− PMN, and in gp91phox−/− mice, the PMN were substituted with WT PMN. The lungs and BAL fluid were collected from the mice 4 h after HS/R (HS) or sham operation (SM) for the detection of NLRP3-ASC association and caspase-1 cleavage in the lungs, and IL-1β secretion in the BAL fluid. Global gp91phox knockout significantly prevented HS/R-induced caspase-1 activation, whereas, deficiency of PMN gp91phox attenuated the process. The images are representative of five independent studies. The graph shows the mean and SEM from five mice. *p < 0.01 compared with the groups labeled with no asterisk. B. Effect of deficiency of NAD(P)H oxidase on HMGB1-induced inflammasome activation in MLVEC. WT and gp91phox−/− MLVEC were co-incubated with PMN that were isolated from the blood of WT or gp91phox−/− (gp91−/−) mice subjected sham (SM) or HS in the presence or absence of HMGB1 (0.5 μg/ml) for 4 h. The association of NLRP3 and ASC and caspase-1 cleavage in MLVEC and IL-1β release in the cell culture media were then detected. While deficiency of PMN NAD(P)H oxidase caused an attenuation in activation of inflammasome in EC in response to HMGB1, deficiency of EC endogenous NAD(P)H oxidase prevented the HMGB1-induced activation of inflammasome. The images are representative of five independent studies. The graph shows the mean and SEM from five independent experiments. ** p < 0.01 compared with all other groups; *p < 0.01 compared with the groups labeled with no asterisk. C. ROS production in live MLVEC. WT and gp91phox−/− (gp91−/−) MLVEC were stained with the cell permeable ROS detection reagent H2DFFDA (10 μM) for 10 min. The cells were then washed with HBSS for three times followed by incubation in the growth medium in the presence or absence of HMGB1 (0.5 μg/ml) and/or H2O2 (250 μM) for up to 2 h. ROS production was detected by fluorescence microscopy at different time points as indicated. The images are representative of four independent studies.
The role of NAD(P)H oxidase in mediating HMGB1 activation of endothelial inflammasome was further addressed by utilizing a PMN-EC co-culture approach. MLVEC from WT or gp91phox −/− mice were co-cultured with PMN that were isolated from WT or gp91phox−/− mice in the presence of HMGB1 for 4 h. As shown in Figure 4B, deficiency of gp91phox −/− in EC markedly attenuated HMGB1-induced inflammasome activation and IL-1β release (lane 3, 6, and 9) as compared to that in WT EC (lane 2, 5, and 8); HS-activated WT PMN enhanced HMGB1-induced inflammasome activation and IL-1β release in WT EC (Figure 4B, lane 5). These results indicate that EC NAD(P)H oxidase is an essential component required for HS/R-induced inflammasome activation, whereas, PMN NAD(P)H oxidase importantly contributes to an augmented activation of EC inflammasome following HS/R.
In order to demonstrate that exogenous ROS, such as PMN NAD(P)H oxidase-derived oxidants, can enhance endothelial NAD(P)H oxidase activation and therefore an increased intra-EC production of ROS, the production of ROS in live EC was visualized using H2DFFDA and directly detected by fluorescence microscopy. The EC were observed for 2 h after treatment with HMGB1 and/or hydrogen peroxide (H2O2). Figure 4C shows that HMGB1-induced ROS production in WT MLVEC is accelerated and amplified by adding H2O2, and the ROS production in the gp91phox deficient MLVEC in response to HMGB1/H2O2 is markedly diminished, indicating that the observed ROS production in the EC is derived mainly from NAD(P)H oxidase and amplified by exogenous ROS.
NAD(P)H oxidase mediates HS-induced TXNIP-NALP3 interaction
A recent report showed that thioredoxin (TRX)-interacting protein (TXNIP) is a redox-sensitive component of inflammasome activation, and ROS can cause TXNIP to associate with NLRP3, which leads to inflammasome activation (19). Based on the data presented above, we hypothesized that HS activation of endothelial inflammasome might be mediated by ROS-induced association of TXNIP and NLRP3. To test this hypothesis, we first detected the association of TXNIP and NLRP3 in the lung tissue at 4 h following HS/R. As demonstrated in Figure 5A, HS/R induced the association of TXNIP and NLRP3 in the lung (lane 1), and this effect of HS/R largely depends on NAD(P)H oxidases, since lack of PMN (lane 2), PMN NAD(P)H oxidase (lane 4), or global NAD(P)H oxidase (lane 5 and lane 6) decreased the binding of TXNIP and NLRP3 (Figure 5A).
Figure 5. NAD(P)H oxidase mediates HS/HMGB1-induced TXNIP-NALP3 interaction.
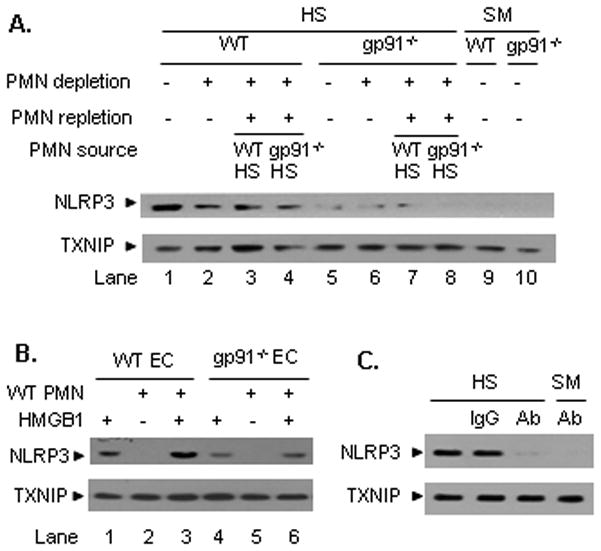
A. In vivo HS/R-induced association of NLRP3 and TXNIP requires NAD(P)H oxidase. PMN depletion in WT and gp91phox−/− (gp91−/−) mice was performed as described in Materials and Methods 16 h before HS/R. PMN repletion was performed in neutropenic mice during resuscitation phase using tail vein injection of PMN that were isolated from blood of WT mice subjected to HS at 2 h after resuscitation. The lungs were harvested from the mice 4 h after HS/R or sham operation (SM) for the detection of NLRP3-TXNIP association using coimmunoprecipitation and immunoblotting. The images are representative of five independent experiments. B. In vitro HMGB1-induced association of NLRP3 and TXNIP requires NAD(P)H oxidase. WT and gp91phox−/− (gp91−/−) MLVEC were co-incubated with PMN (5 × 105 cells), which were isolated from the blood of WT mice at 2 h after HS/R, in the presence or absence of HMGB1 (0.5 μg/ml) for 4 h. The association of NLRP3 and TXNIP in MLVEC was then detected. HS-activated PMN enhanced the HMGB1-induced association of NLRP3 and TXNIP, however, deficiency of NAD(P)H oxidase in MLVEC markedly attenuated the association in response to HMGB1. The images are representatives of three independent studies. C. WT (C57BL/6) mice received anti-HMGB1 antibody (600 μg per mouse) or control non-specific IgG by i.p. injection 10 min before HS/R. Lung tissue was then recovered at 4 h after HS/R. The association of NLRP3 and TXNIP in the lung tissue was detected using immunoprecipitation and immunoblotting. The images are representative of three independent experiments.
This result has also been recapitulated in EC-PMN co-culture system as shown in Figure 5B. HMGB1 stimulation for 4 h induced association of TXNIP-NLRP3 in WT MLVEC (lane 1), which was amplified when the EC co-incubated with WT HS-activated PMN (lane 3). However, the HMGB1-induced association of TXNIP and NLRP3 was markedly decreased in gp91phox −/− EC (lane 4 and 6).
The role of HMGB1 in mediating HS/R-induced association of TXNIP-NLRP3 in the lung was further confirmed in vivo utilizing neutralizing antibody against HMGB1. As shown in Figure 5C, treatment with anti-HMGB1 antibody resulted in a significant decrease in the HS/R-induced TXNIP-NLRP3 association in the lung as compared to non-specific IgG-treated animals.
TXNIP-NLRP3 interaction mediates HMGB1-induced inflammasome activation
To further elucidate the role of TXNIP in activating endothelial NLRP3 inflammasome, we knocked down TXNIP in MLVEC using siRNA techniques. At 48 h after transfection of TXNIP siRNA into MLVEC, the TXNIP protein could not be detected in the cells (Figure 6A). Knockdown of TXNIP in MLVEC dramatically reduced HMGB1-induced NLRP3-ASC association and caspases-1 cleavage in the EC, as well as IL-1β release, as compared to that in the MLVEC treated with non-specific control siRNA (Figure 6B).
Figure 6. TXNIP-NLRP3 interaction mediates HMGB1-induced inflammasome activation.
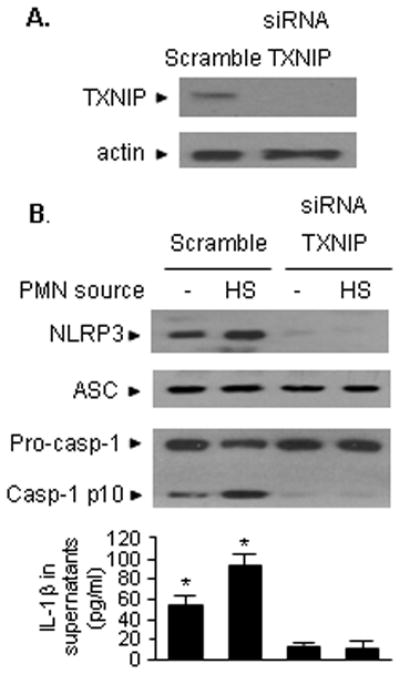
A. TXNIP in MLVEC was knocked down using siRNA techniques. At 48 h after transfection of TXNIP siRNA into MLVEC, the TXNIP protein could not be detected in the cells. B. MLVEC were transfected with TXNIP siRNA as described in the Materials and Methods 48 h prior to treatment with HMGB1 (0.5 μg/ml) for 4 h. NLRP3-ASC association and caspase-1 cleavage in the EC were then detected, as well as measurement of IL-1β in the media. Knockdown of TXNIP in MLVEC prevented HMGB1-induced NLRP3-ASC association and caspase-1 cleavage and IL-1β release, as compared to that in the MLVEC treated with non-specific control siRNA. The images are representatives of three independent studies. The graph shows the mean and SEM from three independent experiments. *p < 0.01 compared with the groups labeled with no asterisk.
Discussion
Pulmonary EC are activated in a very early stage in lung inflammation to be triggered by sepsis, trauma or shock (8, 22, 32), and play a critical role in promoting the development of ALI. Lung endothelium is an important source of IL-1β in response to HS. Conversely, lung EC are also targets of IL-1β, and release a range of inflammatory molecules in response to IL-1β. Thus, IL-1β through interaction with lung EC forms a feedback mechanism to amplify lung inflammation following HS. The present study demonstrates that the global ischemia/reperfusion injury initiated by resuscitated HS activates lung endothelial inflammasomes through an HMGB1-NAD(P)H oxidase-ROS-TXNIP-NLRP3 pathway. Despite the known central role of IL-1β in the development of SIRS, therapies targeting IL-1β, including IL-1 receptor antagonists (IL-1ra) and soluble IL-1R (40–42), have not been effective (43, 44). Thus, targeting DAMPs and ROS simultaneously may present a novel therapeutic strategy for controlling IL-1β secretion in the clinical settings that involve hemorrhage.
HMGB1 is a mediator of organ injury in animal models of infection and endotoxemia (23, 45–49). A recent study showed that HMGB1 administration induced rapid PMN recruitment in vivo, which was dependent on the interaction between RAGE and Mac-1 in the PMN (50). In addition, intratracheal administration of HMGB1 alone can induce organ dysfunction in the form of ALI (45); while, blockage of HMGB1 by administering anti-HMGB1 antibodies prevented hemorrhage-induced increases in pulmonary levels of proinflammatory cytokines, including IL-1β, and PMN infiltration in the lung as well as lung permeability (28). However, no role had been ascribed to HMGB1 in the activation of the inflammasome in a setting of HS/R, a process which is not associated with exposure to bacteria or bacterial products. The findings from the current in vivo and in vitro studies support a role for HMGB1 in mediating HS/R-induced EC inflammasome activation. As shown in Figures 2–6, neutralizing antibody against HMGB1 effectively attenuated HS/R-induced inflammasome activation in the lung and IL-1β secretion in BAL fluid, and direct stimulation of MLVEC with HMGB1 led to activation of the NLRP3-inflammasome and IL-1β release.
RAGE had been originally identified as a receptor for HMGB1 in neurites and malignant cells (51–53). However, recent studies have suggested that both TLR4 and TLR2 are important in mediating HMGB1-induced inflammatory responses (54–56). In the present study, the effect of HMGB1 is mainly mediated through TLR4 signaling, since TLR4 deficiency markedly blocked HMGB1-induced inflammasome activation, caspase-1 cleavage, and IL-1β release in the lung and MLVEC. However, TLR2 and RAGE seem also to be involved in signaling HMGB1 activation of the inflammasome, as in TLR2−/− and RAGE−/− mice inflammasome activation by HMGB1 is partially attenuated. These results may indicate a synergistic collaboration between signaling through TLR4, TLR2, and RAGE in mediating HS/R activation of inflammasome. The mechanism of the cross-talk among these receptor signaling pathways is not clear yet, and an extended study is needed. The importance of caspase-1 in mediating HMGB1/TLR4 signaling-induced IL-1β processing is evident by the observation as shown in Figure 1. Genetic deletion of caspase-1 prevented HS/R-induced IL-1β release in BAL fluid. More interestingly, the lack of caspase-1 even decreased the association of ASC and NLRP3 in the lung, suggesting a role of casepase-1 in keeping the integrity of inflammasome, although caspase-1 does not directly link the binding of ASC and NLRP3.
The present study shows that the endogenous endothelial NAD(P)H oxidase is essential for the HMGB1-induced activation of inflammasome in MLVEC, since gp91phox deficiency was consistently associated with a decreased activation of the inflammasome in both in vivo and in vitro experiments, as evidenced by Figures 4 and 5. However, it is noticeable that gp91phox deficiency did not completely block the HMGB1-induced inflammasome activation, caspase-1 cleavage and IL-1β release as shown in Figure 4B, as well as the association between TXNIP and NLRP3 as shown in Figure 5. These observations suggest that NAD(P)H oxidase is a major, but not a single, source of ROS in EC. Nevertheless, the importance of NAD(P)H oxidase in the development of acute lung injury has been reported with the evidence that either NAD(P)H oxidase inhibitor or genetic deletion of NAD(P)H oxidase components significantly decreases lung inflammation and damage in a setting of HS or sepsis (5, 31, 57–59). We have recently reported a mechanism of EC NAD(P)H oxidase activation by HS/R (22). We showed that HMGB1 is a key factor mediating HS/R-induced EC NAD(P)H oxidase activation, and both Rac1 and p38 MAPK are involved in the signaling pathway (22). The present study also revealed an important role for PMN NAD(P)H oxidase in enhancing inflammasome activation, which is believed to result from an enhanced activation of EC NAD(P)H oxidase by PMN-derived ROS. In a recent report, we also showed that ROS derived from PMN NAD(P)H oxidase are an important mediator in amplifying EC NAD(P)H oxidase activation, and this process is Rac1-dependent but p38 MAPK-independent (22). In deed, we observed in the present study that oxidants enhanced ROS production in MLVEC in response to HMGB1, as shown in Figure 4C. Regarding the mechanism of HS-induced activation of PMN NAD(P)H oxidase, our previously studies have shown that HMGB1 mediates HS/R activation of MyD88-IRAK4- p38 mitogen-activated protein kinase (p38 MAPK) and MyD88-IRAK4-Akt signaling pathways, and in turn, both of the signaling pathways contribute to the activation of PMN NAD(P)H oxidase (31).
ROS has also been suggested as an activator of the inflammasome (16, 60, 61). A recent report showed that exogenous ROS (H2O2) are responsible for mediating uric acid crystals-induced interaction between TXNIP and NLRP3 and subsequent activation of inflammasome in a monocyte/macrophage model (19). In the present study, however, we elucidated in pulmonary EC how HS/R-induced endogenous ROS initiate inflammasome activation. We found that the association of TXNIP and NLRP3 is ROS-sensitive and plays an important role in mediating inflammasome activation in lung EC. As shown in Figure 5, HS/R and HMGB1 induced an increase in the association of TXNIP with NLRP3, and NAD(P)H oxidase-derived ROS are required for this association, because deficiency of gp91 of NAD(P)H oxidase in EC significantly decreased TXNIP and NLRP3 association. Also, the association of TXNIP and NLRP3 seems essential for the subsequent caspase-1 cleavage and IL-1β secretion, since silencing of TXNIP significantly attenuated HMGB1-induced inflammasome assembly, caspase-1 cleavage, and subsequent secretion of IL-1β (Figure 6). TXNIP is a protein with many functions. Although it was originally defined as a TRX-binding protein that regulates the antioxidant function of TRX, its function is actually more diverse. It has been reported that ROS promote dissociation of TXNIP from TRX, which, therefore, allows TXNIP to associate with NLRP3 (19).
In summary, the present study demonstrates a novel mechanism connecting the insult of HS/R and activation of lung EC NLRP3 inflammasome. HMGB1, acting through TLR4 and a synergistic collaboration from the signaling of TLR2 and RAGE, activates EC NAD(P)H oxidase, in turn, the created ROS promote the association of TXNIP to NLRP3, and subsequent activation of inflammasome and IL-1β secretion. PMN-derived ROS exhibit a role in enhancing EC NAD(P)H oxidase activation, and therefore an amplified inflammasome activation in response to HS/R (Figure 7).
Figure 7. Model of HS/R activation of EC NLRP3 inflammasome.
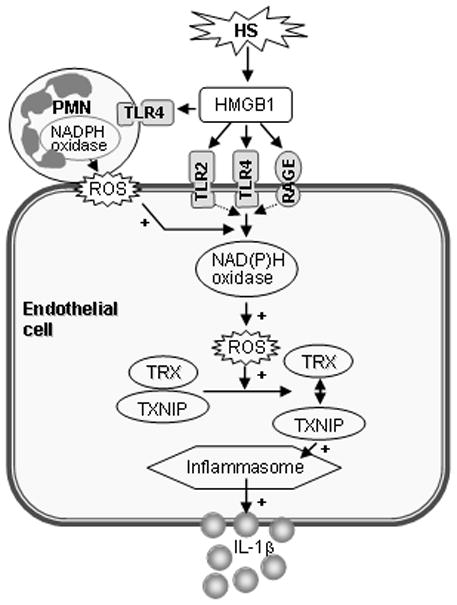
HMGB1, acting through TLR4, and a synergistic collaboration from the signaling of TLR2 and RAGE, activates EC NAD(P)H oxidase, in turn, the created ROS promote the association of TXNIP with NLRP3, and subsequent activation of the inflammasome and IL-1β secretion. PMN-derived ROS play a role in enhancing EC NAD(P)H oxidase activation (+), and therefore help to produce an amplified inflammasome activation in response to HS/R.
Abbreviations used in this paper
- ALI
Acute lung injury
- ASC
apoptosis-associated speck-like protein containing a CARD domain
- DAMPs
damage-associated molecular pattern molecules
- EC
endothelial cells
- HMGB1
high-mobility group box 1
- HS/R
hemorrhagic shock/resuscitation
- MOF
multi-organ failure
- NLRs
NOD-like receptors
- MLVEC
mouse lung vascular endothelial cell
- PMN
polymorphonuclear neutrophils
- ROS
reactive oxygen species
- SIRS
Systemic inflammatory response syndrome
- TRX; TXNIP
thioredoxin-interacting protein
Footnotes
This work was supported by the National Institutes of Health Grant R01-HL-079669 (J.F. and M.A.W.), National Institutes of Health Center Grant P50-GM-53789 (T.R.B. and J.F.) and a VA Merit Award (J.F.).
References
- 1.Zanetti G, Baumgartner JD, Glauser MP. Sepsis and septic shock. Schweiz Med Wochenschr. 1997;127(12):489–99. [PubMed] [Google Scholar]
- 2.Glauser MP, Zanetti G, Baumgartner JD, Cohen J. Septic shock: pathogenesis. Lancet. 1991;338(8769):732–6. doi: 10.1016/0140-6736(91)91452-z. [DOI] [PubMed] [Google Scholar]
- 3.Fowler AA, Hamman RF, Good JT, Benson KN, Baird M, Eberle DJ, Petty TL, Hyers TM. Adult respiratory distress syndrome: risk with common predispositions. Ann Intern Med. 1983;98(5 Pt 1):593–7. doi: 10.7326/0003-4819-98-5-593. [DOI] [PubMed] [Google Scholar]
- 4.Liu Y, Yuan Y, Li Y, Zhang J, Xiao G, Vodovotz Y, Billiar TR, Wilson MA, Fan J. Interacting neuroendocrine and innate and acquired immune pathways regulate neutrophil mobilization from bone marrow following hemorrhagic shock. J Immunol. 2009;182(1):572–80. doi: 10.4049/jimmunol.182.1.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fan J, Li Y, Vodovotz Y, Billiar TR, Wilson MA. Hemorrhagic shock-activated neutrophils augment TLR4 signaling-induced TLR2 upregulation in alveolar macrophages: role in hemorrhage-primed lung inflammation. Am J Physiol Lung Cell Mol Physiol. 2006;290(4):L738–L746. doi: 10.1152/ajplung.00280.2005. [DOI] [PubMed] [Google Scholar]
- 6.Fan J, Marshall JC, Jimenez M, Shek PN, Zagorski J, Rotstein OD. Hemorrhagic shock primes for increased expression of cytokine-induced neutrophil chemoattractant in the lung: role in pulmonary inflammation following lipopolysaccharide. J Immunol. 1998;161(1):440–7. [PubMed] [Google Scholar]
- 7.Orfanos SE, Mavrommati I, Korovesi I, Roussos C. Pulmonary endothelium in acute lung injury: from basic science to the critically ill. Intensive Care Med. 2004;30(9):1702–14. doi: 10.1007/s00134-004-2370-x. [DOI] [PubMed] [Google Scholar]
- 8.Matthay MA, Zimmerman GA, Esmon C, Bhattacharya J, Coller B, Doerschuk CM, Floros J, Gimbrone MA, Jr, Hoffman E, Hubmayr RD, Leppert M, Matalon S, Munford R, Parsons P, Slutsky AS, Tracey KJ, Ward P, Gail DB, Harabin AL. Future research directions in acute lung injury: summary of a National Heart, Lung, and Blood Institute working group. Am J Respir Crit Care Med. 2003;167(7):1027–35. doi: 10.1164/rccm.200208-966WS. [DOI] [PubMed] [Google Scholar]
- 9.Roumen RM, Hendriks T, van der Ven-Jongekrijg J, Nieuwenhuijzen GA, Sauerwein RW, van der Meer JW, Goris RJ. Cytokine patterns in patients after major vascular surgery, hemorrhagic shock, and severe blunt trauma. Relation with subsequent adult respiratory distress syndrome and multiple organ failure. Ann Surg. 1993;218(6):769–76. doi: 10.1097/00000658-199312000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roumen RM, Redl H, Schlag G, Zilow G, Sandtner W, Koller W, Hendriks T, Goris RJ. Inflammatory mediators in relation to the development of multiple organ failure in patients after severe blunt trauma. Crit Care Med. 1995;23(3):474–80. doi: 10.1097/00003246-199503000-00010. [DOI] [PubMed] [Google Scholar]
- 11.Zhu XL, Zellweger R, Zhu XH, Ayala A, Chaudry IH. Cytokine gene expression in splenic macrophages and Kupffer cells following haemorrhage. Cytokine. 1995;7(1):8–14. doi: 10.1006/cyto.1995.1002. [DOI] [PubMed] [Google Scholar]
- 12.Shenkar R, Abraham E. Effects of hemorrhage on cytokine gene transcription. Lymphokine Cytokine Res. 1993;12(4):237–47. [PubMed] [Google Scholar]
- 13.Shenkar R, Coulson WF, Abraham E. Hemorrhage and resuscitation induce alterations in cytokine expression and the development of acute lung injury. Am J Respir Cell Mol Biol. 1994;10(3):290–7. doi: 10.1165/ajrcmb.10.3.8117448. [DOI] [PubMed] [Google Scholar]
- 14.Abraham E, Richmond NJ, Chang YH. Effects of hemorrhage on interleukin-1 production. Circ Shock. 1988;25(1):33–40. [PubMed] [Google Scholar]
- 15.Rao DA, Tracey KJ, Pober JS. IL-1alpha and IL-1beta are endogenous mediators linking cell injury to the adaptive alloimmune response. J Immunol. 2007;179(10):6536–46. doi: 10.4049/jimmunol.179.10.6536. [DOI] [PubMed] [Google Scholar]
- 16.Tschopp J, Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10(3):210–5. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 17.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140(6):821–32. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 18.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–26. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 19.Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11(2):136–40. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 20.Babior BM. The NADPH oxidase of endothelial cells. IUBMB Life. 2000;50(4–5):267–9. doi: 10.1080/713803730. [DOI] [PubMed] [Google Scholar]
- 21.Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86(5):494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 22.Xiang M, Yin L, Li Y, Xiao G, Vodovotz Y, Billiar TR, Wilson MA, Fan J. Hemorrhagic Shock Activates Lung Endothelial NAD(P)H Oxidase via Neutrophil NADPH Oxidase. Am J Respir Cell Mol Biol. 2011;44:333–340. doi: 10.1165/rcmb.2009-0408OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285(5425):248–51. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 24.Yang H, Ochani M, Li J, Qiang X, Tanovic M, Harris HE, Susarla SM, Ulloa L, Wang H, DiRaimo R, Czura CJ, Roth J, Warren HS, Fink MP, Fenton MJ, Andersson U, Tracey KJ. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci U S A. 2004;101(1):296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296(5566):301–5. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 26.Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. 2004;4(6):469–78. doi: 10.1038/nri1372. [DOI] [PubMed] [Google Scholar]
- 27.Zeh HJ, 3rd, Lotze MT. Addicted to death: invasive cancer and the immune response to unscheduled cell death. J Immunother. 2005;28(1):1–9. doi: 10.1097/00002371-200501000-00001. [DOI] [PubMed] [Google Scholar]
- 28.Kim JY, Park JS, Strassheim D, Douglas I, Diaz Del Valle F, Asehnoune K, Mitra S, Kwak SH, Yamada S, Maruyama I, Ishizaka A, Abraham E. HMGB1 contributes to the development of acute lung injury after hemorrhage. Am J Physiol Lung Cell Mol Physiol. 2005 doi: 10.1152/ajplung.00359.2004. [DOI] [PubMed] [Google Scholar]
- 29.Yang R, Harada T, Mollen KP, Prince JM, Levy RM, Englert JA, Gallowitsch-Puerta M, Yang L, Yang H, Tracey KJ, Harbrecht BG, Billiar TR, Fink MP. Anti-HMGB1 neutralizing antibody ameliorates gut barrier dysfunction and improves survival after hemorrhagic shock. Mol Med. 2006;12(4–6):105–14. doi: 10.2119/2006-00010.Yang. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peltz ED, Moore EE, Eckels PC, Damle SS, Tsuruta Y, Johnson JL, Sauaia A, Silliman CC, Banerjee A, Abraham E. Hmgb1 Is Markedly Elevated within Six Hours of Mechanical Trauma in Humans. Shock. 2008 doi: 10.1097/shk.0b013e3181997173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fan J, Li Y, Levy RM, Fan JJ, Hackam DJ, Vodovotz Y, Yang H, Tracey KJ, Billiar TR, Wilson MA. Hemorrhagic shock induces NAD(P)H oxidase activation in neutrophils: role of HMGB1-TLR4 signaling. J Immunol. 2007;178(10):6573–80. doi: 10.4049/jimmunol.178.10.6573. [DOI] [PubMed] [Google Scholar]
- 32.Li Y, Xiang M, Yuan Y, Xiao G, Zhang J, Jiang Y, Vodovotz Y, Billiar TR, Wilson MA, Fan J. Hemorrhagic Shock Augments Lung Endothelial Cell Activation: Role of Temporal Alterations of TLR4 and TLR2. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1670–R1680. doi: 10.1152/ajpregu.00445.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsung A, Hoffman RA, Izuishi K, Critchlow ND, Nakao A, Chan MH, Lotze MT, Geller DA, Billiar TR. Hepatic ischemia/reperfusion injury involves functional TLR4 signaling in nonparenchymal cells. J Immunol. 2005;175(11):7661–8. doi: 10.4049/jimmunol.175.11.7661. [DOI] [PubMed] [Google Scholar]
- 34.Kaczorowski DJ, Mollen KP, Edmonds R, Billiar TR. Early events in the recognition of danger signals after tissue injury. J Leukoc Biol. 2008;83(3):546–52. doi: 10.1189/jlb.0607374. [DOI] [PubMed] [Google Scholar]
- 35.Assier E, Jullien V, Lefort J, Moreau JL, Di Santo JP, Vargaftig BB, Lapa e Silva JR, Theze J. NK cells and polymorphonuclear neutrophils are both critical for IL-2-induced pulmonary vascular leak syndrome. J Immunol. 2004;172(12):7661–8. doi: 10.4049/jimmunol.172.12.7661. [DOI] [PubMed] [Google Scholar]
- 36.Fan J, Frey RS, Malik AB. TLR4 signaling induces TLR2 expression in endothelial cells via neutrophil NADPH oxidase. J Clin Invest. 2003;112(8):1234–43. doi: 10.1172/JCI18696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cotter MJ, Norman KE, Hellewell PG, Ridger VC. A novel method for isolation of neutrophils from murine blood using negative immunomagnetic separation. Am J Pathol. 2001;159(2):473–81. doi: 10.1016/S0002-9440(10)61719-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tiruppathi C, Freichel M, Vogel SM, Paria BC, Mehta D, Flockerzi V, Malik AB. Impairment of store-operated Ca2+ entry in TRPC4(−/−) mice interferes with increase in lung microvascular permeability. Circ Res. 2002;91(1):70–6. doi: 10.1161/01.res.0000023391.40106.a8. [DOI] [PubMed] [Google Scholar]
- 39.Lamkanfi M. Emerging inflammasome effector mechanisms. Nat Rev Immunol. 11(3):213–20. doi: 10.1038/nri2936. [DOI] [PubMed] [Google Scholar]
- 40.Fisher CJ, Jr, Opal SM, Lowry SF, Sadoff JC, LaBrecque JF, Donovan HC, Lookabaugh JL, Lemke J, Pribble JP, Stromatt SC, et al. Role of interleukin-1 and the therapeutic potential of interleukin-1 receptor antagonist in sepsis. Circ Shock. 1994;44(1):1–8. [PubMed] [Google Scholar]
- 41.Fisher CJ, Jr, Dhainaut JF, Opal SM, Pribble JP, Balk RA, Slotman GJ, Iberti TJ, Rackow EC, Shapiro MJ, Greenman RL, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. Jama. 1994;271(23):1836–43. [PubMed] [Google Scholar]
- 42.Opal SM, Fisher CJ, Jr, Dhainaut JF, Vincent JL, Brase R, Lowry SF, Sadoff JC, Slotman GJ, Levy H, Balk RA, Shelly MP, Pribble JP, LaBrecque JF, Lookabaugh J, Donovan H, Dubin H, Baughman R, Norman J, DeMaria E, Matzel K, Abraham E, Seneff M. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: a phase III, randomized, double-blind, placebo-controlled, multicenter trial. The Interleukin-1 Receptor Antagonist Sepsis Investigator Group. Crit Care Med. 1997;25(7):1115–24. doi: 10.1097/00003246-199707000-00010. [DOI] [PubMed] [Google Scholar]
- 43.Abraham E. Why immunomodulatory therapies have not worked in sepsis. Intensive Care Med. 1999;25(6):556–66. doi: 10.1007/s001340050903. [DOI] [PubMed] [Google Scholar]
- 44.Huber TS, Gaines GC, Welborn MB, 3rd, Rosenberg JJ, Seeger JM, Moldawer LL. Anticytokine therapies for acute inflammation and the systemic inflammatory response syndrome: IL-10 and ischemia/reperfusion injury as a new paradigm. Shock. 2000;13(6):425–34. doi: 10.1097/00024382-200006000-00002. [DOI] [PubMed] [Google Scholar]
- 45.Abraham E, Arcaroli J, Carmody A, Wang H, Tracey KJ. HMG-1 as a mediator of acute lung inflammation. J Immunol. 2000;165(6):2950–4. doi: 10.4049/jimmunol.165.6.2950. [DOI] [PubMed] [Google Scholar]
- 46.Erlandsson Harris H, Andersson U. Mini-review: The nuclear protein HMGB1 as a proinflammatory mediator. Eur J Immunol. 2004;34(6):1503–12. doi: 10.1002/eji.200424916. [DOI] [PubMed] [Google Scholar]
- 47.Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, Janson A, Kokkola R, Zhang M, Yang H, Tracey KJ. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med. 2000;192(4):565–70. doi: 10.1084/jem.192.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Czura CJ, Tracey KJ. Targeting high mobility group box 1 as a late-acting mediator of inflammation. Crit Care Med. 2003;31(1 Suppl):S46–50. doi: 10.1097/00003246-200301001-00007. [DOI] [PubMed] [Google Scholar]
- 49.Wang H, Yang H, Czura CJ, Sama AE, Tracey KJ. HMGB1 as a late mediator of lethal systemic inflammation. Am J Respir Crit Care Med. 2001;164(10 Pt 1):1768–73. doi: 10.1164/ajrccm.164.10.2106117. [DOI] [PubMed] [Google Scholar]
- 50.Orlova VV, Choi EY, Xie C, Chavakis E, Bierhaus A, Ihanus E, Ballantyne CM, Gahmberg CG, Bianchi ME, Nawroth PP, Chavakis T. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. Embo J. 2007;26(4):1129–39. doi: 10.1038/sj.emboj.7601552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, Nagashima M, Lundh ER, Vijay S, Nitecki D, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem. 1995;270(43):25752–61. doi: 10.1074/jbc.270.43.25752. [DOI] [PubMed] [Google Scholar]
- 52.Huttunen HJ, Fages C, Rauvala H. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J Biol Chem. 1999;274(28):19919–24. doi: 10.1074/jbc.274.28.19919. [DOI] [PubMed] [Google Scholar]
- 53.Huttunen HJ, Fages C, Kuja-Panula J, Ridley AJ, Rauvala H. Receptor for advanced glycation end products-binding COOH-terminal motif of amphoterin inhibits invasive migration and metastasis. Cancer Res. 2002;62(16):4805–11. [PubMed] [Google Scholar]
- 54.Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279(9):7370–7. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 55.Park JS, Gamboni-Robertson F, He Q, Svetkauskaite D, Kim JY, Strassheim D, Sohn JW, Yamada S, Maruyama I, Banerjee A, Ishizaka A, Abraham E. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am J Physiol Cell Physiol. 2006;290(3):C917–24. doi: 10.1152/ajpcell.00401.2005. [DOI] [PubMed] [Google Scholar]
- 56.Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, Yang H, Li J, Tracey KJ, Geller DA, Billiar TR. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005;201(7):1135–43. doi: 10.1084/jem.20042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fan J, Li Y, Vodovotz Y, Billiar TR, Wilson MA. Neutrophil NAD(P)H oxidase is required for hemorrhagic shock-enhanced TLR2 up-regulation in alveolar macrophages in response to LPS. Shock. 2007;28(2):213–8. doi: 10.1097/shk.0b013e318033ec9d. [DOI] [PubMed] [Google Scholar]
- 58.Impellizzeri D, Esposito E, Mazzon E, Paterniti I, Di Paola R, Bramanti P, Cuzzocrea S. Effect of apocynin, a NADPH oxidase inhibitor, on acute lung inflammation. Biochem Pharmacol. 2010;81(5):636–48. doi: 10.1016/j.bcp.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 59.Al-Mehdi AB, Zhao G, Dodia C, Tozawa K, Costa K, Muzykantov V, Ross C, Blecha F, Dinauer M, Fisher AB. Endothelial NADPH oxidase as the source of oxidants in lungs exposed to ischemia or high K+ Circ Res. 1998;83(7):730–7. doi: 10.1161/01.res.83.7.730. [DOI] [PubMed] [Google Scholar]
- 60.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320(5876):674–7. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007;14(9):1583–9. doi: 10.1038/sj.cdd.4402195. [DOI] [PubMed] [Google Scholar]