

Abstract
Interferon regulatory factor 7 (IRF7) is a potent transcription factor of type I interferons (IFNs) and IFN stimulated genes (ISGs) and is known as the master regulator of type I IFN-dependent immune responses. Because excessive responses could harm the host, IRF7 itself is delicately regulated at the transcriptional, translational, and posttranslational levels. Modification of IRF7 by small ubiquitin-related modifiers (SUMOs) has been shown to regulate IFN expression and antiviral responses negatively, but the specific E3 ligase needed for IRF7 SUMOylation has remained unknown. As reported here, we have identified the tripartite motif–containing (TRIM) protein 28 (TRIM28) as a binding partner of IRF7. We have demonstrated that TRIM28 also interacts with the SUMO E2 enzyme and increases SUMOylation of IRF7 both in vivo and in vitro, suggesting it acts as a SUMO E3 ligase of IRF7. Unlike the common SUMO E3 ligase, protein inhibitor of activated STAT 1(PIAS1), the E3 activity of TRIM28 is specific to IRF7, because it has little effect on IRF7’s close relative IRF3. TRIM28 is therefore, so far as we know, the first IRF7-specific SUMO E3 reported. TRIM28-mediated SUMOylation of IRF7 is increased during viral infection, and SUMOylation of transcription factors usually results in transcriptional repression. Overexpression of TRIM28 therefore inhibits IRF7 transactivation activity, whereas knockdown of TRIM28 has the opposite effect and potentiates IFN production and antiviral responses. Collectively, our results suggest that TRIM28 is a specific SUMO E3 ligase and negative regulator of IRF7.
Interferons (IFNs) are the central components of host innate immune responses to viral infections; their expression is regulated by transcriptional interferon regulatory factors (IRFs), particularly IRF3 and IRF7. Upon viral infection, virus-specific pathogen-associated molecular patterns (PAMPs) are recognized by host pathogen-recognition receptors (PRRs), such as Toll-like receptor (TLR) and RIG-I like–receptor (RLR), that initiate a series of intracellular signaling events, leading to phosphorylation, dimerization, nuclear accumulation of IRF3 and IRF7, and ultimately stimulation of IFN gene transcription (1-5). IRF3 is expressed ubiquitously and constitutively and is involved in early responses to viral infection, whereas IRF7 is expressed at low levels in most cell types except cells of lymphoid origin, but its expression is up-regulated by IFNs and viral infections (1, 6, 7). Studies with knockout mice have revealed that IRF7 is indispensible for induction of type I IFNs in most cell types and thus regarded as the master regulator of type I IFN-dependent responses (2, 3). The critical role of IRF7 in controlling IFN induction is supported by the finding that a variety of viruses encode proteins directed at IRF7 as counter measures to disarm the IFN-dependent host antiviral responses. For example, the open reading frame 45 (ORF45) of Kaposi’s sarcoma–associated herpesvirus (KSHV) inhibits IRF7 by blocking its virus-induced phosphorylation and nuclear translocation; RTA of KSHV acts as a ubiquitin ligase to promote ubiquitination and degradation of IRF7; VP35 of Ebola Zaire virus promotes SUMOylation of IRF7 and suppresses its transactivation activity (8-13).
Although the induction of IFNs plays a pivotal role in host immune defenses against viral infection, uncontrolled and excessive IFN responses would harm the host and have been implicated in autoimmune and other diseases (14, 15). Hosts have therefore evolved elaborate mechanisms involving diverse components of the signaling pathways to control the strength and duration of IFN responses. For examples, A20 negatively regulates RIG-I activity, NLRX1 interacts with MAVS and inhibits MAVS-mediated IFN production, RNF5 works as a ubiquitin E3 ligase to promote MITA degradation, IRAK-M impairs TLR signaling by blocking the formation of IRAK-TRAF6 complex, and ATF4 links integrated cellular stress responses and innate immune responses to down-regulate IRF7 and IFN expressions (16-20). The IRFs themselves are also subjected to multiple layers of regulation, including transcriptional and translational (21, 22) as well as posttranslational modifications and/or associations with other proteins, including phosphorylation (23, 24), acetylation (25), ubiquitination (9, 26-28), and SUMOylation of IRF7 and IRF3 (29).
SUMOylation, a posttranslational conjugation of small ubiquitin-like modifiers (SUMO)s to lysine residues of target protein substrates, has emerged as a central mechanism in modulating cellular functions (30-32). In mammals, at least three SUMO isoforms are known: SUMO1, SUMO2, and SUMO3 (SUMO2 and SUMO3 are 96% identical). Like ubiquitination, SUMOylation requires three-step enzymatic reactions involving the activating enzyme E1 (SAE1/SAE2), conjugating enzyme E2 (Ubc9), and ligase E3 such as PIAS-family proteins (33, 34). SUMOylation usually occurs within the consensus motif, ωKX(E/D) (ω is a hydrophobic residue, X is any residue, and K is the lysine conjugated to SUMO), but exceptions occur (35). SUMOylation of transcription factors and cofactors usually results in transcriptional repression (30, 31, 36, 37). Indeed, we recently found that SUMOylation of IRF7 and IRF3 suppresses their transactivation activities and thus IFN induction (29).
The tripartite motif–containing (TRIM) protein family has more than 60 members in humans. Each member protein characteristically contains a RING domain, one or two B-box domains (cysteine/histidine-rich motifs), and a helical coiled-coil domain in the N-terminal region (38, 39). Many members of the TRIM family are involved in regulation of innate immune responses. For example, TRIM5α is well known for restricting retrovirus replication (40-42); TRIM25 is a RING-finger E3 ubiquitin ligase of RIG-I and is essential for its activation by K63-linked ubiquitination (43); TRIM21 causes the degradation of IRF3 and IRF7 by K48-linked ubiquitination (26, 44); TRIM30α catalyzes K48-linked ubiquitination of both TAB2 and TAB3 and directs them to degradation, thus inhibiting NF-κB activation (45); TRIM19, also known as promyelocytic leukemia protein (PML), is the key constituent protein of the distinct intranuclear PML body (also known as ND10) and is involved in inhibition of a wide range of RNA and DNA viruses (46).
TRIM28, also known as KRAB-associated protein 1 (KAP1) and transcription intermediary factor 1β (TIF1β), has been reported to be a transcriptional corepressor for some transcription factors, in particular for the Krüppel-associated box domain–containing zinc-finger transcription factors (KRAB-ZFPs) (47, 48). TRIM28 interacts with heterochromatin protein 1 (HP1) and is a component of several chromatin-remodeling complexes such as histone methyltransferase SET domain bifurcated 1 (SETDB1), the nucleosome-remodeling and histone-deacetylation (NuRD) complex, and the nuclear-receptor corepressor complex 1 (N-CoR1) (49, 50). TRIM28-mediated corepression relies on the plant homeodomain and bromodomain in the C-terminal region (50). Besides functioning as a transcriptional cofactor, TRIM28 has also been shown to be involved in, for example, DNA double-strand break repair (51), restricting retrovirus replication (52), and regulation of self-renewal of embryonic stem cells (53). In an effort to elucidate the mechanism of IRF7 activation, we attempted to identify cellular proteins associated with IRF7. In the work reported here, we found that TRIM28 interacts with IRF7 and increases its SUMOylation both in vivo and in vitro. We provide further evidence that TRIM28 is a specific SUMO E3 ligase of IRF7 and negatively regulates its activity and IFN-based antiviral responses, supporting the expanding roles of TRIM proteins in regulation of innate immunity.
Materials and Methods
Cell culture and reagents
Human embryonic kidney (HEK)293, HEK293T, and human alveolar epithelial A549 (54) cells were cultured in DMEM supplemented with 10% FBS, 2 mM L-glutamine, and antibiotics. The antibodies used in this study were purchased from Sigma (mouse anti-Flag (M2), anti-hemagglutinin (HA), anti-VSV-G, and anti-β-actin), Santa Cruz Biotech (rabbit anti-IRF7), Cell Signaling (rabbit anti-TRIM28), Abcam (mouse anti-SUMO2), and Clonetech (mouse anti-eGFP). Sendai virus was purchased from Charles River. N-ethylmaleimide, 3×Flag peptides, and 3×HA peptides were purchased from Sigma.
Plasmids
Flag-IRF7 constructs of aa1 to 503 (full length), 1 to 135, 1 to 283, 283 to 503, and 283 to 466 were generated by cloning of PCR amplified fragments into pCMV-TAG3 (Stratagene). We purchased human full-length TRIM28 from Origene and subcloned it into pKH3 vector to generate pKH3-TRIM28. DNA fragments of TRIM28 1 to 835 (full length), 1 to 617, 140 to 835, 140 to 617, 1 to 400, 400 to 835, 1 to 140, 140 to 400, 400 to 617, and 617 to 835 were generated by PCR from pKH3-TRIM28 and cloned into pEGFP or pGEX-5X vectors. pEGFP-PIAS1 was generated by subclone from pcDNA 3.1- PIAS1-HA (10) into pEGFP vector. pFlag-Ubc9 has been described previously (55).
Immunoprecipitation
For immunoprecipitation (IP) of Flag-tagged proteins in HEK293T cells, transfected cells were washed with cold PBS and lysed with whole-cell lysis buffer (WCL, 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 1 mM sodium orthovanadate [Na3VO4], 40 mM β-gylcerophosphate, 1 mM sodium fluoride, 10% glycerol, 5 mM EDTA, 5 μg/ml of aprotinin, 5 μg/ml of leupeptin, 5 mM benzamidine, and 1 mM phenylmethylsulfonyl fluoride). Cell lysates were centrifuged at 10,000 × g for 10 min at 4°C and incubated with EZview red anti-Flag M2 beads for 4 h or overnight at 4°C. After washing with lysis buffer twice and TBS (50 mM Tris-HCl, pH 7.4, 150 mM NaCl) three times, the bound proteins were eluted by incubation with 150 μg/ml 3×Flag peptide in TBS for 1 h at 4°C. For IP of endogenous IRF7, cells were washed with cold PBS and lysed in WCL. Cell lysates were centrifuged at 10,000 × g for 10 min at 4°C and precleared with 20 μl prewashed protein G beads for 2 h at 4°C. Then cell lysates were incubated with 3 μg anti-IRF7 antibody or control IgG overnight at 4°C. After incubation with antibody, 50 μl prewashed protein G beads were added to each sample and incubated for another 4 h at 4°C. After washing with lysis buffer twice and TBS three times, the beads were boiled in Laemmli sample buffer.
In vitro translation and GST pull down
Methionine-labeled IRF7 (35S-IRF7) was produced by in vitro translation with the TNT Coupled Reticulocyte Lysate Kit (Promega). GST-tagged proteins were expressed in Escherichia coli BL21 cells and purified with glutathione sepharose beads. Equal amounts of GST and GST-tagged proteins were mixed with 5 μl of the translation product of IRF7 in 0.5 ml buffer (25 mM Tris-Cl, pH7.5, 1 mM EDTA, 150 mM NaCl, 0.1% NP40, and 10% glycerol). After rotation at 4°C for 4 h, 50 μl glutathione beads were added to each mixture and rotated for an additional 30 minutes. The beads were washed extensively, and the bound proteins were eluted and analyzed with SDS-PAGE. The gel was dried and analyzed by phosphorimager.
In vivo SUMOylation assay
Plasmids expressing Flag-IRF7 (7 μg), HA-SUMO2 (7 μg), and GFP-TRIM28 (7 μg) were cotransfected into HEK293T cells (2 × 106 cells in 100 mm dish). Forty-eight h after transfection, the cells were washed with PBS, harvested, and lysed with 150 μl SDS lysis solution (150 mM Tris-Cl, pH 6.8, 5% SDS, 30% glycerol). After brief sonication, the cell lysates were diluted 1:10 with dilution buffer (PBS with 0.5% NP-40, 1 × complete protease inhibitor, and 20 mM freshly dissolved N-ethylmaleimide). The diluted cell lysates were immunoprecipitated with anti-Flag affinity resins. The immunocomplexes were then analyzed by western blot with anti-HA and anti-Flag antibodies.
In vitro SUMOylation assay
SUMO2, E1, and Ubc9 proteins; SUMO conjugation reaction buffer; and Mg-ATP were purchased from Boston Biochem (Cambridge, MA). Flag-IRF7 ID and HA-TRIM28 were transiently overexpressed in HEK293T cells and purified with the affinity resins. For the in vitro SUMOylation assays, SUMO2 (2 μg), E1 (200 ng), Ubc9 (100 ng), Flag-IRF7 ID (1 μg), and HA-TRIM28 (2 μg) were mixed with different combinations and incubated at 30°C for 3 h. The mixtures were then immunoprecipitated with anti-Flag affinity resins, and the IP complexes were analyzed by western blot with anti-SUMO2 and anti-Flag antibodies.
RNA interference
Hairpin-forming oligonucleotides were designed and cloned into RNAi-Ready pSIREN-RetroQ vector (Clontech) according to the manufacturer’s instructions. Four target sequences against human TRIM28 were initially designed, and the most effective one (5′-CCA AGA GCT GGT GGA ATT C-3′) was chosen for further study. Stock retroviruses were packaged in GP2-293 cells and used to infect HEK293 or A549 cells as described previously (56). Selection of the infected cells with 2 μg/ml puromycin produced stable cells for further analysis. A549 cells transduced with retrovirus-delivered small interfering RNAs (siRNAs) against IRF7 or IRF3 have been described previously (16).
Luciferase reporter assays
Luciferase assays were performed essentially as previously described (8, 16). Briefly, HEK293 cells or HEK293T cells seeded in 24-well plates were transfected by luciferase reporter and pRL-TK internal control plasmids with Lipofectamine 2000 (Invitrogen). Eight h after transfection, cells were infected with Sendai viruses (80 hemagglutinin units/well). Dual luciferase assays (Promega) were performed 24 h after transfection. The relative luciferase activity was expressed in arbitrary units, normalization of firefly luciferase activity to Renilla luciferase activity. Data represent the average of three independent experiments, and error bars represent standard deviation.
Plaque assay
Standard plaque assays were used to determine the titers of vesicular stomatitis virus (VSV). Briefly, HeLa cells were infected with 10-fold serially diluted VSV for 1 h. The inoculum was then replaced with DMEM containing 1% methylcellulose. Twenty-four h after infection, the infected cells were fixed in 5% formaldehyde and stained with 0.1% crystal violet. All samples were assayed in duplicate, and the averages are presented.
IFN ELISA
Human IFNα was measured with commercial ELISA kits according to the manufacturer’s protocols (PBL Biomedical Laboratories). The details were described previously (16).
Mass spectrometry analysis
HEK293T cells were transfected with double-tagged Flag-IRF7-HA expression vector and Flag-luciferase-HA as a control. Two days after transfection, cells were lysed in WCL and immunoprecipitated with anti-Flag resin. The immunocomplexes were eluted with 3×Flag peptide. The resultant eluates were further immunoprecipitated with anti-HA resin. The HA-peptide-eluted immunocomplexes were resolved by SDS-PAGE. After staining with colloidal Coomassie blue, distinct protein bands on the gel were excised and subjected to liquid chromatography–tandem mass spectrometry by the proteomic facility at the Wistar Institute as previously described (56-58). Briefly, samples were digested with trypsin in gels and a portion of the peptide digest was injected onto a nanocapillary reverse-phase HPLC coupled to the nanoelectrospray ionization source of an-ion trap mass spectrometer. This mass spectrometer measures peptide masses and then fragments individual peptides to produce MS/MS spectra of fragments that reflect the peptide sequence. The MS/MS spectra are run against a sequence database by the program SEQUEST and associated software packages for identification of the proteins.
Results
TRIM28 interacts with IRF7
To delineate the mechanisms of IRF7 regulation, we attempted to identify cellular proteins associated with IRF7 by tandem affinity purification. We constructed a double-tagged Flag-IRF7-HA expression vector that was found to be functionally comparable to the untagged version in transient reporter assays (data not shown). We expressed the Flag-IRF7-HA and Flag-luciferase-HA as a control in HEK293T cells and performed sequential IP using anti-Flag and anti-HA affinity resins. The IP complexes were resolved by SDS-PAGE; distinct protein bands were excised and analyzed by LC-MS/MS as previously described (56, 58). A 100-kDa protein was identified as TRIM28/KAP1/TIF1β, and its identity was confirmed by western blot (Fig. 1A). The interaction between ectopically expressed Flag-IRF7 and HA-TRIM28 was confirmed by reciprocal co-IP assays (data not shown). Moreover, the interaction between endogenous TRIM28 and IRF7 was detected in Sendai virus–infected A549 cells that express an appreciable level of IRF7 (Fig. 1B).
FIGURE 1.
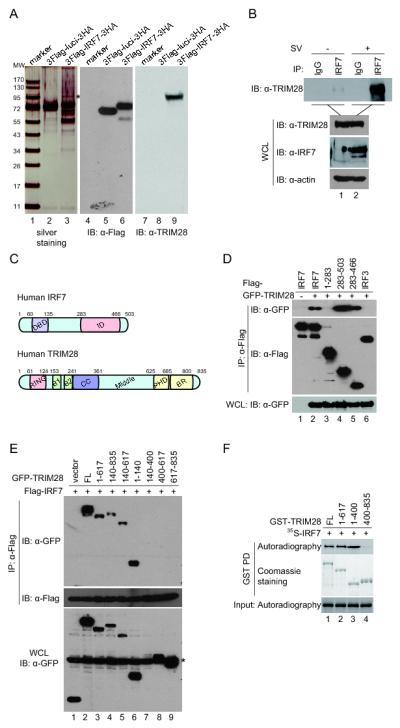
TRIM28 interacts with IRF7. (A) Identification of TRIM28 as an interferon-regulatory factor 7 (IRF7) binding partner. HEK293T cells were transfected with plasmids expressing double-tagged Flag-IRF7-HA and Flag-luciferase-HA as control. The whole-cell lysates were immunoprecipitated with anti-Flag and anti-hemagglutinin (anti-HA) resins sequentially. The eluted IP complexes were analyzed by SDS-PAGE and visualized by silver staining (left panel). The 100-kDa protein band (marked by an asterisk) was identified as tripartite motif–containing (TRIM) protein 28 (TRIM28) by liquid chromatography–tandem mass spectrometry, and its identity was confirmed by western blot. (B) Interaction of endogenous TRIM28 with IRF7. A549 cells were left uninfected or infected with Sendai virus for 24 h. The cell lysates were immunoprecipitated with anti-IRF7 or control serum. The immunoprecipitation (IP) complexes and cell lysates were analyzed by western blot with the antibodies indicated. (C) Schematic representation of domains of human IRF7 and TRIM28. DBD, DNA bind domain; ID, internal inhibitory domain; RING, RING-finger domain; B1 and B2, B-box 1 and B-box 2; CC, coiled-coil domain; Middle, middle region; PHD, plant homeodomain; BR, Bromodomain. (D) Association of TRIM28 with IRF7 ID domain. HEK293T cells were transfected with plasmids expressing GFP-tagged TRIM28 and full length IRF7 or truncation mutants. The cell lysates were immunoprecipitated with anti-Flag affinity resins. The IP complexes and lysates were analyzed by western blot with the indicated antibodies. (E) Association of IRF7 with the RING-finger domain, two B-box domains, and coiled-coil domain of TRIM28. HEK293T cells were transfected with plasmids expressing Flag-IRF7 with full length TRIM28 or truncation mutants. Immunoprecipitation experiments were performed similarly as in (D). The asterisk marks a nonspecific band. (F) Direct association of IRF7 with TRIM28 in vitro. GST-TRIM28 full length or its truncated fusion proteins bound on the glutathione beads were incubated with 35S-labeled full-length IRF7 for 30 min at 4°C. After extensive washes, the bound proteins were resolved by SDS-PAGE and detected by autoradiography. The input GST proteins were revealed by Coomassie staining. The mapping experiments have been repeated at least three times, and representative results are shown.
Mapping the binding domains of IRF7 and TRIM28 revealed that the C-terminal half of IRF7, particularly the aa283–466 region, known as the internal inhibitory domain (ID), bound to TRIM28 as effectively as the full-length IRF7, whereas the N-terminal half, aa1–283, did not bind (Fig. 1C, 1D). The binding is specific because TRIM28 bound to IRF7 but not to IRF3 under the same conditions (Fig. 1D, compare lane 6 and lane 2). TRIM28 has the characteristic tripartite motif, namely, a RING-finger domain, two B-box domains, and a coiled-coil domain (RBCC) in the N-terminal half and a middle region, a plant homeodomain (PHD), and a bromodomain (BR) in the C-terminal half (Fig. 1C). Mapping with a series of truncation mutants revealed that the aa1–617, aa140–835, aa140–617, and aa1–140 domains bound to the IRF7 ID domain, whereas the aa400–617 and aa617–835 did not (Fig. 1E), suggesting that the RING domain itself and the region encompassing the B-box and coiled-coil domain bind to IRF7. Collectively, the results indicate that the two proteins interact with each other through the RBCC region (aa1–400) of TRIM28 and the ID domain (aa283–466) of IRF7.
To determine whether TRIM28 binds to IRF7 directly, we determined whether in vitro translated IRF7 binds to GST-TRIM28 fusion protein using GST pull-down assays. Consistent with the results of co-IP assays, the full-length TRIM28 and the N-terminal RBCC domain (aa1–400) bound to IRF7, but the C-terminal did not (Fig. 1F). Together, these data demonstrated that TRIM28 interacts with IRF7.
TRIM28 specifically increases SUMOylation of IRF7
Because TRIM28 has been shown to have SUMO E3 ligase activity (55, 59), we determined whether TRIM28 affects SUMOylation of IRF7. SUMOylated IRF7 is detectable in cells cotransfected with wild-type SUMO1 or SUMO2 but not in cells transfected with conjugation-deficient SUMO G/A mutants (Fig. 2A). As shown in Fig. 2B, TRIM28 increased SUMOylation of IRF7 by both SUMO1 and SUMO2 (Fig. 2B). Moreover, TRIM28 is specific for IRF7 and has little effect on the closely related IRF3 (Fig. 2C, compare lanes 4 and 3 and lanes 6 and 5), unlike the general SUMO E3 ligase PIAS1, which increases SUMOylation of IRF7 and IRF3 equally (10). The main SUMOylation region in human IRF7 is located in the ID domain (Fig. 2D). We found that TRIM28 and PIAS1 induce SUMOylation of the IRF7 ID domain comparably (Fig. 2E). The ID domain contains several lysine residues predicted by SUMOplot to be possible SUMOylation sites (60). We introduced K to R mutations into these residues and found that the K444/446R mutation abolished the SUMOylation, but other mutations had little or no effect (Fig. 2F), suggesting that K444 and K446 are the critical sites of TRIM28-mediated IRF7 SUMOylation.
FIGURE 2.

TRIM28 increases the SUMOylation of IRF7 in cells. (A) IRF7 was SUMOylated in cells. HEK293T cells were transfected with plasmids expressing Flag-IRF7 plus HA-SUMOs or conjugation-deficient (G/A) mutants. Forty-eight h after transfection, cells were lysed in buffer with high concentration of SDS, diluted, and then immunoprecipitated with anti-Flag affinity resins. The IP complexes were analyzed by western blot with anti-HA antibody, which detected SUMOylated protein, about 20 kDa larger than the unmodified one. We also probed the IP complexes and cell lysates with the indicated antibodies to reveal protein input and overall SUMOylation of cellular proteins. (B) TRIM28 promoted SUMOylation of IRF7. HEK293T cells were transfected with Flag-IRF7, HA-SUMO1, or HA-SUMO2 in the presence or absence of GFP-TRIM28–expression plasmids. The cell lysates were analyzed as described above for detection of the SUMOylated IRF7. (C) TRIM28 specifically increased SUMOylation of IRF7 but not that of IRF3. HEK293T cells were transfected with HA-SUMO2, Flag-IRF7, or Flag-IRF3 in the presence or absence of GFP-TRIM28–expression plasmids. SUMOylated IRF7 or IRF3 were detected as described above. (D) The TRIM28-mediated SUMOylation sites were located mainly in the ID domain of IRF7. HEK293T cells were transfected with HA-SUMO2, full-length Flag-IRF7, or its truncated mutants in the presence or absence of GFP-TRIM28–expression plasmids. SUMOylation assays were performed as described above. (E) TRIM28 induced SUMOylation of IRF7 as efficiently as did PIAS1. HEK293T cells were transfected with Flag-IRF7 ID (aa283–466), HA-SUMO2 in the presence of GFP-TRIM28, GFP-PIAS1, or empty vector–control plasmids. The experiments were performed as above for detection of the SUMOylated IRF7. (F) Identification of the major SUMOylation sites of the IRF7 ID domain. HEK293T cells were transfected with plasmids expressing HA-SUMO2, GFP-TRIM28, and Flag-IRF7 ID or K-to-R substitution mutants. The assays were performed as described above for detection of SUMOylation of IRF7. All the experiments have been repeated at least three times and representative results are shown.
TRIM28 is a RING domain–dependent SUMO E3 ligase of IRF7
The PHD domain of TRIM28 has been shown to possess SUMO E3 activity and promote the intramolecular SUMOylation of the molecule (55). We next determined whether the same domain is involved in IRF7 SUMOylation. As shown in Fig. 3A, expression of the full-length TRIM28 markedly increased the level of SUMOylation of IRF7; surprisingly, deletion of the C-terminal PHD domain had no effect on the increase, but deletion of the N-terminal RING domain abolished the increase (Fig. 3A), suggesting that the RING rather than the PHD domain is required for the specific increase of IRF7 SUMOylation by TRIM28. We noticed that TRIM28 increased the overall cellular SUMOylation, which also depends on the RING domain (Fig. 3A, middle panel, lanes 3, 4, and 5). The minimal region of TRIM28 for increasing IRF7 and overall cellular SUMOylation appears to be aa1–617, which includes the RBCC and the middle domains (Fig. 3A, lane 4). Expression of the C-terminal half of TRIM28 aa 400–835 resulted in distinct pattern of overall SUMO signals (Fig. 3A, middle panel, lane 7), presumably representing the intramolecular SUMOylation mediated by the PHD domain as reported recently (55). This mutant had little effect on the IRF7-specific SUMOylation, however (Fig. 3A, lane 7).
FIGURE 3.
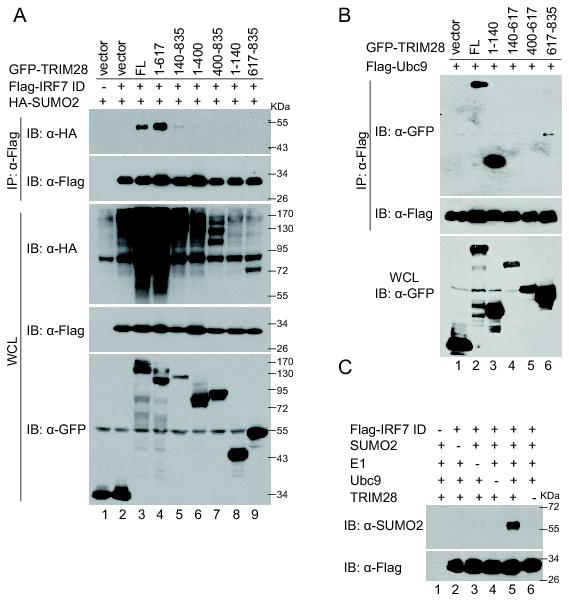
TRIM28 is a SUMO E3 ligase of IRF7. (A) The RING-finger domain of TRIM28 mediated the SUMOylation of IRF7. HEK293T cells were transfected with Flag-IRF7 ID, HA-SUMO2 in the presence of GFP-TRIM28, or its truncated-mutant expression plasmids. The experiments were performed as described in Fig. 2A for detection of SUMOylated IRF7. (B) TRIM28 interacted with Ubc9 mainly through the RING domain. HEK293T cells were transfected with Flag-Ubc9 and GFP-tagged TRIM28 full length and truncation mutants. The cell lysates were immunoprecipitated with anti-Flag affinity resins. The IP complexes and the input cell lysates were analyzed by western blot with antibodies as indicated. (C) TRIM28 catalyzed SUMOylation of IRF7 in vitro. Purified proteins SUMO2 (2 μg), E1 (200 ng), Ubc9 (100 ng), Flag-IRF7 ID (1 μg), and HA-TRIM28 (2 μg) were mixed with the indicated combinations and incubated at 30°C for 3 h. The mixtures were immunoprecipitated with anti-Flag resins, and the IP complexes were then immunoblotted with anti-SUMO2 and anti-Flag. The experiments were repeated at least twice, and representative results are shown.
A SUMO ligase is defined by its ability to bind to E2 Ubc9 and substrates and to increase SUMOylation of the substrate both in vivo and in vitro (61). We have demonstrated that TRIM28 interacts with IRF7 (Fig. 1). Co-IP assays revealed that TRIM28 also interacts with Ubc9, mainly through the RING domain (Fig. 3B), whereas the PHD itself interacts weakly with Ubc9, in agreement with previous reports (55). As shown in Fig. 3C, we found that purified HA-TRIM28 greatly increased the conjugation of SUMO2 to Flag-IRF7 ID domain. The reaction depends on E1, E2, and SUMO molecules (Fig. 3C). Together, these data support the conclusion that TRIM28 is a SUMO E3 ligase of IRF7.
TRIM28-mediated IRF7 SUMOylation is increased during viral infections
SUMOylation of IRF7 and IRF3 has been shown to increase after viral infection (62). We next determined how TRIM28-mediated IRF7 SUMOylation reacts to viral infection. As shown in Fig. 4A, the basal level of IRF7 SUMOylation was low and was increased by a Sendai-virus infection (compare lanes 3 and 4 to lane 2, top panel). Ectopic expression of TRIM28 increased SUMOylation of IRF7 drastically (compare lanes 5, 6, and 7 to lanes 2, 3, and 4, respectively) but increased that of IRF3 (compare lanes 11, 12, and 13 to lanes 8, 9, and 10, respectively) only moderately in the presence of Sendai-virus infection. In the absence of Sendai-virus infection, expression of TRIM28 increased SUMOylation of IRF7 but not IRF3 (compare lanes 5 and 2 and lanes 11 and 8), confirming the specificity to IRF7 and also implying that other SUMO E3s are responsible for IRF3. Furthermore, we found that the HA-TRIM28 purified from Sendai-virus–infected cells showed greater E3 ligase activity toward IRF7 in the in vitro SUMO assays (Fig. 4B). These results demonstrated that TRIM28 increases IRF7 SUMOylation during viral infections.
FIGURE 4.
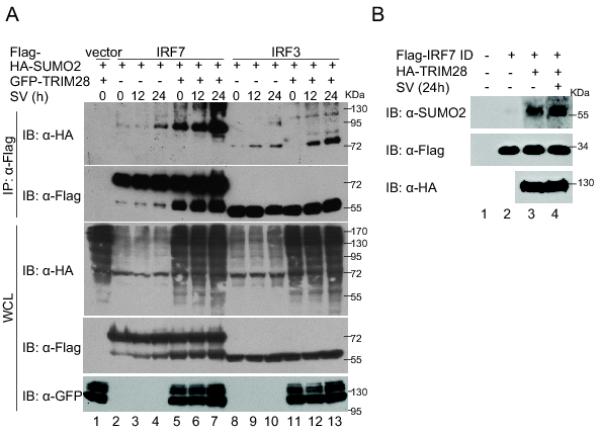
TRIM28-mediated IRF7 SUMOylation is increased during viral infections. (A) TRIM28 specifically increased virus-induced SUMOylation of IRF7 but not that of IRF3. HEK293T cells were transfected with indicated plasmids (each 7 μg) and infected with Sendai virus for different lengths of time before cell harvest. Forty-eight h after transfection, in vivo SUMOylation assays were performed, similar to those described in Fig. 2. (B) Sendai-virus infection increased the SUMO E3 activity of TRIM28. HEK293T cells were transfected with 10 μg of HA-TRIM28 plasmid. After 24 h, the transfected cells were infected with Sendai virus or mock-infected for 24 h before cell harvest. HA-TRIM28 proteins were purified from HEK293T cells and used in in vitro SUMOylation assays (2 μg SUMO2, 100 ng E1, 200 ng Ubc9, 1 μg Flag-IRF7 ID, and 2 μg HA-TRIM28). After 3 h incubation at 30°C, the mixtures were immunoprecipitated with anti-Flag affinity resins. The IP complexes were analyzed by immunoblotting with anti-SUMO2 and anti-Flag. All the experiments were repeated at least three times, and representative results are shown.
TRIM28 negatively regulates IRF7
Because SUMOylation negatively regulates IRF7 (29), we next determined the effect of TRIM28 on IRF7 transactivation activity. Luciferase reporter assays indicated that ectopic expression of TRIM28 inhibits IRF7-induced IFNα1, IFNβ, and ISRE promoter activities in a dose-dependent manner (Fig. 5A, 5B, and 5C).
FIGURE 5.

TRIM28 negatively regulated IRF7 activity. (A), (B), and (C) TRIM28 inhibited the transactivation activity of IRF7. HEK293T cells seeded in 24-well plates were transfected with the IFNα1-luc (A), IFNβ-luc (B), or ISRE-luc (C) luciferase promoter reporter (100 ng per well), pRL-TK (10 ng, as an internal control), pCR3.1-IRF7 (20 ng), and increasing amounts of TRIM28-expression plasmids (50 ng, 100 ng, and 250 ng). Eight h after transfection, cells were infected with Sendai virus or left untreated. Dual luciferase assays were performed 16 h after viral infection. (D) TRIM28 RING-finger domain was required for inhibition on IRF7 transactivation activity. HEK293T cells were transfected with IFNα1 promoter reporter (100 ng), pRL-TK (10 ng), pCR3.1-IRF7 (20 ng), and TRIM28 or its truncated-mutants expression plasmids (100 ng). Experiments were performed as in (A). (E) Mutation of the main SUMOylation sites (K444 and K446) potentiated IRF7 transactivation activity. HEK293T cells were transfected with IFNα1 promoter reporter (100 ng), pRL-TK (10 ng), and Flag-IRF7 or its K to R mutants expression plasmids (20 ng). Luciferase assays were performed as described above. Data from luciferase experiments represent the average of at least three independent experiments, and error bars represent standard deviation.
TRIM28 (KAP-1) is also known as a corepressor, particularly for KAB-containing zinc-finger transcription factors (50). The transcriptional suppression by TRIM28 relies on the C-terminal PHD and BR domains that recruit HP1 and repression complexes such as SETDB1, NuRD, and N-CoR1 (49, 50). We found that deletion of the PHD and BR domain (aa1–617) had little effect on the inhibition of IRF7 by TRIM28 but that further deletion of the RING domain (aa140–617) abolished the inhibition (Fig. 5D). Deletion of RING-finger domain from the full-length TRIM28 (aa140–835) also compromised the inhibition, suggesting that the RING domain–mediated SUMOylation of IRF7 is involved in inhibition and that this inhibitory activity is separable from the corepressor function of TRIM28 that requires the PHD and BR domains (Fig. 5D). Furthermore, we found that K-to-R mutation of SUMOylation sites in the IRF7 ID domain by TRIM28 led to an increase in IRF7 transactivation activity (Fig. 5E). These results indicated that TRIM28 RING-finger–domain mediated SUMOylation negatively regulates IRF7 activity.
Knockdown of TRIM28 potentiates IFN expression and antiviral responses
To determine the effect of endogenous TRIM28 on virus-induced IFN induction, we knocked down TRIM28 expression by siRNAs. Reduction of TRIM28 expression (Fig. 6A, lower panel, compare lanes 3, 4 to lanes 1, 2) resulted in lower level of SUMOylation of IRF7 (Fig. 6A, upper panel, compare lane 4 to 2). Luciferase reporter assays revealed that knockdown of TRIM28 correspondingly potentiated IRF7 transactivation (Fig. 6B and 6C). Mutation of the SUMOylation sites (K444 and K446) increased IRF7 transactivation activity in the control cells, but the increase was no longer apparent in cells in which TRIM28 was knocked down (Fig. 6D), suggesting that TRIM28 contributes to inhibition of IRF7 by SUMOylation on K444 and K446. Although IRF7 has been shown to be the master regulator of induction of type I IFNs in mice, because knockout of IRF7 but not of IRF3 abolishes expression of IFN genes (2), whether the same is true in human cells remained to be determined. As shown in Figure 6E, knockdown of IRF7 by siRNAs caused more severe defects in induction of IFN than knockdown of IRF3, suggesting that inactivation of IRF7 had more profound effects on induction of type I IFNs than did inactivation of IRF3. The results were in agreement with those from studies with knockout mice (2). We next knocked down TRIM28 expression in A549 cells, which express detectable amounts of IRF7 (Fig. 6F, lower panel) and found that abolition of TRIM28 expression increased Sendai virus–induced IFNα productions (Fig. 6F, upper panel). Consequently, knockdown of TRIM28 in A549 cells weakened infection of IFN-sensitive VSV (Fig. 6G and 6H). Together, these results suggest that TRIM28 acts as a negative regulator of type I IFNs induction.
FIGURE 6.

Knockdown of TRIM28 potentiated IFN expression and antiviral responses. (A) Knockdown of TRIM28 expression by siRNAs impaired SUMOylation of IRF7. HEK293 cells were transduced with retroviral vectors expressing siTRIM28 or siControl siRNAs. After selection with puromycin (2 μg/ml) for 2 weeks, the stably transduced cells were then transfected with plasmids expressing HA-SUMO2 and Flag-IRF7 for in vivo SUMOylation assays as described in Figure 2. (B) and (C) Knockdown of TRIM28 potentiated transactivation activity of IRF7. HEK293 siControl or HEK293 siTRIM28 cells were transfected with the IFNα1 (B) or IFNβ (C) promoter reporters (100 ng), pRL-TK (10 ng) and IRF7 (20 ng). Eight h after transfection, cells were infected with Sendai virus or left untreated. Dual luciferase assays were performed 16 h after infection. (D) TRIM28 contributed to the inhibition of IRF7 by SUMOylation on K444 and K446. HEK293 siControl cells or siTRIM28 cells were transfected with IFNα1 promoter reporter (100 ng), pRL-TK (10 ng), and Flag-IRF7 or its K to R mutants expression plasmids (20 ng). Luciferase assays were performed as described above. (E) Knockdown of IRF7 had more profound effect on induction of type I IFN than knockdown of IRF3. A549 cells stably transduced with siControl, siIRF7, or siIRF3 siRNAs were infected with Sendai virus for 24 h. The culture medium was then collected and used for measurement of IFNα by ELISA. (F) Knockdown of TRIM28 potentiated IFNα production. A549 siControl or A549 siTRIM28 cells were treated with Sendai virus for 24 h. The culture medium was collected after treatment and used for measurement of IFNα by ELISA. (G) and (H) Knockdown of TRIM28 inhibited VSV replication. A549 siControl and A549 siTRIM28 cells were infected with increasing amounts of VSV. Twenty-four h after infection, cells lysates were tested with anti-VSV-G (E), and cell medium was used for plaque assays (F). Data represent the average of at least three independent experiments, and error bars represent standard deviation.
Discussion
We have identified TRIM28 as an IRF7-binding protein and demonstrated that it specifically increases SUMOylation of IRF7 both in vivo and in vitro. We further confirmed the interaction between TRIM28 and Ubc9. A SUMO E3 ligase is defined by its ability to bind to both the E2 Ubc9 and its substrates and to increase SUMOylation of the substrate both in vivo and in vitro (61). Our data therefore support the conclusion that TRIM28 is a SUMO E3 ligase of IRF7. TRIM28 acts as a SUMO E3 ligase specific to IRF7 and has little effect on the closely related IRF3. This specificity distinguishes it from the general SUMO E3 ligase PIAS1, which displays no preference between IRF7 and IRF3 (10).
Some of the TRIM-family proteins have been shown to function as ubiquitin E3 ligases and to participate in regulation of innate immunity. For example, TRIM25 catalyses K63-linked ubiquitination of RIG-I (43), TRIM21 mediates K48-linked ubiquitination of IRF3 (44), and TRIM30α promotes ubiquitination and degradation of TAB2 and TAB3 (45). In addition, SUMOylation of some TRIM proteins such as PML (TRIM19) has been extensively studied (63-65). Recently, some TRIM proteins, including TRIM28, were reported to act as SUMO E3 ligases whose activities seem to rely on both the RING-finger and the B-box domains (59). Previously, the PHD domain of TRIM28 was reported to act as a SUMO E3 ligase that catalyzes its SUMOylation intramolecularly. Our data provide further evidence that TRIM28 is a SUMO E3 ligase that is specific to IRF7. Interestingly, we found that the C-terminal PHD domain is dispensable but that the N-terminal RING domain is required for the IRF7-specific SUMOylation, suggesting distinct SUMO E3 activities encoded by TRIM28. In agreement with a recent report (59), both the RING-finger and the B-box domains of TRIM28 are indispensible for the SUMO E3 activity. In addition, our data suggest that the coiled-coil domain and middle region are also required for SUMOylation of IRF7. So far as we know, TRIM28 is the first IRF7-specific SUMO E3 to be reported. Up to now, considerably fewer SUMO E3 ligases than ubiquitin E3 ligases have been discovered; the latter number in the hundreds in mammals. Among the known SUMO E3 ligases, some contain a RING or RING-like domain, but the defining features of SUMO E3 ligases remained poorly characterized. We speculate that additional substrate-specific (for example, IRF3-specific) SUMO E3 will be found in the future.
SUMOylation of IRF7 inhibits its transactivation activity and consequently IFN induction. Consistently, we found that overexpression of TRIM28 inhibits whereas depletion of TRIM28 potentiates IRF7 activation and expression of IFN genes. TRIM28-induced SUMOylation of IRF7 is higher after viral infection, reinforcing our previous results that SUMOylation of IRF7 represents a postactivation attenuation mechanism of IFN production. Suppression of IRF7 by TRIM28 is complicated by its intrinsic suppressor activity. TRIM28 (under its alternate name, KAP1) was identified as a transcriptional corepressor through its ability to repress genes by recruiting HP1 and repression complexes such as SETDB1, NuRD, and N-CoR1 (49, 50). The N-terminal RBCC domain of TRIM28 is responsible for interactions with transcriptional factors, whereas the C-terminal PHD and BR are required for suppression of gene expression, and mutations of either the PHD or BR domain compromise its association with the silencing partners and relieve repression (50). We have demonstrated that repression of IRF7 by TRIM28 is mediated by the RING domain–dependent SUMOylation of IRF7 that requires the N-terminal RBCCM domain, an effect distinct from the general repressor activity that requires the PHD and BR domains in the C-terminal region.
That SUMOylation of transcription factors leads to transcriptional suppression has been well established (30, 31, 36, 37). We recently demonstrated that SUMOylation of IRF7 and IRF3 suppresses their transactivation activities and that the general SUMO E3 ligase PIAS1 promotes SUMOylation of IRF7 and IRF3 (10, 29). Although the exact mechanism of SUMOylation-mediated suppression of IRF7 has not been elucidated in detail, we believe that the general mechanisms underlying SUMO-mediated transcriptional suppression are probably applicable to the case of IRF7. In particular, K63-linked ubiquitination on lysines 444, 446, and 452 of (human) IRF7 by TRAF6 is known to be involved in IRF7 activation (27). Differential modification of these sites is expected to alter IRF7 activity. Indeed, K48-linked ubiquitination by KSHV-encoded E3 ligase RTA mediates degradation of IRF7 and transcriptional suppression (9). These residues are also identified as the main sites of SUMOylation (Fig. 2F). Because both ubiquitination and SUMOylation are reversible and conceivably competitive, SUMOylation of these lysine residues would depress IRF7 activation. We found that lysine 444 and lysine 446 are the main sites of SUMOylation in the presence of TRIM28. In agreement with our hypothesis, a K444/446R mutation resulted in a higher transactivation activity.
SUMOylation of IRF7 becomes higher after viral infection, but the underlying mechanism remains poorly understood. We have shown that TRIM28-mediated IRF7 is also increased by viral infections, suggesting that TRIM28 is involved in this process. TRIM28 is a nuclear protein, whereas IRF7 is mostly cytoplasmic before viral infection. Viral infection–induced nuclear accumulation of IRF7, which would cause their interaction to occur more efficiently, should account for a portion of the increase of its SUMOylation. In addition, we found, with in vitro assays, that the E3 ligase activity of TRIM28 was increased by Sendai-virus infection, although how the E3 activity is regulated by viral infection is unclear. Determining how the E3 ligase activity of TRIM28 is regulated during viral infection and possibly modulated by certain viral factors will be interesting.
Besides its functions as transcriptional cofactor and SUMO E3 ligase, TRIM28 has recently been shown to be involved in other functions such as DNA double-strand break repair (51), restriction of retrovirus replication (52), and regulation of self-renewal of embryonic stem cells (53), although the mechanistic details have not yet been elucidated. Also interesting will be elucidating its RING finger–dependent SUMO E3 activity and determining whether IRF7 or other possible substrates are involved in these functions. Our findings, that TRIM28 acts as a specific SUMO E3 ligase of IRF7 and negatively regulates its activity and IFN-based antiviral responses, support the expanding roles of TRIM proteins in regulation of innate immune responses through posttranslational modifications of the critical regulatory components by ubiquitin and ubiquitin-like-molecules.
Acknowledgments
We thank members of the Zhu laboratory for reading the manuscript and for helpful discussion. We also thank Anne B. Thistle at the Florida State University for excellent editorial assistance.
This work was supported by National Institutes of Health grant R01DE016680.
Abbreviations used in this article
- Br
bromodomain
- HA
hemagglutinin
- ID
inhibitory domain
- HEK
Human embryonic kidney
- HP1
heterochromatin protein 1
- IFN
interferon
- IP
immunoprecipitation
- IRF
interferon regulatory factor
- KSHV
Kaposi’s sarcoma-associated herpesvirus
- ORF
open reading frame
- PAMP
pathogen-associated molecular pattern
- PHD
plant homeodomain
- PIAS1
protein inhibitor of activated STAT 1
- PML
promyelocytic leukemia protein
- PRR
pathogen-recognition receptor
- RBCC
a RING-finger domain, two B-box domains, and a coiled-coil domain
- RIG-I
retinoic acid-inducible gene I
- RLR
retinoic acid-inducible gene I–like receptor
- siRNAs
small interfering RNAs
- SUMO
small ubiquitin-related modifier
- TRIM
tripartite motif–containing protein
- VSV
vesicular stomatitis virus
References
- 1.Hiscott J. Triggering the innate antiviral response through IRF-3 activation. J Biol Chem. 2007;282:15325–15329. doi: 10.1074/jbc.R700002200. [DOI] [PubMed] [Google Scholar]
- 2.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 3.Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- 4.Honda K, Taniguchi T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol. 2006;6:644–658. doi: 10.1038/nri1900. [DOI] [PubMed] [Google Scholar]
- 5.Ozato K, Tailor P, Kubota T. The interferon regulatory factor family in host defense: mechanism of action. J Biol Chem. 2007;282:20065–20069. doi: 10.1074/jbc.R700003200. [DOI] [PubMed] [Google Scholar]
- 6.Tamura T, Y. H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol. 2008;26:535–584. doi: 10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- 7.Ning S, Pagano JS, Barber GN. IRF7: activation, regulation, modification and function. Genes Immun. 2011 doi: 10.1038/gene.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu FX, King SM, Smith EJ, Levy DE, Yuan Y. A Kaposi’s sarcoma-associated herpesviral protein inhibits virus-mediated induction of type I interferon by blocking IRF-7 phosphorylation and nuclear accumulation. Proc Natl Acad Sci USA. 2002;99:5573–5578. doi: 10.1073/pnas.082420599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu Y, Wang SE, Hayward GS. The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity. 2005;22:59–70. doi: 10.1016/j.immuni.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 10.Chang TH, Kubota T, Matsuoka M, Jones S, Bradfute SB, Bray M, Ozato K. Ebola Zaire virus blocks type I interferon production by exploiting the host SUMO modification machinery. PLoS Pathog. 2009;5:e1000493. doi: 10.1371/journal.ppat.1000493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buettner N, V. C, Martínez-Sobrido L, Weber F, Waibler Z, Kochs G. Thogoto virus ML protein is a potent inhibitor of the interferon regulatory factor-7 transcription factor. J Gen Virol. 2010;91:220–227. doi: 10.1099/vir.0.015172-0. [DOI] [PubMed] [Google Scholar]
- 12.Wu L, Fossum E, Joo CH, Inn KS, Shin YC, Johannsen E, Hutt-Fletcher LM, Hass J, Jung JU. Epstein-Barr virus LF2: an antagonist to type I interferon. J Virol. 2009;83:1140–1146. doi: 10.1128/JVI.00602-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joo CH, Shin YC, Gack M, Wu L, Levy D, Jung JU. Inhibition of interferon regulatory factor 7 (IRF7)-mediated interferon signal transduction by the Kaposi’s sarcoma-associated herpesvirus viral IRF homolog vIRF3. J Virol. 2007;81:8282–8292. doi: 10.1128/JVI.00235-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sozzani S, Bosisio D, Scarsi M, Tincani A. Type I interferons in systemic autoimmunity. Autoimmunity. 2010;43:196–203. doi: 10.3109/08916930903510872. [DOI] [PubMed] [Google Scholar]
- 15.Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol. 2005;23:307–336. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- 16.Liang Q, Deng H, Sun CW, Townes TM, Zhu F. Negative regulation of IRF7 activation by activating transcription factor 4 suggests a cross-regulation between the IFN responses and the cellular integrated stress responses. J Immunol. 2011;186:1001–1010. doi: 10.4049/jimmunol.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin R, Yang L, Nakhaei P, Sun Q, Sharif-Askari E, Julkunen I, Hiscott J. Negative regulation of the retinoic acid-inducible gene I-induced antiviral state by the ubiquitin-editing protein A20. J Biol Chem. 2006;281:2095–2103. doi: 10.1074/jbc.M510326200. [DOI] [PubMed] [Google Scholar]
- 18.Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, Accavitti-Loper MA, Madden VJ, Sun L, Ye Z, Lich JD, Heise MT, Chen Z, Ting JP. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature. 2008;451:573–577. doi: 10.1038/nature06501. [DOI] [PubMed] [Google Scholar]
- 19.Zhong B, Zhang L, Lei C, Li Y, Mao AP, Yang Y, Wang YY, Zhang XL, Shu HB. The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity. 2009;30:397–407. doi: 10.1016/j.immuni.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 20.Kobayashi K, Hernandez LD, Galan JE, Janeway CA, Jr., Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 21.Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol. 2008;26:535–584. doi: 10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- 22.Colina R, Costa-Mattioli M, Dowling RJ, Jaramillo M, Tai LH, Breitbach CJ, Martineau Y, Larsson O, Rong L, Svitkin YV, Makrigiannis AP, Bell JC, Sonenberg N. Translational control of the innate immune response through IRF-7. Nature. 2008;452:323–328. doi: 10.1038/nature06730. [DOI] [PubMed] [Google Scholar]
- 23.Paz S, Sun Q, Nakhaei P, Romieu-Mourez R, Goubau D, Julkunen I, Lin R, Hiscott J. Induction of IRF-3 and IRF-7 phosphorylation following activation of the RIG-I pathway. Cell Mol Biol (Noisy-le-grand) 2006;52:17–28. [PubMed] [Google Scholar]
- 24.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 25.Caillaud A, Prakash A, Smith E, Masumi A, Hovanessian AG, Levy DE, Marie I. Acetylation of interferon regulatory factor-7 by p300/CREB-binding protein (CBP)-associated factor (PCAF) impairs its DNA binding. J Biol Chem. 2002;277:49417–49421. doi: 10.1074/jbc.M207484200. [DOI] [PubMed] [Google Scholar]
- 26.Higgs R, Lazzari E, Wynne C, Ni Gabhann J, Espinosa A, Wahren-Herlenius M, Jefferies CA. Self protection from anti-viral responses--Ro52 promotes degradation of the transcription factor IRF7 downstream of the viral Toll-Like receptors. PLoS One. 2010;5:e11776. doi: 10.1371/journal.pone.0011776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ning S, Campos AD, Darnay BG, Bentz GL, Pagano JS. TRAF6 and the three C-terminal lysine sites on IRF7 are required for its ubiquitination-mediated activation by the tumor necrosis factor receptor family member latent membrane protein 1. Mol Cell Biol. 2008;28:6536–6546. doi: 10.1128/MCB.00785-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ning S, Pagano JS. The A20 deubiquitinase activity negatively regulates LMP1 activation of IRF7. J Virol. 2010;84:6130–6138. doi: 10.1128/JVI.00364-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kubota T, Matsuoka M, Chang TH, Tailor P, Sasaki T, Tashiro M, Kato A, Ozato K. Virus infection triggers SUMOylation of IRF3 and IRF7, leading to the negative regulation of type I interferon gene expression. J Biol Chem. 2008;283:25660–25670. doi: 10.1074/jbc.M804479200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gill G. Post-translational modification by the small ubiquitin-related modifier SUMO has big effects on transcription factor activity. Curr Opin Genet Dev. 2003;13:108–113. doi: 10.1016/s0959-437x(03)00021-2. [DOI] [PubMed] [Google Scholar]
- 31.Hay RT. SUMO: a history of modification. Mol Cell. 2005;18:1–12. doi: 10.1016/j.molcel.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 32.Johnson ES. Protein modification by SUMO. Annu Rev Biochem. 2004;73:355–382. doi: 10.1146/annurev.biochem.73.011303.074118. [DOI] [PubMed] [Google Scholar]
- 33.Palvimo JJ. PIAS proteins as regulators of small ubiquitin-related modifier (SUMO) modifications and transcription. Biochem Soc Trans. 2007;35:1405–1408. doi: 10.1042/BST0351405. [DOI] [PubMed] [Google Scholar]
- 34.Shuai K, Liu B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat Rev Immunol. 2005;5:593–605. doi: 10.1038/nri1667. [DOI] [PubMed] [Google Scholar]
- 35.Zhu J, Zhu S, Guzzo CM, Ellis NA, Sung KS, Choi CY, Matunis MJ. Small ubiquitin-related modifier (SUMO) binding determines substrate recognition and paralog-selective SUMO modification. J Biol Chem. 2008;283:29405–29415. doi: 10.1074/jbc.M803632200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gill G. Something about SUMO inhibits transcription. Curr Opin Genet Dev. 2005;15:536–541. doi: 10.1016/j.gde.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 37.Garcia-Dominguez M, Reyes JC. SUMO association with repressor complexes, emerging routes for transcriptional control. Biochim Biophys Acta. 2009;1789:451–459. doi: 10.1016/j.bbagrm.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 38.Ozato K, Shin DM, Chang TH, Morse HC., 3rd TRIM family proteins and their emerging roles in innate immunity. Nat Rev Immunol. 2008;8:849–860. doi: 10.1038/nri2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nisole S, Stoye JP, Saib A. TRIM family proteins: retroviral restriction and antiviral defence. Nat Rev Microbiol. 2005;3:799–808. doi: 10.1038/nrmicro1248. [DOI] [PubMed] [Google Scholar]
- 40.Sayah DM, Sokolskaja E, Berthoux L, Luban J. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature. 2004;430:569–573. doi: 10.1038/nature02777. [DOI] [PubMed] [Google Scholar]
- 41.Yap MW, Nisole S, Lynch C, Stoye JP. Trim5alpha protein restricts both HIV-1 and murine leukemia virus. Proc Natl Acad Sci U S A. 2004;101:10786–10791. doi: 10.1073/pnas.0402876101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sawyer SL, Wu LI, Emerman M, Malik HS. Positive selection of primate TRIM5alpha identifies a critical species-specific retroviral restriction domain. Proc Natl Acad Sci U S A. 2005;102:2832–2837. doi: 10.1073/pnas.0409853102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 44.Higgs R, Ni Gabhann J, Ben Larbi N, Breen EP, Fitzgerald KA, Jefferies CA. The E3 ubiquitin ligase Ro52 negatively regulates IFN-beta production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. J Immunol. 2008;181:1780–1786. doi: 10.4049/jimmunol.181.3.1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi M, Deng W, Bi E, Mao K, Ji Y, Lin G, Wu X, Tao Z, Li Z, Cai X, Sun S, Xiang C, Sun B. TRIM30 alpha negatively regulates TLR-mediated NF-kappa B activation by targeting TAB2 and TAB3 for degradation. Nat Immunol. 2008;9:369–377. doi: 10.1038/ni1577. [DOI] [PubMed] [Google Scholar]
- 46.Everett RD, Chelbi-Alix MK. PML and PML nuclear bodies: implications in antiviral defence. Biochimie. 2007;89:819–830. doi: 10.1016/j.biochi.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 47.Moosmann P, Georgiev O, Le Douarin B, Bourquin JP, Schaffner W. Transcriptional repression by RING finger protein TIF1 beta that interacts with the KRAB repressor domain of KOX1. Nucleic Acids Res. 1996;24:4859–4867. doi: 10.1093/nar/24.24.4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Friedman JR, Fredericks WJ, Jensen DE, Speicher DW, Huang XP, Neilson EG, Rauscher FJ., 3rd KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev. 1996;10:2067–2078. doi: 10.1101/gad.10.16.2067. [DOI] [PubMed] [Google Scholar]
- 49.Ayyanathan K, Lechner MS, Bell P, Maul GG, Schultz DC, Yamada Y, Tanaka K, Torigoe K, Rauscher FJ., 3rd Regulated recruitment of HP1 to a euchromatic gene induces mitotically heritable, epigenetic gene silencing: a mammalian cell culture model of gene variegation. Genes Dev. 2003;17:1855–1869. doi: 10.1101/gad.1102803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schultz DC, Friedman JR, Rauscher FJ., 3rd Targeting histone deacetylase complexes via KRAB-zinc finger proteins: the PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2alpha subunit of NuRD. Genes Dev. 2001;15:428–443. doi: 10.1101/gad.869501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Noon AT, Shibata A, Rief N, Lobrich M, Stewart GS, Jeggo PA, Goodarzi AA. 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nat Cell Biol. 2010;12:177–184. doi: 10.1038/ncb2017. [DOI] [PubMed] [Google Scholar]
- 52.Rowe HM, Jakobsson J, Mesnard D, Rougemont J, Reynard S, Aktas T, Maillard PV, Layard-Liesching H, Verp S, Marquis J, Spitz F, Constam DB, Trono D. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature. 2010;463:237–240. doi: 10.1038/nature08674. [DOI] [PubMed] [Google Scholar]
- 53.Hu G, Kim J, Xu Q, Leng Y, Orkin SH, Elledge SJ. A genome-wide RNAi screen identifies a new transcriptional module required for self-renewal. Genes Dev. 2009;23:837–848. doi: 10.1101/gad.1769609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Giard DJ, Aaronson SA, Todaro GJ, Arnstein P, Kersey JH, Dosik H, Parks WP. In vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors. J Natl Cancer Inst. 1973;51:1417–1423. doi: 10.1093/jnci/51.5.1417. [DOI] [PubMed] [Google Scholar]
- 55.Ivanov AV, Peng H, Yurchenko V, Yap KL, Negorev DG, Schultz DC, Psulkowski E, Fredericks WJ, White DE, Maul GG, Sadofsky MJ, Zhou MM, Rauscher FJ., 3rd PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol Cell. 2007;28:823–837. doi: 10.1016/j.molcel.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kuang E, Tang Q, Maul GG, Zhu F. Activation of p90 ribosomal S6 kinase by ORF45 of Kaposi’s sarcoma-associated herpesvirus and its role in viral lytic replication. J Virol. 2008;82:1838–1850. doi: 10.1128/JVI.02119-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu FX, Yuan Y. The ORF45 protein of Kaposi’s sarcoma-associated herpesvirus is associated with purified virions. J Virol. 2003;77:4221–4230. doi: 10.1128/JVI.77.7.4221-4230.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu FX, Chong JM, Wu L, Yuan Y. Virion proteins of Kaposi’s sarcoma-associated herpesvirus. J Virol. 2005;79:800–811. doi: 10.1128/JVI.79.2.800-811.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chu Y, Yang X. SUMO E3 ligase activity of TRIM proteins. Oncogene. 2011;30:1108–1116. doi: 10.1038/onc.2010.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xue Y, Zhou F, Fu C, Xu Y, Yao X. SUMOsp: a web server for sumoylation site prediction. Nucleic Acids Res. 2006;34:W254–257. doi: 10.1093/nar/gkl207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Geiss-Friedlander R, Melchior F. Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol. 2007;8:947–956. doi: 10.1038/nrm2293. [DOI] [PubMed] [Google Scholar]
- 62.Chang TH, Kubota T, Matsuoka M, Jones S, Bradfute SB, Bray M, Ozato K. Ebola Zaire virus blocks type I interferon production by exploiting the host SUMO modification machinery. PLoS Pathogens. 2009;5:e1000493. doi: 10.1371/journal.ppat.1000493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Muller S, Matunis MJ, Dejean A. Conjugation with the ubiquitin-related modifier SUMO-1 regulates the partitioning of PML within the nucleus. EMBO J. 1998;17:61–70. doi: 10.1093/emboj/17.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kamitani T, Kito K, Nguyen HP, Wada H, Fukuda-Kamitani T, Yeh ET. Identification of three major sentrinization sites in PML. J Biol Chem. 1998;273:26675–26682. doi: 10.1074/jbc.273.41.26675. [DOI] [PubMed] [Google Scholar]
- 65.Duprez E, Saurin AJ, Desterro JM, Lallemand-Breitenbach V, Howe K, Boddy MN, Solomon E, de The H, Hay RT, Freemont PS. SUMO-1 modification of the acute promyelocytic leukaemia protein PML: implications for nuclear localisation. J Cell Sci. 1999;112(Pt 3):381–393. doi: 10.1242/jcs.112.3.381. [DOI] [PubMed] [Google Scholar]