

Abstract
This review discusses the application of cellular biology, molecular biophysics, and computational simulation to understand membrane-mediated mechanisms by which oxysterols regulate cholesterol homeostasis. Side-chain oxysterols, which are produced enzymatically in vivo, are physiological regulators of cholesterol homeostasis and primarily serve as cellular signals for excess cholesterol. These oxysterols regulate cholesterol homeostasis through both transcriptional and non-transcriptional pathways; however, many molecular details of their interactions in these pathways are still not well understood. Cholesterol trafficking provides one mechanism for regulation. The current model of cholesterol trafficking regulation is based on the existence of two distinct cholesterol pools in the membrane: a low and a high availability/activity pool. It is proposed that the low availability/activity pool of cholesterol is integrated into tightly packing phospholipids and relatively inaccessible to water or cellular proteins, while the high availability cholesterol pool is more mobile in the membrane and is present in membranes where the phospholipids are not as compressed. Recent results suggest that that oxysterols may promote cholesterol egress from membranes by shifting cholesterol from the low to the high activity pools. Furthermore, molecular simulations suggest a potential mechanism for oxysterol “activation” of cholesterol through its displacement in the membrane. This review discusses these results as well as several other important interactions between oxysterols and cholesterol in cellular and model lipid membranes.
Keywords: Oxysterols, cholesterol, sterols, homeostasis, molecular dynamics
1. Introduction
Membranes are an essential component of cells, serving to partition the inside of the cell from its extracellular environment as well as compartmentalizing different organelles within the cell. The structures of biological membranes are derived primarily from their constituent phospholipids attached to a glycerol backbone. Phospholipids are a diverse group, but all retain a polar or charged headgroup and one or more long hydrophobic tails. This basic structure provides an amphiphilic character that allows self-assembly into stable membrane bilayers.
Membranes not only serve to separate cellular compartments but also to regulate cellular behavior, often through protein-membrane interactions [1]. Membrane influence on protein function has been described for many proteins; e.g., the fluidity and cholesterol composition of the membrane environment regulates the activity of adenylate cyclase [2], membrane structure regulates G-protein activity [3], and phospholipid chain length alters the behavior of diacylglycerol kinase through hydrophobic matching of the membrane to the protein [1]. Membrane structure is also known to govern protein sorting and association. Reversible protein association with membranes to form enzymatically active complexes is controlled in part through the composition and structure of membranes [4]. Integral membrane protein distribution within the cell is regulated by hydrophobic matching of the protein to membranes of different thicknesses [5].
Membrane lipids themselves also serve as signaling molecules: phosphatidylinositols function as messengers in a number of membrane-cytoskeletal interactions [6], and sphingolipids signal in apoptosis, the cell cycle, and differentiation [7, 8]. More common lipids also signal through movement and trafficking. For example, movement of phosphaditylserine marks apoptotic cells [9] and cholesterol depletion alters protein localization and trafficking [10, 11].
Phospholipids are responsible for forming the basic structure of biomembranes, but sterols, while not necessary for eukaryotic membrane structure, modulate membrane structure and are essential for membrane function. All sterols are built around four rings fused in a trans configuration, which provide a rigid, planar, and hydrophobic structure. Decoration of this base steroid ring structure with different functional groups accounts for the diversity among sterol species. In contrast to the wide variety of phospholipid species found in biological membranes, most membranes contain only a single dominant sterol. Cholesterol is the primary sterol found in mammalian cells, and it is required for mammalian cell function and viability. While cholesterol is largely hydrophobic, its hydroxyl group confers an essential amphiphilic character that allows it to readily incorporate into phospholipid bilayers, extensively modulating their structure and behavior.
Cholesterol primarily serves as a structural component of cellular membranes. When incorporated into phospholipid bilayers, cholesterol aligns so that its polar hydroxyl group is near the interface with the aqueous environment while its hydrophobic body is buried in the bilayer [12, 13]. The interaction of cholesterol with neighboring phospholipids alters membrane structure. The alignment and ordering of nearby phospholipid tails causes membrane condensation, decreasing the area of the membrane and increasing the thickness [13]. Cholesterol also broadens the liquid-to-solid phase transition, inducing an intermediate liquid-ordered phase that retains lateral mobility while increasing lipid order [14-16]. These changes result in a mechanically stronger membrane with decreased permeability due to tighter packing among lipids [10, 14].
Because cholesterol is such an important part of mammalian membranes, cells expend significant amounts of energy to control their cholesterol levels through a variety of mechanisms including de novo synthesis, intracellular transfer and storage of cholesterol, and elimination of cholesterol through efflux and metabolic pathways [17, 18]. Orchestration of these cholesterol homeostatic pathways is accomplished through a variety of mechanisms. Oxygenated derivatives of cholesterol (see Figure 1) are produced by either the attack of cholesterol by reactive oxygen species (e.g., 7-ketocholesterol, 7α-hydroxycholesterol, and 7β-hydroxycholesterol) or by enzymatic reactions (e.g., 24(S)-hydroxycholesterol, 25-hydroxycholesterol, and 27-hydroxycholesterol) and play a central role in regulation of cholesterol homeostatic pathways. This review focuses on the importance of oxysterols in understanding cholesterol homeostasis, discusses the role of membranes themselves in oxysterol signaling, and demonstrates how techniques that provide different scales of resolution can improve our understanding of the molecular basis of cellular cholesterol homeostasis.
Figure 1.

An overview of common biological oxysterols, both those produced from attack on cholesterol by reactive oxygen species (7-keto-, 7α-hydroxy-, and 7β-hydroxycholesterol) and those produced enzymatically by the cell (25(S)-hydroxy-, 25-hydroxy-, and 27-hydroxycholesterol).
2. The role of oxysterols in cholesterol homeostasis
The side-chain oxysterols, such as 24-, 25-, and 27-hydroxycholesterol (HC; see Figure 1) are produced enzymatically in vivo and are physiologic regulators of cholesterol homeostasis. These oxysterols principally serve as signals for excess cholesterol in the cell. When cholesterol is in excess, some of that cholesterol is oxygenated by endogenous hydroxylases [19, 20]. Inhibition of oxysterol production through either direct disruption of hydroxylases [21] or disruption of cholesterol trafficking to the hydroxylases [22] interferes with the ability of cells to appropriately respond to elevated levels of cholesterol, indicating that the feedback effects of cholesterol on regulatory pathways are mediated in part through cholesterol conversion into side-chain oxysterols.
2.1. Pathways of homeostasis
Oxysterols regulate cholesterol homeostasis through transcriptional and non-transcriptional mechanisms (see Figure 2), both of which are necessary for tight control of cellular cholesterol levels. Transcriptional regulation allows the cell to efficiently respond to changes in ambient cholesterol concentration by altering the production of new proteins. Modulation of protein levels, however, occurs over a relatively slow timescale (e.g. hours). By contrast, non-transcriptional regulation, which can directly modify the behavior of proteins, allows for a cellular response to changes in cholesterol concentration over a faster time scale (e.g. minutes).
Figure 2.
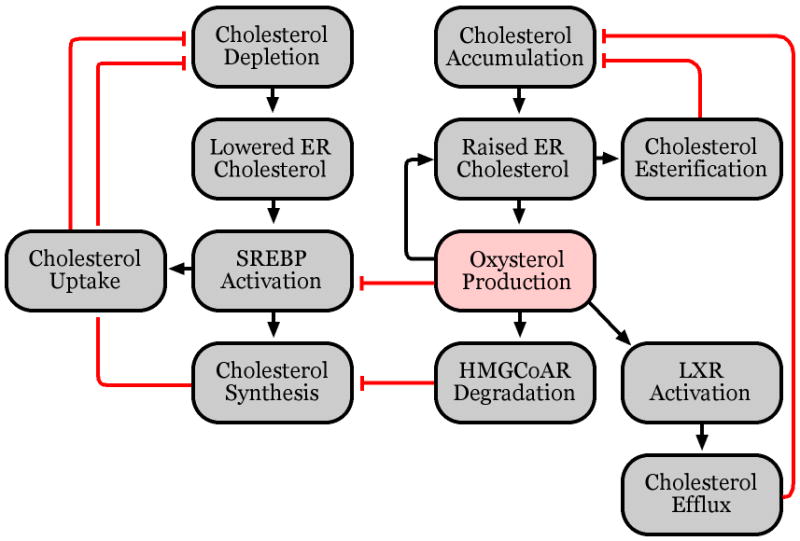
Pathways of cholesterol homeostasis.
Two major pathways have been described in which oxysterols are known to play a role as transcriptional regulators. Side-chain oxysterols are the physiological ligands of the liver X receptors (LXRs) [23, 24]. LXRs up-regulate the expression of a number of genes involved in export of excess cholesterol from the cell, including bile acid synthesis proteins such as Cyp7A1 [25], and the ABCA1 transporter which transfers excess cholesterol to circulating apolipoproteins [26]. Side-chain oxysterols also serve to suppress the maturation of the sterol response element binding-protein (SREBP) family of transcription factors [27, 28]. Activated SREBPs up-regulate the expression of genes involved in cholesterol synthesis and uptake, including HMGCoA reductase, the rate-limiting enzyme in the cholesterol synthesis pathway, and the LDL receptor, which mediates the uptake of cholesterol-rich LDL [17, 29-33]. In response to elevated cholesterol concentrations, oxysterols are produced that suppress the transcription of genes responsible for cholesterol synthesis and uptake while up-regulating the transcription of genes involved in cholesterol export.
Oxysterols can also affect rapid changes in cholesterol levels through post-transcriptional mechanisms. In response to cholesterol loading, cholesterol is rapidly converted to oxysterols, which promote the ubiquitination of HMGCoA reductase and targets it for proteasome-dependent degradation [21, 30, 34, 35]. Oxysterols also regulate cholesterol trafficking between the plasma membrane and internal compartments, most importantly the endoplasmic reticulum (ER) [36]. Most cholesterol regulatory enzymes are present in the ER, so modulation of this trafficking affects the cell’s response to changes in total cholesterol levels. In particular, acyl-CoA cholesterol acyl transferase (ACAT) conjugates excess ER cholesterol with fatty acids to produce cholesteryl esters that are stored in lipid droplets [37]. ACAT-dependent Cholesterol esterification is largely controlled by the ER cholesterol concentration. Thus, oxysterols, which stimulate transfer of cholesterol from the plasma membrane to the ER, indirectly promote cholesterol esterification by ACAT [36, 37]. These effects occur on a much faster time scale than oxysterol-dependent transcriptional regulation through the SREBP and LXR pathways, allowing the cell to quickly shut down cholesterol synthesis and shuttle excess cholesterol to storage compartments.
2.2. Mechanisms of homeostasis
While oxysterols are known to act in these pathways, in many cases, the mechanisms through which they act are less well understood. The most straightforward mechanism of action for oxysterols is activation of the LXR pathway. Side-chain oxysterols were identified as activators of LXRs at physiological concentrations [24]. Activation requires an intact LXR ligand binding domain, and co-incubation of LXR with oxysterol partially protects LXR from proteolytic digestion. Furthermore, activation is oxysterol specific, with minor structural or stereochemical changes causing large differences in activation [24, 38]. This suggests a simple model where oxysterols bind specifically and directly to LXR as ligands, causing conformational changes which result in activation.
Side-chain oxysterol suppression of SREBP activation appears to be more complex [17, 29]. When first synthesized, SREBPs are inactive; they must undergo proteolytic maturation to become active transcription factors. This activation occurs through transport of SREBPs from the ER to the Golgi complex where Golgi-resident proteases subsequently cleave it to produce an active fragment. Transport is facilitated by the SREBP cleavage-activating protein (Scap), which binds to the SREBP regulatory domain to form an SREBP-Scap complex. Scap also contains a sterol sensing domain (SSD), which causes conformational changes in Scap in the presence of cholesterol. When ER cholesterol levels are low, Scap binds to vesicular packaging proteins, initiating the transfer of the complex to the Golgi. Under cholesterol-rich conditions, Scap instead binds to the ER-resident Insig proteins, blocking vesicular transport and SREBP maturation [33].
While cholesterol itself can suppress SREBP activation, side-chain oxysterols are significantly more potent [27, 28]. Cholesterol directly binds to the Scap SSDs, causing a conformational change that induces binding to Insig [33]. Oxysterols also induce Scap/Insig binding, but do not bind Scap [39]. They do, however, interact with Insig, as measured through competitive binding experiments [39]. We have observed that the effects of oxysterols on SREBP suppression are not stereospecific; enantiomeric oxysterols suppress SREBP activation as effectively as natural oxysterols [38]. We propose that the oxysterols do not exert their effects through direct oxysterol-protein interactions but rather via a non-stereospecific mechanism. While this mechanism has not yet been conclusively identified, oxysterols are known to perturb the behavior of membranes, making oxysterol/membrane interactions a promising candidate.
Regulated degradation of HMGCoA reductase acts through a remarkably similar mechanism. Like Scap, HMGCoA reductase contains a SSD that induces conformational changes in the presence of cholesterol [30, 35]. This exposes ubiquitination sites which, once ubiquitinated, target it to the proteasome where it is degraded [30, 35]. The proteolytic inactivation of HMGCoA reductase is dependent on the production of oxysterols [21]. Additionally, ubiquitinase recruitment is dependent on binding of HMGCoA reductase to Insig, the same regulatory protein involved in SREBP regulation [40]. This suggests that oxysterols may act similarly in both pathways through non-stereospecific modulation of Insig interactions with other proteins.
Oxysterol regulation of cholesterol trafficking is even less well understood. Membrane and soluble protein carriers for cholesterol movement within the cell are required to explain the regulation and homeostasis of cholesterol. However, individual pathways, driving forces, and regulatory elemtents are not known for cholesterol trafficking between the plasma membrane and the ER. Cholesterol movement between the two compartments is modulated by oxysterols on a timescale of minutes, suggesting that it is assisted by some active process rather than passively diffusing [41, 42]. If additional cholesterol is directly removed from or added to the plasma membrane, the ER cholesterol concentration responds extremely rapidly. In particular, increases in the plasma membrane cholesterol concentration quickly result in much larger increases in the ER cholesterol concentration: an increase of 20% in the plasma membrane can result in as much as a five-fold increase in ER cholesterol content. This non-linear response suggests an alteration of both transport kinetics and compartment capacity in the regulation of cholesterol subcellular distributions.
The current model of how this trafficking is regulated is based on the hypothesis that there exist two distinct cholesterol pools in the membrane: a low and a high activity pool [41, 42]. It is proposed that the low activity pool consists of cholesterol that is sequestered within the phospholipids and relatively inaccessible to other molecules, while the high activity pool of cholesterol that is more accessible and mobile in the non-condensed phospholipids of the membrane. Distribution between the high and low activity pools is determined by the ability of the phospholipids to condense with cholesterol, which in turn is dependent on the phospholipid composition of the membrane. Thus, raising the plasma membrane cholesterol concentration can saturate the ability of the membrane to accommodate cholesterol in the condensed phospholipids, whereupon excess cholesterol transitions into the high activity pool where it is more available for trafficking to the ER. Measurements of cholesterol availability to external sources such as cholesterol oxidase, cyclodextrin, and perfringolysin O show increases in cholesterol availability that correspond with ER trafficking measurements [42, 43].
This model suggests that oxysterols drive cholesterol trafficking by shifting cholesterol from the low to the high activity pools. The cholesterol availability model may also explain the oxysterol effects on SREBP suppression and HMGCoA reductase degradation. Suppression of SREBP activation and an increase in availability of ER cholesterol to perfringolysin O occurs very abruptly at the same cholesterol concentration, indicating that cholesterol availability may be directly related to signaling in these pathways [43].
3. Oxysterol behavior in model membranes
Cellular models provide a physiologically relevant, intact environment for study of cholesterol homeostasis. While these models allow for the study of complex interactions between pathways required for a complete understanding of cholesterol homeostasis, they can also interfere with a detailed stepwise understanding of individual pathways. To resolve this issue, simpler models are used which mimic relevant parts of the complex system. Cellular studies have suggested that some of the effects of oxysterols on biological pathways may be mediated through oxysterol perturbation of membrane properties and oxysterol activation of cholesterol within the membrane. Experiments using liposomes and membrane monolayers allow direct investigation of oxysterol effects on membranes and membrane components without interference from protein components present in a cell.
Liposome experiments have been performed using a carboxyfluorescein (CF) assay originally developed for investigation of pore-forming molecules. Liposomes prepared with a concentration of CF high enough to cause self-quenching were incubated with oxysterols. When treated with a pore-forming molecule like melittin, the pores formed in the liposomes allow CF to leak out, lowering the concentration in the liposomes below that necessary for self-quenching, and producing a fluorescent signal. This “dequenching” signal was observed upon treatment with oxysterols including 25-HC and 27-HC [38]. Side-chain oxysterols are known to increase membrane permeability to ions and small polar molecules, suggesting that oxysterols might be permeabilizing the membrane, allowing CF to leak out [44, 45]. This hypothesis was tested by first treating liposomes with oxysterols and subsequently treating them with cyclodextrin to remove the added oxysterol from the liposomes. It was found that the cyclodextrin treatment fully reversed the dequenching caused by the oxysterols themselves [38]. The reversibility of the signal rules out the possibility that CF is simply leaking out of the liposomes due to pore formation. Rather, the addition of oxysterols likely induces membrane expansion, increasing liposome volume and dilution of the CF, whereas removal of the oxysterols reversed the expansion and led to requenching of the CF.
However, these assays provide indirect evidence for membrane expansion. More direct evidence for oxysterol effects on membranes comes from Langmuir film balance experiments on oxysterol/phospholipid monolayers. Such model membrane systems have been widely used to study the effects of cholesterol and cholesterol derivatives on monolayer behavior, and recently several groups have extended these experiments to oxysterol interactions. With cholesterol and most cholesterol derivatives, as the mole fraction of the sterol in the phospholipid monolayer increases, the mean molecular area decreases in a non-linear fashion [38, 46, 47]. The reduction in mean molecular area as compared to what would be expected from a mixture of two non-interacting components, indicates that these sterols condense membranes. In contrast, oxysterols increase the mean molecular area as compared with an ideal mixture. These effects are highly dependent on the phospholipid composition of the membrane. Cholesterol’s condensing effects are stronger in membranes containing saturated lipids such as dipalmitoylphosphatidylcholine (DPPC), while the side-chain oxysterols show larger expansive effects in membranes containing unsaturated lipids such as dioleolyphosphatidylcholine (DOPC) and 1-palmitoyl-2-oleoyl-phosphatidylcholine (POPC). Furthermore, the expansive effect of side-chain oxysterols has been shown to be non-enantioselective, as oxysterol enantiomers have identical effects as their natural counterparts.
4. Molecular simulations of oxysterol-membrane interactions
Cellular studies have demonstrated that side-chain oxysterols influence cholesterol accessibility in membranes and biophysical experiments have shown that they change membrane structure. However, these experiments cannot provide the molecular details of how oxysterols cause these effects. In order to obtain atomic scale resolution, computational models can be used to simulate small systems. The large-scale changes in these simulated systems can be directly compared with experimental data to provide insight into the molecular details which are only available from the simulations. Membrane simulations have been performed for decades, but the increasing speed and power of the available computational resources have allowed simulations to become larger and more complex. While cholesterol/phospholipid membrane simulations have been performed by multiple laboratories over the last fifteen years, oxysterols/phospholipid membranes have only recently begun to be simulated. Recently, our group published results from simulations of membranes containing various concentrations of cholesterol, 25-HC, and POPC, a simple phospholipid [12, 48]. Simulations of POPC alone, POPC with low (18%) and high (30%) concentrations of cholesterol or 25-HC, and POPC with low concentrations (15%) of both cholesterol and 25-HC were performed.
4.1. Testing simulations against experimental observations
Because simulations are necessarily imperfect models of real-world systems, before molecular and structural conclusions can be drawn from the simulation, mesoscopic system properties must first be compared with experimental data. The first and most straightforward comparison is between experimental and simulation membrane area measurements. Reasonable agreement between mean molecular area for simulated and experimental pure phospholipid bilayers was demonstrated [12, 48]. While direct comparisons between areas for sterol-containing bilayers could not be made, general agreement between simulated bilayers and experimental monolayers was found for the effects of cholesterol and 25-HC on membrane area. In particular, cholesterol was found to have a generally condensing effect that was stronger at low concentrations, while 25-HC had an expansive effect that was roughly linear up to 30 mol percent [12, 48]. Partitioning of membrane area using solvent-accessible surface area showed that most of the changes in membrane area was due to changes in phospholipid area induced by the cholesterol or 25-HC present in the membrane [12].
Mass density profiles for the simulations were also performed, allowing estimation of changes in membrane thickness. As demonstrated experimentally, cholesterol thickens the bilayer, increasing the distance between the two phosphate peaks [12, 48]. 25-HC has the opposite effect, thinning the bilayer [12, 48]. Volume measurements, calculated as the product of the membrane area and thickness, show that the marginal volumes of cholesterol and 25-HC membranes are roughly similar, indicating that changes in area are compensated by changes in thickness [48].
4.2. Molecular details of membrane-oxysterol interactions
The tail order of membrane phospholipids is used as a reporter of membrane organization and accessible in both experimental NMR measurements and molecular simulation. As has been shown experimentally, the addition of cholesterol causes dose-dependent increases in lipid tail order along the length of the chain, which can be attributed to tail packing around the rigid cholesterol rings [48]. The effects of 25-HC are very different, with increases in tail order near the lipid head group and decreases in order near the ends of the tails, consistent with experimental results indicating a loss of membrane compression due to disordering of the lipid tails by the oxysterols.
The increase in cholesterol availability to external acceptors seen in oxysterol-containing membranes suggests structural changes in cholesterol interactions with phospholipids induced by oxysterols. The simulations containing both cholesterol and 25-HC were examined for any structural changes in phospholipids or cholesterol that could be indicative of increased cholesterol accessibility. It was found that the average position of cholesterol within the membrane was shifted slightly into the aqueous phase in the presence of 25-HC [12]. This was not merely an effect of a higher total sterol concentration because higher cholesterol concentrations did not show the same shift. This shift in cholesterol position was accompanied by an increase in cholesterol exposure to water and an increase in hydrogen bonding between cholesterol hydroxyl groups and water. Thus oxysterols appear to alter cholesterol accessibility by allowing it to protrude more from the membrane.
The effects of oxysterols on membrane order and cholesterol accessibility may be attributable to the orientation and position of sterols in the bilayer (see Figure 3). Cholesterol adopts primarily parallel orientations under all conditions, with the long axis of the sterol rings roughly oriented with the membrane normal [12, 48]. 25-HC shows two preferred orientations: a parallel orientation like that of cholesterol, and an interfacial orientation with the ring axis tilted and the sterol tail angled towards the bilayer surface, allowing both hydroxyl groups of the molecule to interact with both water and neighboring phospholipid headgroups [12, 48]. Each of these oxysterol orientations may be relevant to their effects. The parallel orientations result in burial of the 25-hydroxyl group. These buried polar groups self-associate in the generally hydrophobic bilayer interior and produce a cross-leaflet network of hydrogen bonded oxysterols that could contribute to membrane thinning. The interfacial orientations similarly connect neighboring phospholipids with hydrogen bonds and could directly interfere with cholesterol-phospholipid interactions.
Figure 3.
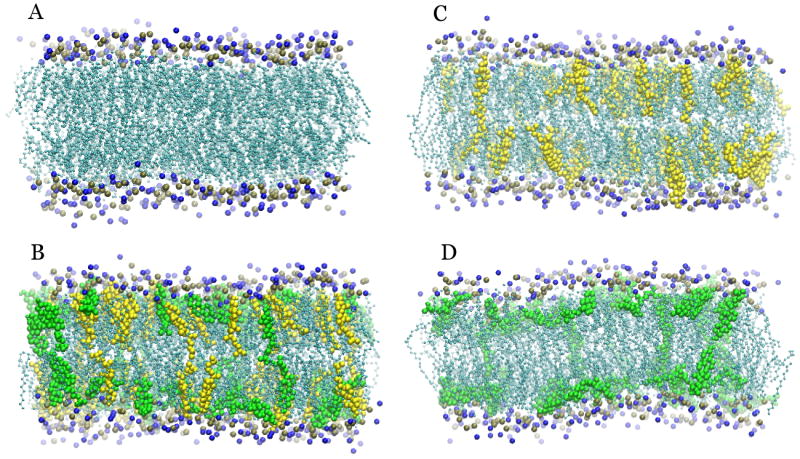
Differences in the preferred orientations of cholesterol (yellow) and 25-HC (green) can clearly be seen, with cholesterol preferring upright and 25-HC tilted orientations. The increased exposure of sterols in the mixed bilayer can be seen as sterols shift into the phospholipid headgroup (blue and ochre) region.
5. Conclusions
The biological importance of oxysterols in the regulation of cholesterol homeostasis has become increasingly well established. Oxysterols, in particular the side-chain oxysterols, have been identified as physiological effectors in LXR activation and SREBP activation. Furthermore, they are known to have effects on HMGCoA reductase degradation and cholesterol trafficking. The precise mechanisms through which they act, however, have yet to be fully elucidated.
When investigating how oxysterols can perturb cholesterol homeostasis, multiple scales of analysis are useful. Cells provide the most complete environment for experimentation, where oxysterol effects on specific pathways can be conclusively demonstrated under physiological conditions. However, while these experiments can provide clues to how these effects are produced, it is difficult to obtain clear evidence for mechanisms at the cellular level. Biochemical and biophysical studies using simple membranes and purified proteins allow experimentation and testing of potential mechanisms at the expense of potentially limiting applicability to the intact system. Meanwhile, well-validated molecular simulations provide detailed structural information on very limited systems. Because each of these types of studies provides distinct types of information on oxysterol effects, their combined use has provided a fuller understanding of oxysterol effects on cholesterol disposition in bilayer membranes.
Based on the more comprehensive understanding derived from these complementary studies, oxysterol effects on membranes and membrane lipids have been identified as a likely molecular basis for oxysterol signaling. Two membrane effects of side-chain oxysterols provide potential mechanisms of action: oxysterol perturbation of gross membrane properties, and oxysterol activation of cholesterol. Side-chain oxysterols have been found to change membrane properties in ways distinct from cholesterol. They cause lateral membrane expansion, thin the bilayer significantly, and increase membrane permeability. These kinds of structural changes can influence protein behavior: thickness differences between cellular membranes are thought to help sort membrane proteins to their appropriate location [5] and membrane structural changes can alter signaling and behavior in proteins such as adenylate cyclase, G-proteins, diacylglycerol kinase, and others [1-4, 49].
The activation of cholesterol by side-chain oxysterols has been identified in cellular systems by measuring cholesterol availability to cholesterol oxidase attack. Structural changes in cholesterol/membrane interactions have also been seen in simulated membranes that appear to be indicative of cholesterol activation. Trafficking of plasma membrane cholesterol to the ER is dependent on the membrane cholesterol concentration, with higher concentrations shifting cholesterol into the active pool that is rapidly transferred to internal compartments. This implies that the sharp increase in the ER cholesterol pool upon addition of 25-HC reflects its ability to increase the availability of cholesterol. A similar mechanism may be important for regulation of SREBP activation and HMGCoAR degradation. Oxysterol activation of cholesterol in the ER membrane could free cholesterol to bind to and activate the sterol sensing domains of these regulatory proteins.
A potential limitation of this model for oxysterol signaling is that, in cells, the bulk concentrations of oxysterols are orders of magnitude lower that what is used in the oxysterol simulations or the oxysterol cellular experiments. If those membrane effects are concentration dependent, then this lowered concentration would seem to rule out membranes as physiological effectors, though they may still be relevant when oxysterols are added to membranes in large amounts. On the other hand, while the bulk concentrations of oxysterols are extremely low, the localization of oxysterols within the cell remains to be determined. It is possible that oxysterols may concentrate in specific cellular compartments, where they could exert regulatory effects. Furthermore, it is unclear whether the membrane effects, in particular cholesterol activation, are strongly concentration dependent. If oxysterols essentially act as catalysts for cholesterol transfer from a complexed to an active pool, then depletion of that active pool by transfer out of the membrane or through binding to a sensing protein would allow that pool to be replenished by further oxysterol-induced cholesterol activation.
Further work is necessary to fully understand the mechanisms of oxysterol action in the many cholesterol homeostatic pathways in which it is involved. The lipid dependence of oxysterol effects is not clear. It is known that ER and plasma membranes have different active cholesterol thresholds, but any distinctions in how oxysterols affect them are not yet known. Both direct examination using isolated membranes and simulations of oxysterols with different lipid composition could provide further insight.
While membrane simulations have been well validated, NMR spectroscopy of liposomes or bicelles could provide further structural evidence for oxysterol-induced cholesterol activation. The structural details of the complexed and active pools of cholesterol are also still unclear. Simulations of membranes with higher cholesterol concentrations should allow both pools to be clearly observed. Finally, other sterols are also of interest. Most work to date has been done with 25-HC, but 27-HC is known to have slightly different effects, and the structural reasons are not known. Also of interest is LY295427, a sterol-derived molecule that inhibits the regulatory effects of side-chain oxysterols by an unknown mechanism. Analysis of the behavior of these sterol compounds in model membranes and dynamic simulations may provide further insight into the molecular mechanisms by which oxysterols exert their physiological effects.
Highlights.
A review of oxysterol action in cholesterol homeostasis with particular emphasis on molecular details of interaction.
Cellular, molecular, and computational approaches provide multiscale insight into oxysterol interaction with biological membranes.
Potential mechanisms for oxysterol “activation” of cholesterol are discussed.
Acknowledgments
This work was supported by the National Institutes of Health grant R01 HL067773. Computational resources were provided by the Texas Advanced Computing Center through Teragrid grants TG-MCB060053 and TG-MCA08X003 as well as the National Biomedical Computation Resource (NIH P41 RR0860516).
Abbreviations
- ACAT
acyl-CoA cholesterol acyl transferase
- CF
carboxyfluorescein
- DOPC
dioleolyphosphatidylcholine
- DPPC
dipalmitoylphosphatidylcholine
- HC
hydroxycholesterol
- LXR
liver X receptor
- POPC
1-palmitoyl-2-oleoyl-phosphatidylcholine
- Scap
SREBP cleavage-activating protein
- SREBP
sterol response element binding protein
References
- 1.Lee A. How lipids affect the activities of integral membrane proteins. Biochimica et biophysica acta. 2004;1666:62–87. doi: 10.1016/j.bbamem.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 2.Houslay M. Regulation of adenylate cyclase (EC 4.6.1.1) activity by its lipid environment. Proceedings of the Nutrition Society. 1985;44:157–165. doi: 10.1079/pns19850034. [DOI] [PubMed] [Google Scholar]
- 3.Yang Q, Alemany R, Casas J, Kitajka K, Lanier S, Escribá P. Influence of the membrane lipid structure on signal processing via G protein-coupled receptors. Molecular pharmacology. 2005;68:210–217. doi: 10.1124/mol.105.011692. [DOI] [PubMed] [Google Scholar]
- 4.McIntosh T, Simon S. Roles of bilayer material properties in function and distribution of membrane proteins. Annual review of biophysics and biomolecular structure. 2006;35:177–198. doi: 10.1146/annurev.biophys.35.040405.102022. [DOI] [PubMed] [Google Scholar]
- 5.Lundbaek JA, Andersen OS, Werge T, Nielsen C. Cholesterol-induced protein sorting: an analysis of energetic feasibility. Biophysical journal. 2003;84:2080–2089. doi: 10.1016/S0006-3495(03)75015-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Czech MP. PIP2 and PIP3: complex roles at the cell surface. Cell. 2000;100:603–606. doi: 10.1016/s0092-8674(00)80696-0. [DOI] [PubMed] [Google Scholar]
- 7.Futerman A, Hannun Y. The complex life of simple sphingolipids. EMBO reports. 2004;5:777–782. doi: 10.1038/sj.embor.7400208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hannun Y, Obeid L. Principles of bioactive lipid signalling: lessons from sphingolipids. Nature reviews Molecular cell biology. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 9.Fadeel B, Xue D. The ins and outs of phospholipid asymmetry in the plasma membrane: roles in health and disease. Critical reviews in biochemistry and molecular biology. 2009;44:264–277. doi: 10.1080/10409230903193307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ikonen E. Cellular cholesterol trafficking and compartmentalization. Nature reviews Molecular cell biology. 2008;9:125–138. doi: 10.1038/nrm2336. [DOI] [PubMed] [Google Scholar]
- 11.Ridsdale A, Denis M, Gougeon P-Y, Ngsee J, Presley J, Zha X. Cholesterol is required for efficient endoplasmic reticulum-to-Golgi transport of secretory membrane proteins. Molecular biology of the cell. 2006;17:1593–1605. doi: 10.1091/mbc.E05-02-0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olsen B, Schlesinger P, Ory D, Baker N. 25-Hydroxycholesterol increases the availability of cholesterol in phospholipid membranes. Biophysical journal. 2011;100:948–956. doi: 10.1016/j.bpj.2010.12.3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohvo-Rekilä H, Ramstedt B, Leppimäki P, Slotte P. Cholesterol interactions with phospholipids in membranes. Progress in lipid research. 2002;41:66–97. doi: 10.1016/s0163-7827(01)00020-0. [DOI] [PubMed] [Google Scholar]
- 14.Simons K, Vaz W. Model systems, lipid rafts, and cell membranes. Annual review of biophysics and biomolecular structure. 2004;33:269–295. doi: 10.1146/annurev.biophys.32.110601.141803. [DOI] [PubMed] [Google Scholar]
- 15.Feigenson G. Phase boundaries and biological membranes. Annual review of biophysics and biomolecular structure. 2007;36:63–77. doi: 10.1146/annurev.biophys.36.040306.132721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Meer G, Voelker D, Feigenson G. Membrane lipids: where they are and how they behave. Nature reviews Molecular cell biology. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horton J, Goldstein J, Brown M. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. The Journal of clinical investigation. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ory D. Nuclear receptor signaling in the control of cholesterol homeostasis: have the orphans found a home? Circulation research. 2004;95:660–670. doi: 10.1161/01.RES.0000143422.83209.be. [DOI] [PubMed] [Google Scholar]
- 19.Chiang J. Regulation of bile acid synthesis: pathways, nuclear receptors, and mechanisms. Journal of hepatology. 2004;40:539–551. doi: 10.1016/j.jhep.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 20.Norlin M, Wikvall K. Enzymes in the conversion of cholesterol into bile acids. Current molecular medicine. 2007;7:199–218. doi: 10.2174/156652407780059168. [DOI] [PubMed] [Google Scholar]
- 21.Lange Y, Ory D, Ye J, Lanier M, Hsu F-F, Steck T. Effectors of rapid homeostatic responses of endoplasmic reticulum cholesterol and 3-hydroxy-3-methylglutaryl-CoA reductase. The Journal of biological chemistry. 2008;283:1445–1455. doi: 10.1074/jbc.M706967200. [DOI] [PubMed] [Google Scholar]
- 22.Frolov A, Zielinski S, Crowley J, Dudley-Rucker N, Schaffer J, Ory D. NPC1 and NPC2 regulate cellular cholesterol homeostasis through generation of low density lipoprotein cholesterol-derived oxysterols. The Journal of biological chemistry. 2003;278:25517–25525. doi: 10.1074/jbc.M302588200. [DOI] [PubMed] [Google Scholar]
- 23.Janowski B, Grogan M, Jones S, Wisely B, Kliewer S, Corey E, Mangelsdorf D. Structural requirements of ligands for the oxysterol liver X receptors LXRα and LXRβ. Proceedings of the National Academy of Sciences. 1999;96:266–271. doi: 10.1073/pnas.96.1.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature. 1996;383:728–731. doi: 10.1038/383728a0. [DOI] [PubMed] [Google Scholar]
- 25.Björkhem I. Are side-chain oxidized oxysterols regulators also in vivo? Journal of lipid research. 2009;50(Suppl):S213–S218. doi: 10.1194/jlr.R800025-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tall AR. Cholesterol efflux pathways and other potential mechanisms involved in the athero-protective effect of high density lipoproteins. Journal of internal medicine. 2008;263:256–273. doi: 10.1111/j.1365-2796.2007.01898.x. [DOI] [PubMed] [Google Scholar]
- 27.Goldstein J, Rawson R, Brown M. Mutant mammalian cells as tools to delineate the sterol regulatory element-binding protein pathway for feedback regulation of lipid synthesis. Archives of biochemistry and biophysics. 2002;397:139–148. doi: 10.1006/abbi.2001.2615. [DOI] [PubMed] [Google Scholar]
- 28.Humphries GM, McConnell HM. Potent immunosuppression by oxidized cholesterol. Journal of immunology (Baltimore, Md : 1950) 1979;122:121–126. [PubMed] [Google Scholar]
- 29.Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 30.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 31.Shimano H, Horton JD, Hammer RE, Shimomura I, Brown MS, Goldstein JL. Overproduction of cholesterol and fatty acids causes massive liver enlargement in transgenic mice expressing truncated SREBP-1a. The Journal of clinical investigation. 1996;98:1575–1584. doi: 10.1172/JCI118951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yabe D, Brown M, Goldstein J. Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:12753–12758. doi: 10.1073/pnas.162488899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang T, Espenshade P, Wright M, Yabe D, Gong Y, Aebersold R, Goldstein J, Brown M. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110:489–500. doi: 10.1016/s0092-8674(02)00872-3. [DOI] [PubMed] [Google Scholar]
- 34.Gil G, Faust JR, Chin DJ, Goldstein JL, Brown MS. Membrane-bound domain of HMG CoA reductase is required for sterol-enhanced degradation of the enzyme. Cell. 1985;41:249–258. doi: 10.1016/0092-8674(85)90078-9. [DOI] [PubMed] [Google Scholar]
- 35.Kuwabara P, Labouesse M. The sterol-sensing domain: multiple families, a unique role? Trends in genetics : TIG. 2002;18:193–201. doi: 10.1016/s0168-9525(02)02640-9. [DOI] [PubMed] [Google Scholar]
- 36.Lange Y, Ye J, Rigney M, Steck T. Regulation of endoplasmic reticulum cholesterol by plasma membrane cholesterol. Journal of Lipid Research. 1999;40:2264–2270. [PubMed] [Google Scholar]
- 37.Chang T-Y, Li B-L, Chang C, Urano Y. Acyl-coenzyme A:cholesterol acyltransferases. American Journal of Physiology - Endocrinology And Metabolism. 2009;297:E1–E9. doi: 10.1152/ajpendo.90926.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gale S, Westover E, Dudley N, Krishnan K, Merlin S, Scherrer D, Han X, Zhai X, Brockman H, Brown R, Covey D, Schaffer J, Schlesinger P, Ory D. Side chain oxygenated cholesterol regulates cellular cholesterol homeostasis through direct sterol-membrane interactions. The Journal of biological chemistry. 2009;284:1755–1764. doi: 10.1074/jbc.M807210200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Radhakrishnan A, Ikeda Y, Kwon HJ, Brown M, Goldstein J. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:6511–6518. doi: 10.1073/pnas.0700899104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sever N, Yang T, Brown M, Goldstein J, DeBose-Boyd R. Accelerated degradation of HMG CoA reductase mediated by binding of insig-1 to its sterol-sensing domain. Molecular cell. 2003;11:25–33. doi: 10.1016/s1097-2765(02)00822-5. [DOI] [PubMed] [Google Scholar]
- 41.Lange Y, Steck T. Cholesterol homeostasis and the escape tendency (activity) of plasma membrane cholesterol. Progress in lipid research. 2008;47:319–332. doi: 10.1016/j.plipres.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lange Y, Ye J, Steck T. How cholesterol homeostasis is regulated by plasma membrane cholesterol in excess of phospholipids. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:11664–11667. doi: 10.1073/pnas.0404766101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sokolov A, Radhakrishnan A. Accessibility of cholesterol in endoplasmic reticulum membranes and activation of SREBP-2 switch abruptly at a common cholesterol threshold. The Journal of biological chemistry. 2010;285:29480–29490. doi: 10.1074/jbc.M110.148254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holmes RP, Yoss NL. 25-Hydroxysterols increase the permeability of liposomes to Ca2+ and other cations. Biochimica et biophysica acta. 1984;770:15–21. doi: 10.1016/0005-2736(84)90067-1. [DOI] [PubMed] [Google Scholar]
- 45.Theunissen JJ, Jackson RL, Kempen HJ, Demel RA. Membrane properties of oxysterols. Interfacial orientation, influence on membrane permeability and redistribution between membranes. Biochimica et biophysica acta. 1986;860:66–74. doi: 10.1016/0005-2736(86)90499-2. [DOI] [PubMed] [Google Scholar]
- 46.Mintzer E, Charles G, Gordon S. Interaction of two oxysterols, 7-ketocholesterol and 25-hydroxycholesterol, with phosphatidylcholine and sphingomyelin in model membranes. Chemistry and physics of lipids. 2010;163:586–593. doi: 10.1016/j.chemphyslip.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 47.Stottrup B, Keller S. Phase behavior of lipid monolayers containing DPPC and cholesterol analogs. Biophysical journal. 2006;90:3176–3183. doi: 10.1529/biophysj.105.072959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Olsen B, Schlesinger P, Baker N. Perturbations of membrane structure by cholesterol and cholesterol derivatives are determined by sterol orientation. Journal of the American Chemical Society. 2009;131:4854–4865. doi: 10.1021/ja8095224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Edidin M. The state of lipid rafts: from model membranes to cells. Annual review of biophysics and biomolecular structure. 2003;32:257–283. doi: 10.1146/annurev.biophys.32.110601.142439. [DOI] [PubMed] [Google Scholar]