

Abstract
Women are born with a finite population of ovarian follicles, which are slowly depleted during their reproductive years until reproductive failure (menopause) occurs. The rate of loss of primordial follicles is determined by genetic and environmental influences, but certain toxic exposures can accelerate this process. Ionizing radiation reduces preantral follicle numbers in rodents and humans in a dose-dependent manner. Cigarette smoking is linked to menopause occurring 1–4 yr earlier than with nonsmokers, and components of smoke, polycyclic aromatic hydrocarbons, can cause follicle depletion in rodents or in ovaries in vitro. Chemotherapeutic agents, such as alkylating drugs and cisplatin, also cause loss of preantral ovarian follicles. Effects depend on dose, type, and reactivity of the drug, and the age of the individual. Evidence suggests DNA damage may underlie follicle loss induced by one common alkylating drug, cyclophosphamide. Occupational exposures have also been linked to ovarian damage. In an industrial setting, 2-bromopropane caused infertility in men and women, and it can induce ovarian follicle depletion in rats. Solvents, such as butadiene, 4-vinylcyclohexene, and their diepoxides, can also cause specific preantral follicle depletion. The mechanism(s) underlying effects of the latter compound may involve alterations in apoptosis, survival factors such as KIT/Kit Ligand, and/or the cellular signaling that maintains primordial follicle dormancy. Estrogenic endocrine disruptors may alter follicle formation/development and impair fertility or normal development of offspring. Thus, specific exposures are known or suspected of detrimentally impacting preantral ovarian follicles, leading to early ovarian failure.
Keywords: atresia, fertility, follicle, ovary, toxicity, toxicology
The rate of ovarian follicle loss is accelerated by multiple toxic exposures, including drugs and environmental toxicants, leading to earlier ovarian failure, loss of fertility, and other detrimental health effects.
INTRODUCTION
Accelerated loss of preantral follicles leads to earlier reproductive failure in women. Thus, it is of interest to better understand how preantral follicles may be adversely impacted by exposures to xenobiotic chemicals. Furthermore, identification of the types of exposures that impact the primordial follicle population is necessary to protect women from permanent effects on the ovarian reserve, leading to early loss of reproductive function. At present, this is difficult because of a lack of methods for quantifying preantral follicle numbers in living individuals. This review will discuss the establishment and development of ovarian preantral follicles and how environmental, occupational, and pharmaceutical exposures can cause ovarian damage and irreversible follicle loss, resulting in acceleration toward amenorrhea, infertility, and early ovarian failure.
PRIMORDIAL FOLLICLE FORMATION
During embryogenesis, primordial germ cells migrate to the genital ridge. Bone morphogenic proteins (BMPs) 4, 8b, and 2 play a critical role in the differentiation of primordial germ cells from the epiblast, as determined from characterizing mutant or knockout mice [1–3]. Once in the genital ridge, germ cells are called oogonia, and they proliferate to establish a large pool of germ cells. During the mitotic phase, germ cells proliferate to form cysts or nests, which are groups of oogonia surrounded by somatic cells destined to become the pregranulosa cells of primordial follicles. These groups of cells are linked by intercellular bridges [4, 5] and are thought to undergo mitosis synchronously, thereby forming these clusters of gametes [6, 7], although an in vitro study suggested that germ cell cysts form by aggregation [8]. The cysts become surrounded by a basement membrane, separating them from the surrounding cells [9, 10]. Groups of oogonia begin to enter into meiosis starting around Gestational Day 13.5 in mice, but some oogonia can remain as late as a few days after birth [11, 12]. In humans, the timing in which oocytes are arrested in the diplotene phase of meiosis ranges from 2 to 7 mo of fetal development [13, 14].
Formation of individual primordial follicles occurs 2–4 days after birth in rodents, and shortly before birth in humans [15–18]. In fact, some primordial follicles can form before birth, and a few of those begin developing as soon as they are formed, leading to the first wave of growing follicles [19]. Follicle formation involves rearrangement of the pregranulosa cells and partial breakdown and reformation of the basement membrane into entities that contain only single gametes [9]. Further loss of gametes occurs as follicle formation progresses, possibly due to elimination of improperly formed follicles or incomplete support from too few pregranulosa cells. Loss of oocytes is thought to be necessary for proper follicle formation, and apoptosis is thought to be the underlying process by which certain of the oocytes are eliminated [20]. Knockout or overexpressing mice with altered expression of genes involved in apoptosis have been shown to modify the follicle pool in mice [21–24]. The timing of the events surrounding primordial follicle formation in mammalian species is a topic that still requires further study for better elucidation of the underlying processes.
Establishment of the pool of primordial follicles appears to have multiple points at which oogonia and oocytes are lost [25]. From ∼6 million to 7 million germ cells formed at its peak during embryogenesis, follicle numbers at birth drop to ∼1 million to 2 million in girls, which is reduced further to ∼300 000 by puberty [26]. Particular decreases in gamete numbers are observed during the end of the mitotic period as well as during the formation of primordial follicles [7, 19, 20]. The reason for this is unknown, but it has been suggested that incorrectly developing or abnormal germ cells may be eliminated, leaving only the best. Alternatively, those gametes that are lost might have served as support cells and are eliminated once they are no longer needed, as happens in invertebrates [7].
PRIMORDIAL FOLLICLE ACTIVATION
The primordial follicle consists of an oocyte, arrested in prophase I of meiosis, surrounded by a single layer of flattened pregranulosa cells. Once primordial follicles have been formed, they remain quiescent for months or years until they are individually activated to initiate growth and development into primary follicles. Why some primordial follicles enter the growth phase early while others are quiescent for decades is largely a mystery. After primordial follicle development is initiated, the follicle is destined to either ovulate or undergo atresia, which is a natural process of follicle elimination and removal. Once the available pool of primordial follicles reaches a critical lower limit, ovarian function ceases and women enter menopause [16, 26].
One of the initial events in primordial follicle activation (primordial to primary follicle transition) is a change in the granulosa cells from a flattened to a cuboidal morphology while the oocyte increases in diameter [16]. Primordial follicle recruitment is distinct from other stages of folliculogenesis in that it can proceed without extraovarian hormonal input [27–29]. The spontaneous initiation of primordial follicle growth can also occur rapidly in vitro in cultured ovarian tissue (Fig. 1) [30, 31]. Because of this important characteristic, whole neonatal ovarian cultures have been used extensively to study the role of a variety of signaling factors that can affect primordial follicle activation. There are multiple activator and suppressor pathways that can converge to regulate activation of primordial follicles.
FIG. 1.
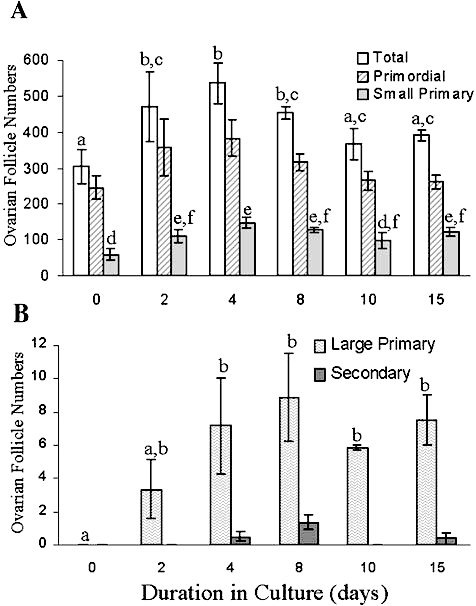
Onset of development of preantral follicles during culture of ovaries from PND4 Fischer 344 rats. Ovaries were collected and cultured in vitro for up to 15 days. Tissue samples were collected at various times and processed for histological evaluation. Ovarian follicles were classified and counted as primordial, small primary follicles (A), and large primary and secondary preantral follicles (B). Values represent means ± SEM; means within the same follicle type with no common letters over the columns are significantly different from each other, P < 0.05; n = 4–7 ovaries per time point in three to four experiments. Reproduced from Devine et al. [31] with permission from Elsevier.
The number of growth factors, hormones, and cytokines thought to be responsible for the regulation of the transition from the primordial to the primary follicle is ever increasing (Table 1). The molecular signaling pathways used by these various factors are not well understood, but each plays an important role in the control of the primordial to primary transition and/or the maintenance of primordial quiescence. Factors implicated in the stimulation of the transition to the primary follicle stage are KIT ligand (KITLG) [32, 33], basic platelet-derived growth factor (PDGFB) [34], leukemia inhibitory factor (LIF) [35], fibroblast growth factor 7/keratinocyte growth factor (FGF7) [36], glial cell line-derived neurotrophic factor (GDNF) [37], BMP4 and BMP7 [38, 39], insulin [40, 41], and neurotrophins (BDNF and NGF) [42, 43]. Conversely, anti-Müllerian hormone (AMH) [44], activin A [45], and stromal-derived factor-1/chemokine (C-X-C motif) ligand 4 (SDF-1/CXCL4) [46] have been shown to inhibit primordial follicle transition. Interestingly, many of these stimulating and inhibiting factors do not independently prevent or allow primordial follicle activation. This emphasizes the fact that this critical process requires a balance between stimulatory and inhibitory factors for optimal primordial to primary follicle transition.
TABLE 1.
Signaling factors that regulate the primordial to small primary follicle transition.

PREANTRAL FOLLICLE DEVELOPMENT
Once follicular activation occurs, a number of changes are triggered. Granulosa cells become cuboidal and begin dividing more rapidly. They produce and secrete proteins that form a glycoprotein coat around the oocyte, called the zona pellucida. Growth and development of the oocyte are tightly linked to the proliferation and differentiation of the granulosa cells that support it. Signals from the oocyte, such as GDF9 and BMP15, permit granulosa cells to divide and survive while not directly in contact with the oocyte [47, 48]. It is thought that the KITLG being produced is altered from a membrane-bound form to a secreted protein. At the same time, theca cells are recruited from the interstitium by mechanisms as yet unknown.
Until formation of the antral cavity, the gonadotropin hormones, follicle-stimulating hormone (FSH) and luteinizing hormone (LH), are not required for follicle development. This is demonstrated by the secondary follicles that develop from primordial or primary follicles when Postnatal Day 4 (PND4) ovaries are cultured in vitro in the absence of FSH or LH [49, 50]. Using individual cultured rat or mouse ovaries or follicles (reviewed in Skinner [15] and Demeestere et al. [51]), increased follicle development has been observed with activin A [52–54], keratinocyte growth factor or fibroblast growth factor 7 (FGF7) [55], KITLG [56], GDF9 [57], GH [53], and IGF1 [53, 58, 59]. In contrast, AMH is thought to inhibit follicle growth. Thus, this process is complex, with multiple stages and different signals for each follicle stage. As yet, none of these factors alone or in combination completely explains preantral follicle development, and further research is needed to better characterize the required growth factors needed for proper follicular development.
XENOBIOTIC EFFECTS ON OVARIAN FUNCTION
The destruction of primordial and preantral follicles can be caused by a variety of xenobiotic chemicals. In general, if preantral follicles are targeted, this results in a chemical-induced destruction of the oocytes. If extensive loss of primordial follicles has occurred, the effect may be irreversible ovarian failure (i.e., early menopause). Detection of primordial follicle destruction can be quite problematic because unless larger follicles are also destroyed, this type of damage can cause a delayed effect on cyclicity that is undetected until no follicles are left to be recruited from the primordial pool for development [60]. There are no known biomarkers derived from primordial follicles that could be used to detect such damage. Evidence shows that premature loss of ovarian function by elective oophorectomy results in an increase in a woman's lifetime risk for cardiovascular disease, cognitive impairment or dementia, symptoms of parkinsonism, and osteoporosis, compared with women who went through natural menopause [61, 62]. Therefore, premature loss of ovarian function before natural menopause by xenobiotics may also be associated with a decrease in women's long-term health.
SPECIFIC AGENTS THAT TARGET PRIMORDIAL/ PREANTRAL FOLLICLES
Ionizing Radiation and Chemotherapeutic Agents
Numerous clinical studies, reviews, and meta-analyses have examined the effects of anticancer treatments (i.e., radiation and chemotherapy) on female fertility and their maintenance of reproductive cyclicity. In general, these treatments frequently result in irreversible loss of ovarian function. Often, these studies have been complicated by the diverse exposures and durations of anticancer therapies used, small patient populations, and short follow-up [63–65]. Although detrimental effects on ovaries are of most concern in young cancer patients, acceleration of ovarian follicle loss by radiotherapy and/or chemotherapy is of concern to all premenopausal women [66]. Overall, reports suggest that total body irradiation or irradiation in the pelvic region correlates strongly with loss of menstrual cyclicity and/or fertility. Chemotherapy, which can involve multiple drugs, has been more complicated to evaluate. However, alkylating agents, especially bifunctional drugs, have been found to be linked to the highest risk for loss of cyclicity and fertility. Rodent studies have provided more clear-cut evidence of ovarian follicle loss, leading to infertility. Toxicity could be temporary and rapid if the drugs cause effects only on growing follicles. In contrast, damage to and loss of primordial follicles are irreversible. Continued investigations are needed to monitor current and emerging anticancer treatment regimens to determine the best and safest treatments.
The Childhood Cancer Survivor Studies, one cohort started in 1993 and another started in 2007, examined long-term effects of cancer treatments in survivors who had been treated as children. One of the primary effects identified in patients treated after age 18 yr, when compared to untreated siblings, was a 13-fold increase in the risk of undergoing premature menopause (i.e., before 40 yr of age). Risk was increased with increasing age and increasing doses of radiation or alkylating agents [65, 67]. In women who have undergone these treatments, fewer primordial follicles were observed in ovarian samples from autopsies and fewer antral follicles were observed by ultrasound of survivors than in age-matched control subjects [68–70]. Investigations are continuing to examine the current dosing regimens and their detrimental effects.
Ionizing radiation
Ionizing radiation is highly correlated with loss of ovarian follicles, cyclicity, and fertility. Although not a chemical entity, this type of exposure induces numerous effects, depending on the type of radiation and the dose. Multiple studies have demonstrated radiation-induced loss of ovarian follicles in rats, mice, and nonhuman primates [71–74]. Results in women are also relatively clear and consistent [75, 76]. Women who are exposed to total body irradiation or irradiation of the abdomen or pelvic area are more likely to suffer irreversible ovarian damage and arrest of menstruation than those exposed in other places, and results support a clear dose-response relationship [65, 67, 77]. Radiotherapy given in a fractionated protocol is safer than a single, higher exposure [78]. The median lethal dose (LD50) of primordial follicles has been reported to be between 2 and 6–18 Gray (Gy) [76]. Risk of ovarian failure is dependent on the age at exposure (younger girls or women are more resistant), the dose, whether or not the pelvic area is being exposed, and the fractionation of doses [64, 79, 80]. Wallace et al. [81] developed a predictive model to estimate the number of remaining follicles in patients relative to the dose of ionizing radiation given. Multiple methods have been proposed to protect ovaries from damage during ionizing radiation therapy. Cryopreservation of ovarian tissue before exposures has been proposed. Ovarian transposition—surgically moving ovaries away from their normal location—before irradiation leads to decreased effects on ovarian function, and gonadotropin-releasing hormone analogs may also protect against ovarian damage [82, 83]. Improvements in irradiation protocols and instrumentation will likely further improve its safety and effectiveness, but it is currently the most significant risk factor for undergoing premature ovarian failure in cancer patients [63, 67].
Cisplatin
Detrimental effects on gonads and reproductive function have been observed in cancer patients given cisplatin during a chemotherapy regimen [77, 84]. A few studies have been performed in rodents to study the impact of cisplatin exposures on ovarian function and follicle numbers. In rats, two doses of either 4.5 or 6 mg/kg cisplatin induced reductions in healthy antral follicle numbers, circulating AMH, AMH-positive follicles, and increases in the numbers of follicles with evidence of apoptosis [85]. These doses are, however, in the range of the LD50 in rats [86]. Furthermore, increased numbers of atretic medium and large follicles were observed after 2 or 4 wk of 1–2 mg/kg per day cisplatin. Single doses of 5 mg/kg cisplatin in rats induced significant decreases in primordial follicle numbers as well [87, 88]. These results suggest that cisplatin can have detrimental effects on both growing and primordial ovarian follicles, leading to acute effects as well as permanent reductions in ovarian reserve.
Busulfan (Myleran)
Busulfan has been used for chronic myeloid leukemia but now is used mostly as a conditioning agent to eliminate bone marrow cells before bone marrow transplantation [77, 78]. Cancer chemotherapy containing busulfan has been shown to impair fertility in women [66, 77, 78]. Animal studies examining the ovarian toxicity of busulfan have been relatively limited. A single i.p. dose of 10 mg/kg busulfan in SECXC57BL F1 mice induced eventual depletion of all follicles [89]. Sprague-Dawley rats dosed orally with busulfan for 4 wk had decreased numbers of embryos and preantral follicles at 0.5 and 1.5 mg/kg per day, respectively [90]. In FVB/NJNarl mice, 36 mg/kg busulfan i.p. caused significant depletion of primordial, primary, and secondary follicles [91]. Collectively, these results provide relatively firm evidence that busulfan can damage both primordial and growing preantral follicles.
Cyclophosphamide and other alkylating drugs
Alkylating agents are reactive molecules that form covalent bonds with critical cellular macromolecules. The mechanism of action of these types of drugs is thought to involve adduct formation, as well as intrastrand and interstrand DNA cross-links, blocking the rapid replication machinery that permits the proliferation of cancer cells. When cancer treatments are separated by a mechanism of action, alkylating agents have been shown to be highly correlated with arrested menstruation and ovarian failure [65, 92]. Monofunctional agents (those with only one reactive group) are less correlated with ovarian failure than bifunctional agents [63, 65]. This may relate to the ease of repair associated with the variable types of DNA damage caused by these different agents (i.e., adducts versus DNA double-strand cross-links).
Cyclophosphamide (CPA) is a bifunctional alkylating agent given for treatment of multiple cancers, lupus erythematosus, and other autoimmune problems. Other drugs, such as iphosphamide, can also produce the same active metabolite; thus, its mechanism and effect on the reproductive system are likely similar to CPA. Plowchalk and Mattison [93] demonstrated in C57BL/6N mice that phosphoramide mustard (PM) is the active metabolite of CPA that causes ovarian toxicity. Multiple reviews have reported increased odds ratios for ovarian failure or amenorrhea in patients given CPA [63–65]. Cyclophosphamide is given alone, as well as in combination with other drugs, so the evidence for CPA-induced ovarian toxicity is relatively unambiguous. Fertility was reduced in cancer patients whose treatments included CPA, and the risk for such effects was dose dependent [79]. Amenorrhea is often observed rapidly after treatments, suggesting a direct impact on growing and antral follicles. Furthermore, evidence supports loss of primordial follicles, because there is also an increased risk of undergoing early reproductive failure [63, 77, 94]. As with radiation, dose and age at which exposures occur influence whether or not individuals develop amenorrhea or infertility [95, 96].
Multiple reports have demonstrated primordial follicle loss in mice in response to CPA exposures [97, 98]. Rats were also sensitive to CPA-induced follicle loss, although most reports described impacts on larger follicles. A single dose of CPA caused loss of primordial follicles in a dose-dependent manner in BALB/c mice, with 75 mg/kg inducing 50% loss. By giving a single i.p. dose of 75 mg/kg CPA to BALB/c mice and then mating them at various times after exposures, Meirow et al. [99] tested the ability of the surviving follicles to produce healthy offspring following the exposure to CPA. Results demonstrated that health of all follicle stages was affected by the single dose of CPA, even though not all follicle stages had reduced numbers. Increased numbers of malformed offspring were observed from female mice mated any time between the first and sixth wk after exposure [99]. This timing corresponded to oocytes coming from exposed primordial through antral follicles, based on estimated duration of follicle development [99]. Furthermore, female mice mated 1 wk after CPA exposure, which would correspond to exposure of oocytes in antral follicles, had fewer implantation sites as well as increased malformations of offspring. Malformations, however, may also be due to altered uterine, hypothalamic, or pituitary function, so results must be interpreted with caution.
The role of DNA damage as the mechanism by which alkylating drugs induce follicle loss has also been studied [100]. Bifunctional alkylating drugs, such as CPA, induce DNA adducts, DNA double-strand cross-links, leading to DNA double-strand breaks, to kill cancer cells [101]. To examine whether or not DNA damage occurs in ovarian follicles also, an antibody against phosphohistone H2AFX, which becomes phosphorylated at sites of double-strand breaks, was used to examine whether or not double-strand breaks can be observed in cultured rodent ovaries in response to PM [100]. Punctate staining of phospho-H2AFX was detected specifically in oocytes of cultured mouse and rat ovaries exposed to PM. The effects were concentration dependent, with staining peaking at 24–48 h with 3 and 10 μM. Further, inhibition of H2AFX phosphorylation sensitized ovaries to PM. In contrast, a toxic molecule from cigarette smoke, 9,10-dimethylbenzanthracene (DMBA), which is thought to predominantly produce DNA adducts, did not appear to induce phospho-H2AFX staining in oocytes. In another study, DNA cross-links in granulosa cells were observed and peaked 2 h following a single dose of CPA in immature Sprague-Dawley rats [102]. Taken together, these results support that DNA damage is a mechanism by which bifunctional alkylating drugs cause ovarian damage.
Environmental Agents
Cigarette smoking
Many epidemiological studies performed during the last five decades have shown that cigarette smoke is a reproductive toxicant, demonstrating a strong relationship between smoking and impaired fertility. Women smokers have also been reported to experience a 1- to 4-yr earlier age at the onset of menopause [103–106]. Thus, a significant amount of data exists to demonstrate a relationship between smoking and reduced fertility, but the mechanism is not well understood. There are several possible mechanisms by which cigarette smoke might be involved in the earlier onset of menopause among smokers [105, 107]. Cigarette smoke is a complex mixture of alkaloids (nicotine), polycyclic aromatic hydrocarbons (PAHs), nitroso compounds, aromatic amines, and protein pyrolysates, many of which are reactive and carcinogenic [108]. When components of cigarette smoke were measured in serum and follicular fluid of women exposed to smoke as compared to nonsmokers, levels of the PAH benzo[a]pyrene (BaP) were particularly high [105]. This led the authors to speculate that B[a]P may be a key compound central to the documented adverse effects of cigarette smoke on follicular development, and subsequently fertility. Because of the logical association between early menopause and oocyte destruction, some of the effects of cigarette smoke on fertility are likely to be due to damage to primordial and growing preantral follicles. In animal studies, exposure of female C57BL/6 mice to mainstream cigarette smoke for 8 wk resulted in a reduction in primordial follicle numbers and loss of ovarian volume [109]. In another study, exposure of mice in utero to cigarette smoke resulted in a reduced number of ovarian primordial follicles in female offspring [110].
Polycyclic aromatic hydrocarbons
Because of the prevalence of PAHs in cigarette smoke and in the environment from various combustion processes (including automobile exhaust), exposure to this class of pollutants is ubiquitous. Many animal studies have examined the potential of PAHs to cause ovarian damage or inhibit fertility. Primordial follicle destruction is known to result from dosing of mice and rats with three widely studied PAHs: DMBA, 3-methylcholanthrene (3-MC), and BaP [111, 112]. Because the three PAHs destroy primordial follicles in laboratory animals, it is likely that they contribute to the early onset of menopause in women smokers. In mice treated with PAHs (80 mg/kg BaP, 3-MC, or DMBA), oocyte morphology consistent with necrosis was observed in primordial follicles [113]. In addition to primordial follicles, DMBA reduced numbers of all larger follicle types as well. These changes caused by 3-MC and BaP were seen in the absence of visible effects in the associated granulosa cells. Initial studies in mice and rats examined ovotoxic effects caused by a single high dose of PAHs. The three PAHs, BaP, 3-MC, and DMBA, destroyed oocytes in preantral follicles of Sprague-Dawley rats and in D2 and B6 mice within 14 days following a single i.p. injection, with mice being more susceptible to ovotoxicity than rats [114]. The extent of primordial follicle loss following this high-dose exposure in mice was reported to be 50% within 1–2 days [111]. However, repeated low-dose exposure is the more likely exposure paradigm in women. Therefore, another study was undertaken to determine whether lower doses of these chemicals could produce significant loss of primordial follicles. Female mice were injected repeatedly with various doses of the three PAHs, sufficient to cause 50% loss of primordial follicles after 15 days of daily dosing [112]. Calculating an ovotoxic index using the doses required to cause 50% follicle destruction in both studies, it was determined that relative to a single high-dose exposure, repeated low-dose exposure was more ovotoxic to 250 (DMBA), 120 (3-MC), or 2 (BaP) times a greater extent (Fig. 2) [112]. Thus, these results demonstrate that animal studies designed to more closely mimic human types of exposures may reveal surprising insights as to realistic risk.
FIG. 2.

Initial ovotoxic index (OI) species comparison after 15 days in mice and rats. The dose of each chemical found to cause 50% loss of primordial follicles (A), primary follicles (B), or secondary follicles (ED50; C) in B6C3F1 mice (open bars) and Fischer 344 rats (closed bars) after 15 daily doses was used to calculate the initial OI. The initial OI is expressed as a reciprocal (1/OI) to illustrate the dramatic differences in ovotoxicity between chemicals. Thus, the higher the value, the more ovotoxic the chemical. Note: log scale. OI = (ED50) × (15 days of dosing). ns, not significant (not susceptible at the experimental doses, P < 0.05). Reproduced from Borman et al. [112] with permission from Elsevier.
Following single high-level i.p. injections of PAHs in mice, the relative toxicities causing oocyte destruction in primordial follicles were observed to be DMBA > 3-MC > BaP [111]. This was similar to the relative toxicities determined for these compounds in the experiments exposing mice and rats to multiple doses at lower levels of these compounds [112]. In a subsequent study, B6 mice given single i.p. doses ranging from 1 to 100 mg/kg BaP demonstrated a 50% effective dose (ED50) of 15 mg/kg for oocyte destruction [115]. Collectively, these observations provide support for a cumulative ovotoxic effect of chronic exposures to low doses.
Recent studies have used a PND4 whole-ovary culture system to study the mechanisms involved in DMBA-induced loss of preantral follicles. DMBA in vitro was shown to cause follicle destruction following 6- and 48-h incubations of ovaries from neonatal mice and rats, respectively [116, 117]. DMBA has been shown to upregulate expression of the proapoptotic protein, BAX, in cultured PND4 mouse ovarian primordial follicle oocytes [118]. These observations were supported by the observation that ovaries from BAX-deficient mice are resistant to DMBA-induced primordial follicle destruction [118]. Thus, it appears that the effects of DMBA are mediated by an intracellular pathway involving the proapoptotic branch of the BCL2 family of proto-oncogenes. Therefore, these studies support that DMBA-induced follicle loss is via apoptosis.
An involvement of the aryl hydrocarbon receptor (AHR) in DMBA-induced ovotoxicity has also been investigated. Treatment with the AHR antagonist α-naphthoflavone blocked DMBA-induced expression of BAX in PND4 cultured mouse ovaries. Additionally, AHR-deficient mice were resistant to DMBA-induced primordial follicle destruction [118]. One study that investigated the effects of PAH exposure on pregnant rat dams used a competitive inhibitor of AHR, resveratrol, along with a mixture of BaP and DMBA. Inhibition of AHR completely prevented PAH-induced depletion of primordial and primary follicles in ovaries of female offspring [119]. Further, in vitro exposure of isolated rat follicles with the AHR antagonists resveratrol or 3′,4′-dimethoxy flavone attenuated the detrimental effects of BaP on follicle growth [120].
Occupational Chemicals
Butadiene diepoxide
1,3-Butadiene (BD) and the related olefins isoprene and styrene are released during the manufacture of synthetic rubber and thermoplastic resins. An estimated 9.3 million tons of butadiene were produced worldwide in 2005 [121]. Although BD undergoes rapid destruction in the atmosphere, it is remains present at very low concentrations (0.1–1.0 parts per billion) in urban and suburban areas [122]. This is likely related to the fact that these chemicals have also been reported in cigarette smoke, automobile exhaust, and biomass burning [123–125]. Compounds known to contain epoxide moieties (or which are capable of bioactivation by epoxidation) have been shown to affect reproductive function in laboratory animals. Because of the ability of these compounds to become epoxidated, they have the potential to be ovotoxic and carcinogenic [126]. Animal data indicate that BD can cause ovarian atrophy. A 2-yr chronic BD exposure study by the National Toxicology Program observed ovarian atrophy to be a sensitive end point in female mice [127]. Further, convincing data were provided to support that the diepoxide of BD (BDE) is the most reactive metabolite for causing ovotoxicity, and subsequently ovarian atrophy in mice [128, 129]. In one study, the metabolite of BD, 1,3-butadiene monoepoxide (1.43 mmol/kg), depleted preantral follicles by 98% and growing follicles by 87% in female B6C3F1 mice dosed daily for 30 days compared with control animals [128]. At a much lower dose, 0.14 mmol/kg, BDE depleted preantral follicles by 85% and growing follicles by 63%. Thus, the results of this study support that a diepoxide formed in the metabolism of BD is more potent than the monoepoxide at inducing follicle loss.
4-Vinylcyclohexene and its diepoxide metabolite
The dimerization of BD forms 4-vinylcyclohexene (VCH). VCH and its diepoxide metabolite (VCD) are examples of chemicals in this group (reviewed in Hoyer and Sipes [126, 130]). VCH is an industrial chemical released at low concentrations during the manufacture of rubber tires, plasticizers, and pesticides [123]. Thus, exposures in humans are limited to the occupational setting, where proper protective equipment should ensure these exposures are at a minimum. In spite of the unlikelihood of environmental exposures, this is a good model ovotoxicant that may help elucidate mechanisms of action of other ovarian toxicants. Both VCH and VCD have been shown in mice and rats to 1) produce selective destruction of primordial and primary follicles [131, 132]; 2) cause premature ovarian failure [60, 130, 133]; and 3) increase the risk for development of ovarian tumors [126, 134, 135]. Initial studies in rats determined that VCD causes ovotoxicity by accelerating the natural process of atresia (apoptosis), and this requires 15 days of daily dosing [136, 137]. Mechanistic studies of the ovotoxic action have demonstrated that VCD activates proapoptotic signaling events in the BCL2 and mitogen-activated protein kinase families [138–140]. This effect was selective for preantral follicles isolated from rats dosed repeatedly with VCD. These findings demonstrated a molecular mechanism by which VCD causes follicular atresia via the proapoptotic branch of the Bcl-2 proto-oncogene family. Therefore, because VCD causes a physiological form of ovotoxicity, follicle loss is “silent” and mimics normal follicular atresia, and damage caused by VCD might go unnoticed in exposed individuals who are affected. As a result, because VCD only targets ovarian preantral follicles, chronic exposure in women to low levels of this chemical may represent a risk for early menopause without prior evidence of disrupted menstrual cycles [130].
Previous studies had used small preantral follicle fractions (<100 μm) isolated from VCD-dosed rats (in vivo approach). In order to provide more mechanistic insight into molecular mechanisms by which VCD causes ovotoxicity, investigations of cellular and molecular pathways have used in vitro PND4 rat whole ovaries in culture. Because the PND4 ovary is highly enriched in primordial and small primary follicles (selectively targeted by VCD), this approach lends itself particularly well to these studies [31]. Oligoarray analysis comparing in vivo and in vitro exposure to VCD identified genes whose expression is affected in the target follicle population [141]. A key gene identified in that study was Kit, a tyrosine kinase receptor expressed on the oocyte and theca cells, involved in survival and activation/recruitment of primordial follicles [32]. Following binding of KITL to KIT, expressed in the granulosa cell [142], signaling cascades such as the phosphoinositol-3 kinase pathway are activated (reviewed in Liu et al. [143]). Using the in vitro system, it was shown that VCD induces a decrease in Kit mRNA expression on Day 4 of culture, which precedes both an increase in Kitl mRNA and follicle loss on Day 6 of exposure [141, 144]. Interestingly, VCD also decreased KIT protein by Day 4 of culture (Fig. 3) [145]. Therefore, the initial effect of VCD appears to involve posttranslational modifications as opposed to an impact on gene expression. Further, exogenous KITL in culture partially attenuated VCD-induced follicle loss, whereas there was no effect of exogenous BMP15 or GDF9 [141]. Collectively, these findings support the proposal that VCD directly targets the oocyte and VCD-induced inhibition of the KIT pathway is an early initiating event in its ovotoxic effects.
FIG. 3.

Effect of VCD on KIT protein staining. PND4 F344 rat ovaries were cultured with vehicle control or 30 μM VCD for 2 or 4 days and processed for Western blotting or immunofluorescence staining with confocal microscopy. A) Western blotting for KIT or beta-actin (ACTB) protein on Day 2 and Day 4 of VCD exposure (c, control; v, VCD). B) Quantification of KIT protein staining with normalization to ACTB. C–G) Representative images for immunofluorescence staining of KIT protein on Day 4 are shown. Cy-5 red staining (anti-KIT antibody) for control-treated ovary (C) and VCD-treated ovary, and genomic DNA (green YOYO1 stain) overlay (F) for control-treated ovary (D), immunonegative control (E), and VCD-treated ovary (G) at 40× magnification. Thin arrows indicate primordial follicles; thick arrows indicate small primary follicles; bar = 20 μm. H) Quantification of oocyte pericytoplasmic KIT protein staining on Day 2 and Day 4 of VCD exposure. Values are percentage of control; n = 3. *P < 0.05. By either method, oocyte plasma membrane staining was reduced by VCD on Day 4 of incubation, relative to control. Reproduced from Keating et al. [145] with permission from Elsevier.
2-Bromopropane
In the United States, 2-bromopropane (2-BP) is a contaminant of 1-BP, which is used in spray adhesives and as a precision cleaner and degreaser. It may also be used as an intermediate in the synthesis of pharmaceutical dyes and other chemicals [146]. Because of the detrimental effects on the ozone layer caused by chlorofluorocarbons and the ban on their use, 1-BP and 2-BP were previously used in Asia as substitute propellants and cleaning solvents [146]. In the mid 1990s, an occupational health manager reported that a cluster of women performing small-sized tactile switch assembly in an electronics factory in South Korea were experiencing amenorrhea [147, 148]. 2-BP had been introduced into the electronics plant in early 1994 as a substitute solvent for chlorofluorocarbons. Because there were little or no toxicity data available at that time, the solvent was presumed to be nontoxic, and no personal protective equipment was worn. A total of 16 of 25 exposed women were found to have amenorrhea, as well as high levels of FSH and LH, and 10 women complained of hot flashes. Ovaries in six of these women were found to range from atrophic to almost normal upon follow-up laparoscopic examination [149]. This led to a diagnosis of ovarian failure in these women [148, 150, 151]. Later studies in rats demonstrated both neurological and reproductive effects [146, 152]. In female Wistar rats exposed for 8 wk to 2-BP via inhalation, estrous cyclicity was disrupted, and reduced ovarian and uterine weights were measured [153]. In an additional assessment of those animals, there were decreased ovarian follicles in multiple stages of development, but time course and morphological studies performed in rats suggested that 2-BP initially targets primordial follicles [154–156]. This finding substantiated the cause of amenorrhea, ovarian atrophy, and increased hormone levels in the female workers. Following a detailed evaluation of the available information, the National Toxicology Program panel concluded that reproductive effects observed in female rats are similar to those observed in occupationally exposed women. Because of the consistency and similarity of the animal and human data, they concluded that 2-BP is likely to be an ovarian toxicant in rats and humans [146].
Other ovotoxic agents
The alkylating agents triethylenemelamine (TEM) and isopropyl methanesulfonate (IMS) have been shown to destroy oocytes in small follicles in SECXC57BLF1 mice following a single i.p. injection [89]. This destruction was observed within 3 days of dosing with TEM and IMS. In addition to the chemicals discussed thus far, Dobson and Felton [157] reported a variety of other compounds that were capable of producing significant primordial follicle loss in mice. These chemicals included methyl and ethyl methanesulfonate, and urethane. Additionally, of a number of fungal toxins and antibiotics tested, procarbazine HCl and 4-nitroquinoline-1-oxide were ovotoxic. Finally, dibromochloropropane, N-ethyl-N-nitrosourea, and bleomycin demonstrated primordial follicle killing, with bleomycin being the most potent. In general, all of these ovotoxic chemicals are also known to possess mutagenic-carcinogenic effects. Thus, these studies have further provided a correlation between ovotoxicity and subsequent development of tumorigenesis. How these two events are linked is not clearly understood at this time.
Endocrine Disruptors
Exposure of primordial and preantral follicles to estrogenic compounds can have effects on ovarian follicular development. Environmental awareness is growing about the reproductive consequences of exposure of the embryonic and neonatal ovary to endocrine disruptors [158]. These include both synthetic and natural compounds that mimic the mechanisms of endogenous hormones. The majority of studies that examine the role of endocrine disruptors that target primordial and preantral follicles have concentrated on genistein, bisphenol-A (BPA), and diethylstilbestrol (DES).
Genistein, an isoflavonoid phytoestrogen present in soy products, has been shown to have estrogenic activity and bind to the estrogen receptor (ER) in both in vitro and in vivo studies in mice and rats [158]. It has been proposed that genistein has benefits to human health, including inhibiting carcinogenesis, preventing apoptosis, and having antioxidant properties [159–161]. However, there are also adverse effects reported, depending on the timing of exposure, dose level, and end points examined. Animal experiments have demonstrated that fetal exposure to genistein alters the development of the female reproductive system, resulting in delayed estrous cycles and subfertility [162, 163]. Neonatal genistein treatment in rodents has been shown to inhibit breakdown of oocyte nests and increase oocyte survival, resulting in multioocyte follicles (MOFs) in the ovaries of adult mice [164, 165]. Because mice treated with lavendustin, a tyrosine kinase inhibitor, did not develop MOFs, that property of genistein was determined not be responsible for this effect [166]. Mice lacking ERα (ESR1) still developed MOFs when treated with genistein, but mice that lacked ERβ (ESR2) did not, suggesting that ESR2 plays an important role in this process [166]. The percentage of primordial follicles in 4-mo-old rats was higher following genistein treatment, whereas the percentage of antral follicles was lower, suggesting that adult genistein exposure inhibits the transition of primordial to primary follicles [167].
Bisphenol-A is a high-production volume chemical used in the synthesis of polycarbonate plastics and epoxy resins, such as those used to line cans containing food and beverages and those found in dental sealants [168]. Because BPA is one of the most highly produced chemicals (more than 6.4 billion pounds in 2003) and has been shown to leach from containers of food and beverage products, it is considered a wide source of exposure in humans, and thus a potential health risk [169]. Bisphenol-A has been detected in the serum/plasma and follicular fluid of most women tested, as well as in amniotic fluid and fetal serum on the order of 0.2–20 ng/ml [170–172]. Bisphenol-A has been shown to be a selective ER modulator with tissue- and species-specific effects [173, 174]. Neonatal exposure to BPA resulted in a decrease in primordial follicle numbers and an increase in growing follicles at PND8, indicating a stimulatory effect on early follicular growth [175]. Similar to genistein, postnatal exposure to high doses of BPA has also been shown to increase MOFs [176]. Additionally, when BPA was accidentally released because of improperly washed animal cages and water bottles, the exposure in laboratory animals resulted in meiotic disturbances due to misalignment of the chromosomes on the first meiotic spindle. This exposure ultimately resulted in aneuploidy in the oocytes of the exposed mice, an effect that could be reproduced when mice were intentionally exposed to BPA at a similar dose (20 ng/g body weight; [177]). By exposing knockout mice of ESR1 or ESR2 in utero to BPA, it was determined that loss of ESR2 caused similar effects on aneuploidy and other meiotic disturbances to BPA in oocytes [178]. In contrast, loss of ESR1 did not cause such effects, nor did exposure of knockout mice to BPA cause further increases in meiotic abnormalities.
Diethylstilbestrol, a potent nonsteroidal estrogen, was once widely prescribed to women for the prevention of miscarriage (from the 1940s to the 1970s). However, DES use in pregnant women was banned because of increased frequency of vaginal carcinoma and uterine abnormalities found in the daughters of DES-treated mothers [179]. The potency of ER binding is similar to estradiol, but its lower binding to circulating steroid-binding proteins and its longer half-life in circulation make this drug more bioavailable than estradiol in both the mother and fetus during in utero exposures [180]. Abnormalities in the oviduct and ovary were found in rats and mice exposed to DES perinatally [181]. In addition, mice treated as neonates with DES had an increased occurrence of MOFs [182]. The MOF oocytes from DES-treated neonatal ovaries were also less likely to be fertilized using in vitro fertilization protocols, suggesting that DES detrimentally affects oocyte quality [183]. Diethylstilbestrol has since then been used as a model chemical for the study of estrogenic environmental endocrine disruptors.
CONCLUSION
Only a limited number of exposures are known to cause toxicity to preantral follicles, but those exposures can lead to significant health effects, including infertility, amenorrhea, and alterations in hormone levels. In most cases, effects observed involve decreases in the ovarian reserve of primordial follicles. Mechanisms underlying such ovarian toxicity are poorly understood. Further research is needed both in the basic biology of ovarian function and alterations induced by detrimental exposures in order to better identify what types of exposures pose a risk to women.
Future work that would add significantly to our understanding of ovarian toxicants includes determining what women are exposed to, what effects are detected through epidemiological monitoring, and what mechanisms of action are involved in those exposures that are known to impact ovarian function. Further and continued epidemiological assessments of reproductive effects of environmental, occupational, and medical exposures are needed to characterize ovotoxic effects of current exposures and ensure that new exposures do not negatively impact reproductive function. Studies examining alterations in age at menopause in women with known exposures versus unexposed women will continue to be needed, especially with chemicals identified to impact the ovary in animal studies. More measurements of chemicals in bodily fluids and tissues of humans, especially in relevant samples like follicular fluid, are needed to determine the relevant exposures and levels that need to be studied for reproductive toxicity. A better understanding of the effects of mixtures will be an important area of investigation. Finally, further validation of in vitro approaches to identifying ovotoxicants, including cell, follicle, and whole neonatal ovarian cultures, will be important for both screening of chemicals and examining their mechanism(s) of toxicity. Together, these directions should improve our understanding of causes of early ovarian failure and how to avoid or prevent chemical exposures that have a detrimental impact on reproductive function in women.
Supplementary Material
Footnotes
Supported by National Institutes of Health grants ES09246, 007091, and 06694; the Canadian Institutes of Health Research; and the Natural Sciences and Engineering Research Council of Canada.
REFERENCES
- Ying Y, Zhao GQ. Cooperation of endoderm-derived BMP2 and extraembryonic ectoderm-derived BMP4 in primordial germ cell generation in the mouse. Dev Biol 2001; 232: 484 492. [DOI] [PubMed] [Google Scholar]
- Ying Y, Qi X, Zhao GQ. Induction of primordial germ cells from murine epiblasts by synergistic action of BMP4 and BMP8B signaling pathways. Proc Natl Acad Sci U S A 2001; 98: 7858 7862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohinata Y, Ohta H, Shigeta M, Yamanaka K, Wakayama T, Saitou M. A signaling principle for the specification of the germ cell lineage in mice. Cell 2009; 137: 571 584. [DOI] [PubMed] [Google Scholar]
- Merchant H, Zamboni L. Presence of connections between follicles in juvenile mouse ovaries. Am J Anat 1972; 134: 127 132. [DOI] [PubMed] [Google Scholar]
- Pepling ME, de Cuevas M, Spradling AC. Germline cysts: a conserved phase of germ cell development? Trends Cell Biol 1999; 9: 257 262. [DOI] [PubMed] [Google Scholar]
- Pepling ME, Spradling AC. Female mouse germ cells form synchronously dividing cysts. Development 1998; 125: 3323 3328. [DOI] [PubMed] [Google Scholar]
- Pepling ME, Spradling AC. Mouse ovarian germ cell cysts undergo programmed breakdown to form primordial follicles. Dev Biol 2001; 234: 339 351. [DOI] [PubMed] [Google Scholar]
- Gomperts M, Wylie C, Heasman J. Primordial germ cell migration. Ciba Found Symp 1994; 182: 121 134. [DOI] [PubMed] [Google Scholar]
- Mazaud S, Guyot R, Guigon CJ, Coudouel N, Le Magueresse-Battistoni B, Magre S. Basal membrane remodeling during follicle histogenesis in the rat ovary: contribution of proteinases of the MMP and PA families. Dev Biol 2005; 277: 403 416. [DOI] [PubMed] [Google Scholar]
- Sawyer HR, Smith P, Heath DA, Juengel JL, Wakefield SJ, McNatty KP. Formation of ovarian follicles during fetal development in sheep. Biol Reprod 2002; 66: 1134 1150. [DOI] [PubMed] [Google Scholar]
- Bristol-Gould SK, Kreeger PK, Selkirk CG, Kilen SM, Mayo KE, Shea LD, Woodruff TK. Fate of the initial follicle pool: empirical and mathematical evidence supporting its sufficiency for adult fertility. Dev Biol 2006; 298: 149 154. [DOI] [PubMed] [Google Scholar]
- Hirshfield AN. Heterogeneity of cell populations that contribute to the formation of primordial follicles in rats. Biol Reprod 1992; 47: 466 472. [DOI] [PubMed] [Google Scholar]
- Baker TG. A quantitative and cytological study of germ cells in human ovaries. Proc R Soc Lond B Biol Sci 1963; 158: 417 433. [DOI] [PubMed] [Google Scholar]
- Peters H, Himelstein-Braw R, Faber M. The normal development of the ovary in childhood. Acta Endocrinol (Copenh) 1976; 82: 617 630. [DOI] [PubMed] [Google Scholar]
- Skinner MK. Regulation of primordial follicle assembly and development. Hum Reprod Update 2005; 11: 461 471. [DOI] [PubMed] [Google Scholar]
- Hirshfield AN. Development of follicles in the mammalian ovary. Int Rev Cytol 1991; 124: 43 101. [DOI] [PubMed] [Google Scholar]
- Wagenen GV, Simpson ME. Embryology of the Ovary and Testis Homo sapiens and Macaca mulatta New Haven: Yale University Press; 1965. [Google Scholar]
- Gougeon A. Regulation of ovarian follicular development in primates: facts and hypotheses. Endocr Rev 1996; 17: 121 155. [DOI] [PubMed] [Google Scholar]
- McGee EA, Hsu SY, Kaipia A, Hsueh AJ. Cell death and survival during ovarian follicle development. Mol Cell Endocrinol 1998; 140: 15 18. [DOI] [PubMed] [Google Scholar]
- Tingen CM, Bristol-Gould SK, Kiesewetter SE, Wellington JT, Shea L, Woodruff TK. Prepubertal primordial follicle loss in mice is not due to classical apoptotic pathways. Biol Reprod 2009; 81: 16 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez GI, Robles R, Knudson CM, Flaws JA, Korsmeyer SJ, Tilly JL. Prolongation of ovarian lifespan into advanced chronological age by Bax-deficiency. Nat Genet 1999; 21: 200 203. [DOI] [PubMed] [Google Scholar]
- Bergeron L, Perez GI, Macdonald G, Shi L, Sun Y, Jurisicova A, Varmuza S, Latham KE, Flaws JA, Salter JC, Hara H, Moskowitz MA, Li E, Greenberg A, Tilly JL, Yuan J. Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev 1998; 12: 1304 1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaws JA, Hirshfield AN, Hewitt JA, Babus JK, Furth PA. Effect of bcl-2 on the primordial follicle endowment in the mouse ovary. Biol Reprod 2001; 64: 1153 1159. [DOI] [PubMed] [Google Scholar]
- Ratts VS, Flaws JA, Kolp R, Sorenson CM, Tilly JL. Ablation of bcl-2 gene expression decreases the numbers of oocytes and primordial follicles established in the post-natal female mouse gonad. Endocrinology 1995; 136: 3665 3668. [DOI] [PubMed] [Google Scholar]
- Baker TG, Franchi LL. The fine structure of oogonia and oocytes in the rhesus monkey (Macaca mulatta). Z Zellforsch Mikrosk Anat 1972; 126: 53 74. [DOI] [PubMed] [Google Scholar]
- Faddy MJ, Gosden RG, Gougeon A, Richardson SJ, Nelson JF. Accelerated disappearance of ovarian follicles in mid-life: implications for forecasting menopause. Hum Reprod 1992; 7: 1342 1346. [DOI] [PubMed] [Google Scholar]
- Braw-Tal R, Roth Z. Gene expression for LH receptor, 17 alpha-hydroxylase and StAR in the theca interna of preantral and early antral follicles in the bovine ovary. Reproduction 2005; 129: 453 461. [DOI] [PubMed] [Google Scholar]
- Campbell BK, Telfer EE, Webb R, Baird DT. Evidence of a role for follicle-stimulating hormone in controlling the rate of preantral follicle development in sheep. Endocrinology 2004; 145: 1870 1879. [DOI] [PubMed] [Google Scholar]
- Silva JR, Van den Hurk R, de Matos MH, dos Santos RR, Pessoa C, de Moraes MO, de Figueiredo JR. Influences of FSH and EGF on primordial follicles during in vitro culture of caprine ovarian cortical tissue. Theriogenology 2004; 61: 1691 1704. [DOI] [PubMed] [Google Scholar]
- Wandji SA, Srsen V, Voss AK, Eppig JJ, Fortune JE. Initiation in vitro of growth of bovine primordial follicles. Biol Reprod 1996; 55: 942 948. [DOI] [PubMed] [Google Scholar]
- Devine PJ, Sipes IG, Skinner MK, Hoyer PB. Characterization of a rat in vitro ovarian culture system to study the ovarian toxicant 4-vinylcyclohexene diepoxide. Toxicol Appl Pharmacol 2002; 184: 107 115. [PubMed] [Google Scholar]
- Hutt KJ, McLaughlin EA, Holland MK. Kit ligand and c-Kit have diverse roles during mammalian oogenesis and folliculogenesis. Mol Hum Reprod 2006; 12: 61 69. [DOI] [PubMed] [Google Scholar]
- Nilsson EE, Skinner MK. Kit ligand and basic fibroblast growth factor interactions in the induction of ovarian primordial to primary follicle transition. Mol Cell Endocrinol 2004; 214: 19 25. [DOI] [PubMed] [Google Scholar]
- Nilsson EE, Detzel C, Skinner MK. Platelet-derived growth factor modulates the primordial to primary follicle transition. Reproduction 2006; 131: 1007 1015. [DOI] [PubMed] [Google Scholar]
- Nilsson EE, Kezele P, Skinner MK. Leukemia inhibitory factor (LIF) promotes the primordial to primary follicle transition in rat ovaries. Mol Cell Endocrinol 2002; 188: 65 73. [DOI] [PubMed] [Google Scholar]
- Kezele P, Nilsson EE, Skinner MK. Keratinocyte growth factor acts as a mesenchymal factor that promotes ovarian primordial to primary follicle transition. Biol Reprod 2005; 73: 967 973. [DOI] [PubMed] [Google Scholar]
- Dole G, Nilsson EE, Skinner MK. Glial-derived neurotrophic factor promotes ovarian primordial follicle development and cell-cell interactions during folliculogenesis. Reproduction 2008; 135: 671 682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson EE, Skinner MK. Bone morphogenetic protein-4 acts as an ovarian follicle survival factor and promotes primordial follicle development. Biol Reprod 2003; 69: 1265 1272. [DOI] [PubMed] [Google Scholar]
- Lee WS, Yoon SJ, Yoon TK, Cha KY, Lee SH, Shimasaki S, Lee S, Lee KA. Effects of bone morphogenetic protein-7 (BMP-7) on primordial follicular growth in the mouse ovary. Mol Reprod Dev 2004; 69: 159 163. [DOI] [PubMed] [Google Scholar]
- Yu N, Roy SK. Development of primordial and prenatal follicles from undifferentiated somatic cells and oocytes in the hamster prenatal ovary in vitro: effect of insulin. Biol Reprod 1999; 61: 1558 1567. [DOI] [PubMed] [Google Scholar]
- Kezele PR, Nilsson EE, Skinner MK. Insulin but not insulin-like growth factor-1 promotes the primordial to primary follicle transition. Mol Cell Endocrinol 2002; 192: 37 43. [DOI] [PubMed] [Google Scholar]
- Dissen GA, Garcia-Rudaz C, Ojeda SR. Role of neurotrophic factors in early ovarian development. Semin Reprod Med 2009; 27: 24 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dissen GA, Romero C, Paredes A, Ojeda SR. Neurotrophic control of ovarian development. Microsc Res Tech 2002; 59: 509 515. [DOI] [PubMed] [Google Scholar]
- Durlinger AL, Kramer P, Karels B, de Jong FH, Uilenbroek JT, Grootegoed JA, Themmen AP. Control of primordial follicle recruitment by anti-Mullerian hormone in the mouse ovary. Endocrinology 1999; 140: 5789 5796. [DOI] [PubMed] [Google Scholar]
- Ding CC, Thong KJ, Krishna A, Telfer EE. Activin A inhibits activation of human primordial follicles in vitro. J Assist Reprod Genet 2010; 27: 141 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt JE, Jackson A, Roman SD, Aitken RJ, Koopman P, McLaughlin EA. CXCR4/SDF1 interaction inhibits the primordial to primary follicle transition in the neonatal mouse ovary. Dev Biol 2006; 293: 449 460. [DOI] [PubMed] [Google Scholar]
- Moore RK, Shimasaki S. Molecular biology and physiological role of the oocyte factor, BMP-15. Mol Cell Endocrinol 2005; 234: 67 73. [DOI] [PubMed] [Google Scholar]
- Juengel JL, Bodensteiner KJ, Heath DA, Hudson NL, Moeller CL, Smith P, Galloway SM, Davis GH, Sawyer HR, McNatty KP. Physiology of GDF9 and BMP15 signalling molecules. Anim Reprod Sci 2004; 82–83: 447 460. [DOI] [PubMed] [Google Scholar]
- Desmeules P, Devine PJ. Characterizing the ovotoxicity of cyclophosphamide metabolites on cultured mouse ovaries. Toxicol Sci 2006; 90: 500 509. [DOI] [PubMed] [Google Scholar]
- Nilsson E, Parrott JA, Skinner MK. Basic fibroblast growth factor induces primordial follicle development and initiates folliculogenesis. Mol Cell Endocrinol 2001; 175: 123 130. [DOI] [PubMed] [Google Scholar]
- Demeestere I, Centner J, Gervy C, Englert Y, Delbaere A. Impact of various endocrine and paracrine factors on in vitro culture of preantral follicles in rodents. Reproduction 2005; 130: 147 156. [DOI] [PubMed] [Google Scholar]
- Li R, Phillips DM, Mather JP. Activin promotes ovarian follicle development in vitro. Endocrinology 1995; 136: 849 856. [DOI] [PubMed] [Google Scholar]
- Liu X, Andoh K, Yokota H, Kobayashi J, Abe Y, Yamada K, Mizunuma H, Ibuki Y. Effects of growth hormone, activin, and follistatin on the development of preantral follicle from immature female mice. Endocrinology 1998; 139: 2342 2347. [DOI] [PubMed] [Google Scholar]
- Smitz J, Cortvrindt R, Hu Y, Vanderstichele H. Effects of recombinant activin A on in vitro culture of mouse preantral follicles. Mol Reprod Dev 1998; 50: 294 304. [DOI] [PubMed] [Google Scholar]
- McGee EA, Chun SY, Lai S, He Y, Hsueh AJ. Keratinocyte growth factor promotes the survival, growth, and differentiation of preantral ovarian follicles. Fertil Steril 1999; 71: 732 738. [DOI] [PubMed] [Google Scholar]
- Reynaud K, Cortvrindt R, Smitz J, Driancourt MA. Effects of Kit Ligand and anti-Kit antibody on growth of cultured mouse preantral follicles. Mol Reprod Dev 2000; 56: 483 494. [DOI] [PubMed] [Google Scholar]
- Hayashi M, McGee EA, Min G, Klein C, Rose UM. van DM, Hsueh AJ. Recombinant growth differentiation factor-9 (GDF-9) enhances growth and differentiation of cultured early ovarian follicles. Endocrinology 1999; 140: 1236 1244. [DOI] [PubMed] [Google Scholar]
- Zhao J, Taverne MA, Van Der Weijden GC, Bevers MM, Van den Hurk R. Insulin-like growth factor-I (IGF-I) stimulates the development of cultured rat pre-antral follicles. Mol Reprod Dev 2001; 58: 287 296. [DOI] [PubMed] [Google Scholar]
- Demeestere I, Gervy C, Centner J, Devreker F, Englert Y, Delbaere A. Effect of insulin-like growth factor-I during preantral follicular culture on steroidogenesis, in vitro oocyte maturation, and embryo development in mice. Biol Reprod 2004; 70: 1664 1669. [DOI] [PubMed] [Google Scholar]
- Hooser SB, Douds DP, DeMerell DG, Hoyer PB, Sipes IG. Long-term ovarian and gonadotropin changes in mice exposed to 4-vinylcyclohexene. Reprod Toxicol 1994; 8: 315 323. [DOI] [PubMed] [Google Scholar]
- Shuster LT, Gostout BS, Grossardt BR, Rocca WA. Prophylactic oophorectomy in premenopausal women and long-term health. Menopause Int 2008; 14: 111 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santen RJ, Allred DC, Ardoin SP, Archer DF, Boyd N, Braunstein GD, Burger HG, Colditz GA, Davis SR, Gambacciani M, Gower BA, Henderson VW, Jarjour WN, Karas RH, Kleerekoper M, Lobo RA, Manson JE, Marsden J, Martin KA, Martin L, Pinkerton JV, Rubinow DR, Teede H, Thiboutot DM, Utian WH. Postmenopausal hormone therapy: an Endocrine Society scientific statement. J Clin Endocrinol Metab 2010; 95: s1 s66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne J. Long-term genetic and reproductive effects of ionizing radiation and chemotherapeutic agents on cancer patients and their offspring. Teratology 1999; 59: 210 215. [DOI] [PubMed] [Google Scholar]
- Meirow D, Nugent D. The effects of radiotherapy and chemotherapy on female reproduction. Hum Reprod Update 2001; 7: 535 543. [DOI] [PubMed] [Google Scholar]
- Chemaitilly W, Mertens AC, Mitby P, Whitton J, Stovall M, Yasui Y, Robison LL, Sklar CA. Acute ovarian failure in the childhood cancer survivor study. J Clin Endocrinol Metab 2006; 91: 1723 1728. [DOI] [PubMed] [Google Scholar]
- Molina JR, Barton DL, Loprinzi CL. Chemotherapy-induced ovarian failure: manifestations and management. Drug Saf 2005; 28: 401 416. [DOI] [PubMed] [Google Scholar]
- Sklar CA, Mertens AC, Mitby P, Whitton J, Stovall M, Kasper C, Mulder J, Green D, Nicholson HS, Yasui Y, Robison LL. Premature menopause in survivors of childhood cancer: a report from the childhood cancer survivor study. J Natl Cancer Inst 2006; 98: 890 896. [DOI] [PubMed] [Google Scholar]
- Himelstein-Braw R, Peters H, Faber M. Influence of irradiation and chemotherapy on the ovaries of children with abdominal tumours. Br J Cancer 1977; 36: 269 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicosia SV, Matus-Riley M, Meadows AT. Gonadal effects of cancer therapy in girls. Cancer 1985; 55: 2364 2372. [DOI] [PubMed] [Google Scholar]
- Larsen EC, Muller J, Schmiegelow K, Rechnitzer C, Andersen AN. Reduced ovarian function in long-term survivors of radiation- and chemotherapy-treated childhood cancer. J Clin Endocrinol Metab 2003; 88: 5307 5314. [DOI] [PubMed] [Google Scholar]
- Lee CJ, Yoon YD. Gamma-radiation-induced follicular degeneration in the prepubertal mouse ovary. Mutat Res 2005; 578: 247 255. [DOI] [PubMed] [Google Scholar]
- Jarrell J, Younglai EV, Barr R, O'Connell G, Belbeck L, McMahon A. An analysis of the effects of increasing doses of ionizing radiation to the exteriorized rat ovary on follicular development, atresia, and serum gonadotropin levels. Am J Obstet Gynecol 1986; 154: 306 309. [DOI] [PubMed] [Google Scholar]
- Pesty A, Doussau M, Lahaye JB, Lefevre B. Whole-body or isolated ovary (60)Co irradiation: effects on in vivo and in vitro folliculogenesis and oocyte maturation. Reprod Toxicol 2010; 29: 93 98. [DOI] [PubMed] [Google Scholar]
- Ataya K, Pydyn E, Ramahi-Ataya A, Orton CG. Is radiation-induced ovarian failure in rhesus monkeys preventable by luteinizing hormone-releasing hormone agonists?: preliminary observations. J Clin Endocrinol Metab 1995; 80: 790 795. [DOI] [PubMed] [Google Scholar]
- Adriaens I, Smitz J, Jacquet P. The current knowledge on radiosensitivity of ovarian follicle development stages. Hum Reprod Update 2009; 15: 359 377. [DOI] [PubMed] [Google Scholar]
- Wallace WH, Thomson AB, Kelsey TW. The radiosensitivity of the human oocyte. Hum Reprod 2003; 18: 117 121. [DOI] [PubMed] [Google Scholar]
- Sklar C. Maintenance of ovarian function and risk of premature menopause related to cancer treatment. J Natl Cancer Inst Monogr 2005; 34: 25 27. [DOI] [PubMed] [Google Scholar]
- Thibaud E, Rodriguez-Macias K, Trivin C, Esperou H, Michon J, Brauner R. Ovarian function after bone marrow transplantation during childhood. Bone Marrow Transplant 1998; 21: 287 290. [DOI] [PubMed] [Google Scholar]
- Green DM, Kawashima T, Stovall M, Leisenring W, Sklar CA, Mertens AC, Donaldson SS, Byrne J, Robison LL. Fertility of female survivors of childhood cancer: a report from the childhood cancer survivor study. J Clin Oncol 2009; 27: 2677 2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace WH, Anderson RA, Irvine DS. Fertility preservation for young patients with cancer: who is at risk and what can be offered? Lancet Oncol 2005; 6: 209 218. [DOI] [PubMed] [Google Scholar]
- Wallace WH, Thomson AB, Saran F, Kelsey TW. Predicting age of ovarian failure after radiation to a field that includes the ovaries. Int J Radiat Oncol Biol Phys 2005; 62: 738 744. [DOI] [PubMed] [Google Scholar]
- Davis VJ. Female gamete preservation. Cancer 2006; 107: 1690 1694. [DOI] [PubMed] [Google Scholar]
- Stroud JS, Mutch D, Rader J, Powell M, Thaker PH, Grigsby PW. Effects of cancer treatment on ovarian function. Fertil Steril 2009; 92: 417 427. [DOI] [PubMed] [Google Scholar]
- Wallace WH, Shalet SM, Crowne EC, Morris-Jones PH, Gattamaneni HR, Price DA. Gonadal dysfunction due to cis-platinum. Med Pediatr Oncol 1989; 17: 409 413. [DOI] [PubMed] [Google Scholar]
- Yeh J, Kim B, Liang YJ, Peresie J. Mullerian inhibiting substance as a novel biomarker of cisplatin-induced ovarian damage. Biochem Biophys Res Commun 2006; 348: 337 344. [DOI] [PubMed] [Google Scholar]
- Kociba RJ, Sleight SD. Acute toxicologic and pathologic effects of cis-diamminedichloroplatinum (NSC-119875) in the male rat. Cancer Chemother Rep 1971; 55: 1 8. [PubMed] [Google Scholar]
- Yucebilgin MS, Terek MC, Ozsaran A, Akercan F, Zekioglu O, Isik E, Erhan Y. Effect of chemotherapy on primordial follicular reserve of rat: an animal model of premature ovarian failure and infertility. Aust N Z J Obstet Gynaecol 2004; 44: 6 9. [DOI] [PubMed] [Google Scholar]
- Borovskaya TG, Goldberg VE, Fomina TI, Pakhomova AV, Kseneva SI, Poluektova ME, Goldberg ED. Morphological and functional state of rat ovaries in early and late periods after administration of platinum cytostatics. Bull Exp Biol Med 2004; 137: 331 335. [DOI] [PubMed] [Google Scholar]
- Generoso W, Stout SK, Huff SW. Effects of alkylating chemicals on reproductive capacity of adult female mice. Mutat Res 1971; 13: 171 184. [DOI] [PubMed] [Google Scholar]
- Sakurada Y, Kudo S, Iwasaki S, Miyata Y, Nishi M, Masumoto Y. Collaborative work on evaluation of ovarian toxicity. 5) Two- or four-week repeated-dose studies and fertility study of busulfan in female rats. J Toxicol Sci 2009; 34(suppl 1):SP65–SP72. [DOI] [PubMed] [Google Scholar]
- Tan SJ, Yeh YC, Shang WJ, Wu GJ, Liu JY, Chen CH. Protective effect of a gonadotropin-releasing hormone analogue on chemotherapeutic agent-induced ovarian gonadotoxicity: a mouse model. Eur J Obstet Gynecol Reprod Biol 2010; 149: 182 185. [DOI] [PubMed] [Google Scholar]
- Meirow D. Reproduction post-chemotherapy in young cancer patients. Mol Cell Endocrinol 2000; 169: 123 131. [DOI] [PubMed] [Google Scholar]
- Plowchalk DR, Mattison DR. Phosphoramide mustard is responsible for the ovarian toxicity of cyclophosphamide. Toxicol Appl Pharmacol 1991; 107: 472 481. [DOI] [PubMed] [Google Scholar]
- Howell S, Shalet S. Gonadal damage from chemotherapy and radiotherapy. Endocrinol Metab Clin North Am 1998; 27: 927 943. [DOI] [PubMed] [Google Scholar]
- Costa M, Colia D. Treating infertility in autoimmune patients. Rheumatology (Oxford) 2008; 47 (suppl 3): iii38 iii41. [DOI] [PubMed] [Google Scholar]
- Manger K, Wildt L, Kalden JR, Manger B. Prevention of gonadal toxicity and preservation of gonadal function and fertility in young women with systemic lupus erythematosus treated by cyclophosphamide: the PREGO-Study. Autoimmun Rev 2006; 5: 269 272. [DOI] [PubMed] [Google Scholar]
- Plowchalk DR, Mattison DR. Reproductive toxicity of cyclophosphamide in the C57BL/6N mouse: 1. Effects on ovarian structure and function. Reprod Toxicol 1992; 6: 411 421. [DOI] [PubMed] [Google Scholar]
- Meirow D, Lewis H, Nugent D, Epstein M. Subclinical depletion of primordial follicular reserve in mice treated with cyclophosphamide: clinical importance and proposed accurate investigative tool. Hum Reprod 1999; 14: 1903 1907. [DOI] [PubMed] [Google Scholar]
- Meirow D, Epstein M, Lewis H, Nugent D, Gosden RG. Administration of cyclophosphamide at different stages of follicular maturation in mice: effects on reproductive performance and fetal malformations. Hum Reprod 2001; 16: 632 637. [DOI] [PubMed] [Google Scholar]
- Petrillo SK, Desmeules P, Truong TQ, Devine PJ. Detection of DNA damage in oocytes of small ovarian follicles following phosphoramide mustard exposures of cultured rodent ovaries in vitro. Toxicol Appl Pharmacol 2011; 253: 94 102. [DOI] [PubMed] [Google Scholar]
- Colvin OM. An overview of cyclophosphamide development and clinical applications. Curr Pharm Des 1999; 5: 555 560. [PubMed] [Google Scholar]
- Ataya KM, Valeriote FA, Ramahi-Ataya AJ. Effect of cyclophosphamide on the immature rat ovary. Cancer Res 1989; 49: 1660 1664. [PubMed] [Google Scholar]
- Jick H, Porter J, Morrison AS. Relation between smoking and age of natural menopause. Report from the Boston Collaborative Drug Surveillance Program, Boston University Medical Center. Lancet 1977; 1: 1354 1355. [DOI] [PubMed] [Google Scholar]
- Everson RB, Sandler DP, Wilcox AJ, Schreinemachers D, Shore DL, Weinberg C. Effect of passive exposure to smoking on age at natural menopause. Br Med J (Clin Res Ed) 1986; 293: 792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal MS, Zhu J, Foster WG. Quantification of benzo[a]pyrene and other PAHs in the serum and follicular fluid of smokers versus non-smokers. Reprod Toxicol 2008; 25: 100 106. [DOI] [PubMed] [Google Scholar]
- El-Nemr A, Al-Shawaf T, Sabatini L, Wilson C, Lower AM, Grudzinskas JG. Effect of smoking on ovarian reserve and ovarian stimulation in in-vitro fertilization and embryo transfer. Hum Reprod 1998; 13: 2192 2198. [DOI] [PubMed] [Google Scholar]
- Weinberg CR, Wilcox AJ, Baird DD. Reduced fecundability in women with prenatal exposure to cigarette smoking. Am J Epidemiol 1989; 129: 1072 1078. [DOI] [PubMed] [Google Scholar]
- Stedman RL. The chemical composition of tobacco and tobacco smoke. Chem Rev 1968; 68: 153 207. [DOI] [PubMed] [Google Scholar]
- Tuttle AM, Stampfli M, Foster WG. Cigarette smoke causes follicle loss in mice ovaries at concentrations representative of human exposure. Hum Reprod 2009; 24: 1452 1459. [DOI] [PubMed] [Google Scholar]
- Vahakangas K, Rajaniemi H, Pelkonen O. Ovarian toxicity of cigarette smoke exposure during pregnancy in mice. Toxicol Lett 1985; 25: 75 80. [DOI] [PubMed] [Google Scholar]
- Mattison DR, Thorgeirsson SS. Ovarian aryl hydrocarbon hydroxylase activity and primordial oocyte toxicity of polycyclic aromatic hydrocarbons in mice. Cancer Res 1979; 39: 3471 3475. [PubMed] [Google Scholar]
- Borman SM, Christian PJ, Sipes IG, Hoyer PB. Ovotoxicity in female Fischer rats and B6 mice induced by low-dose exposure to three polycyclic aromatic hydrocarbons: comparison through calculation of an ovotoxic index. Toxicol Appl Pharmacol 2000; 167: 191 198. [DOI] [PubMed] [Google Scholar]
- Mattison DR. Morphology of oocyte and follicle destruction by polycyclic aromatic hydrocarbons in mice. Toxicol Appl Pharmacol 1980; 53: 249 259. [DOI] [PubMed] [Google Scholar]
- Mattison DR. Difference in sensitivity of rat and mouse primordial oocytes to destruction by polycyclic aromatic hydrocarbons. Chem Biol Interact 1979; 28: 133 137. [DOI] [PubMed] [Google Scholar]
- Mattison DR, Shiromizu K, Nightingale MS. Oocyte destruction by polycyclic aromatic hydrocarbons. Am J Ind Med 1983; 4: 191 202. [PubMed] [Google Scholar]
- Rajapaksa KS, Sipes IG, Hoyer PB. Involvement of microsomal epoxide hydrolase enzyme in ovotoxicity caused by 7,12-dimethylbenz[a]anthracene. Toxicol Sci 2007; 96: 327 334. [DOI] [PubMed] [Google Scholar]
- Igawa Y, Keating AF, Rajapaksa KS, Sipes IG, Hoyer PB. Evaluation of ovotoxicity induced by 7, 12-dimethylbenz[a]anthracene and its 3,4-diol metabolite utilizing a rat in vitro ovarian culture system. Toxicol Appl Pharmacol 2009; 234: 361 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matikainen T, Perez GI, Jurisicova A, Pru JK, Schlezinger JJ, Ryu HY, Laine J, Sakai T, Korsmeyer SJ, Casper RF, Sherr DH, Tilly JL. Aromatic hydrocarbon receptor-driven Bax gene expression is required for premature ovarian failure caused by biohazardous environmental chemicals. Nat Genet 2001; 28: 355 360. [DOI] [PubMed] [Google Scholar]
- Jurisicova A, Taniuchi A, Li H, Shang Y, Antenos M, Detmar J, Xu J, Matikainen T. Benito Hernandez A, Nunez G, Casper RF. Maternal exposure to polycyclic aromatic hydrocarbons diminishes murine ovarian reserve via induction of Harakiri. J Clin Invest 2007; 117: 3971 3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal MS. Mulligan Tuttle AM, Casper RF, Lagunov A, Foster WG. Aryl hydrocarbon receptor antagonists attenuate the deleterious effects of benzo[a]pyrene on isolated rat follicle development. Reprod Biomed Online 2010; 21: 100 108. [DOI] [PubMed] [Google Scholar]
- IARC. IARC monographs on the evaluation of carcinogenic risks to humans. 1,3-butadiene, ethylene oxide and vinyl halides (vinyl fluoride, vinyl chloride and vinyl bromide). IARC Monogr Eval Carcinog Risks Hum 2008; 97: 3 471. [PMC free article] [PubMed] [Google Scholar]
- Mumtaz MM, George JD, Gold KW, Cibulas W, Derosa CT. ATSDR evaluation of health effects of chemicals. IV. Polycyclic aromatic hydrocarbons (PAHs): understanding a complex problem. Toxicol Ind Health 1996; 12: 742 995. [DOI] [PubMed] [Google Scholar]
- IARC (International Agency for Research on Cancer). IARC monographs on the evaluation of carcinogenic risks to humans: some industrial chemicals. Lyon, France: 1994. [Google Scholar]
- Melnick RL, Huff J. 1,3-Butadiene: toxicity and carcinogenicity in laboratory animals and humans. Rev Environ Contam Toxicol 1992; 124: 111 144. [DOI] [PubMed] [Google Scholar]
- Grant RL, Haney J, Curry AL, Honeycutt M. A chronic reference value for 1,3-butadiene based on an updated noncancer toxicity assessment. J Toxicol Environ Health B Crit Rev 2010; 13: 460 475. [DOI] [PubMed] [Google Scholar]
- Hoyer PB, Sipes IG. Assessment of follicle destruction in chemical-induced ovarian toxicity. Annu Rev Pharmacol Toxicol 1996; 36: 307 331. [DOI] [PubMed] [Google Scholar]
- National Toxicology Program. NTP toxicology and carcinogenesis studies of 1,3-butadiene (CAS No. 106-99-0) in B6C3F1 mice (inhalation studies). Natl Toxicol Program Tech Rep Ser 1993; 434: 1 389. [PubMed] [Google Scholar]
- Doerr JK, Hooser SB, Smith BJ, Sipes IG. Ovarian toxicity of 4-vinylcyclohexene and related olefins in B6C3F1 mice: role of diepoxides. Chem Res Toxicol 1995; 8: 963 969. [DOI] [PubMed] [Google Scholar]
- Doerr JK, Hollis EA, Sipes IG. Species difference in the ovarian toxicity of 1,3-butadiene epoxides in B6C3F1 mice and Sprague-Dawley rats. Toxicology 1996; 113: 128 136. [DOI] [PubMed] [Google Scholar]
- Hoyer PB, Sipes IG. Development of an animal model for ovotoxicity using 4-vinylcyclohexene: a case study. Birth Defects Res B Dev Reprod Toxicol 2007; 80: 113 125. [DOI] [PubMed] [Google Scholar]
- Smith BJ, Mattison DR, Sipes IG. The role of epoxidation in 4-vinylcyclohexene-induced ovarian toxicity. Toxicol Appl Pharmacol 1990; 105: 372 381. [DOI] [PubMed] [Google Scholar]
- Ito A, Mafune N, Kimura T. Collaborative work on evaluation of ovarian toxicity. 4) Two- or four-week repeated dose study of 4-vinylcyclohexene diepoxide in female rats. J Toxicol Sci 2009; 34(suppl 1):SP53–SP58. [DOI] [PubMed] [Google Scholar]
- Mayer LP, Pearsall NA, Christian PJ, Devine PJ, Payne CM, McCuskey MK, Marion SL, Sipes IG, Hoyer PB. Long-term effects of ovarian follicular depletion in rats by 4- vinylcyclohexene diepoxide. Reprod Toxicol 2002; 16: 775 781. [DOI] [PubMed] [Google Scholar]
- National Toxicology Program. Toxicology and Carcinogenesis Studies of 4-vinyl-1-cyclohexene in F344/a Rats and B6C3F1 Mice (Gavage Studies). 303. Rockville, MD: National Institutes of Health; 1986. [Google Scholar]
- National Toxicology Program. Toxicology and Carcinogenesis studies of 4-vinyl-1-cyclohexene diepoxide in F344/N Rats and B6C3F1 Mice. 362. Rockville, MD: National Institutes of Health; 1989. [PubMed] [Google Scholar]
- Springer LN, McAsey ME, Flaws JA, Tilly JL, Sipes IG, Hoyer PB. Involvement of apoptosis in 4-vinylcyclohexene diepoxide-induced ovotoxicity in rats. Toxicol Appl Pharmacol 1996; 139: 394 401. [DOI] [PubMed] [Google Scholar]
- Borman SM, VanDePol BJ, Kao A, Thompson KE, Sipes IG, Hoyer PB. A single dose of the ovotoxicant 4-vinylcyclohexene diepoxide is protective in rat primary ovarian follicles. Toxicol Appl Pharmacol 1999; 158: 244 252. [DOI] [PubMed] [Google Scholar]
- Hu XM, Christian PJ, Thompson KE, Sipes IG, Hoyer PB. Apoptosis induced in rats by 4-vinylcyclohexene diepoxide is associated with activation of the caspase cascades. Biol Reprod 2001; 65: 87 93. [DOI] [PubMed] [Google Scholar]
- Hu XM, Christian PJ, Sipes IG, Hoyer PB. Expression and redistribution of cellular bad, bax and bcl-xl protein is associated with VCD-induced ovotoxicity in rats. Biol Reprod 2001; 65: 1489 1495. [DOI] [PubMed] [Google Scholar]
- Hu X, Flaws JA, Sipes IG, Hoyer PB. Activation of mitogen-activated protein kinases and AP-1 transcription factor in ovotoxicity induced by 4-vinylcyclohexene diepoxide in rats. Biol Reprod 2002; 67: 718 724. [DOI] [PubMed] [Google Scholar]
- Fernandez SM, Keating AF, Christian PJ, Sen N, Hoying JB, Brooks HL, Hoyer PB. Involvement of the KIT/KITL signaling pathway in 4-vinylcyclohexene diepoxide-induced ovarian follicle loss in rats. Biol Reprod 2008; 79: 318 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail RS, Okawara Y, Fryer JN, Vanderhyden BC. Hormonal regulation of the ligand for c-kit in the rat ovary and its effects on spontaneous oocyte meiotic maturation. Mol Reprod Dev 1996; 43: 458 469. [DOI] [PubMed] [Google Scholar]
- Liu K, Rajareddy S, Liu L, Jagarlamudi K, Boman K, Selstam G, Reddy P. Control of mammalian oocyte growth and early follicular development by the oocyte PI3 kinase pathway: new roles for an old timer. Dev Biol 2006; 299: 1 11. [DOI] [PubMed] [Google Scholar]
- Keating AF, Mark CJ, Sen N, Sipes IG, Hoyer PB. Effect of phosphatidylinositol-3 kinase inhibition on ovotoxicity caused by 4-vinylcyclohexene diepoxide and 7, 12-dimethylbenz[a]anthracene in neonatal rat ovaries. Toxicol Appl Pharmacol 2009; 241: 127 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating AF, Sen N, Sipes IG, Hoyer PB. Dual protective role for glutathione S-transferase class pi against VCD-induced ovotoxicity in the rat ovary. Toxicol Appl Pharmacol 2010; 247: 71 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boekelheide K, Darney SP, Daston GP, David RM, Luderer U, Olshan AF, Sanderson WT, Willhite CC, Woskie S. NTP-CERHR. Expert Panel Report on the reproductive and developmental toxicity of 2-bromopropane. Reprod Toxicol 2004; 18: 189 217. [DOI] [PubMed] [Google Scholar]
- Park J, Shin KS, Kim Y. Occupational reproductive function abnormalities and bladder cancer in Korea. J Korean Med Sci 2010; 25: S41 S45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Jung K, Hwang T, Jung G, Kim H, Park J, Kim J, Park J, Park D, Park S, Choi K, Moon Y. Hematopoietic and reproductive hazards of Korean electronic workers exposed to solvents containing 2-bromopropane. Scand J Work Environ Health 1996; 22: 387 391. [DOI] [PubMed] [Google Scholar]
- Koh JM, Kim CH, Hong SK, Lee KU, Kim YT, Kim OJ, Kim GS. Primary ovarian failure caused by a solvent containing 2-bromopropane. Eur J Endocrinol 1998; 138: 554 556. [DOI] [PubMed] [Google Scholar]
- Kim Y, Park J, Moon Y. Hematopoietic and reproductive toxicity of 2-bromopropane, a recently introduced substitute for chlorofluorocarbons. Toxicol Lett 1999; 108: 309 313. [DOI] [PubMed] [Google Scholar]
- Park J, Kim Y, Park D, Choi K, Park S, Moon Y. An outbreak of hematopoietic and reproductive disorders due to solvents containing 2-bromopropane in an electronic factory, South Korea: epidemiological survey. J Occup Health 1997; 39: 138 143. [Google Scholar]
- Takeuchi Y, Ichihara G, Kamijima M. A review of toxicity of 2-bromopropane: mainly on its reproductive toxicity. J Occup Health 1997; 39: 191. [Google Scholar]
- Kamijima M, Ichihara G, Kitoh J, Tsukamura H, Maeda M, Yu X, Xie Z, Nakajima T, Asaeda N, Hisanaga N, Takeuchi Y. Ovarian toxicity of 2-bromopropane in the non-pregnant female rat. J Occup Health 1997; 39: 144 149. [Google Scholar]
- Sekiguchi S, Asano G, Suda M, Honma T. Influence of 2-bromopropane on reproductive system–short-term administration of 2-bromopropane inhibits ovulation in F344 rats. Toxicol Ind Health 2001; 16: 277 283. [DOI] [PubMed] [Google Scholar]
- Sekiguchi S, Suda M, Zhai YL, Honma T. Effects of 1-bromopropane, 2-bromopropane, and 1,2-dichloropropane on the estrous cycle and ovulation in F344 rats. Toxicol Lett 2002; 126: 41 49. [DOI] [PubMed] [Google Scholar]
- Yu XZ, Kamijima M, Ichihara G, Li W, Kitoh J, Xie Z, Shibata E, Hisanaga N, Takeuchi Y. 2-Bromopropane causes ovarian dysfunction by damaging primordial follicles and their oocytes in female rats. Toxicol Appl Pharmacol 1999; 159: 185 193. [DOI] [PubMed] [Google Scholar]
- Dobson RL, Felton JS. Female germ cell loss from radiation and chemical exposures. Am J Ind Med 1983; 4: 175 190. [PubMed] [Google Scholar]
- Uzumcu M, Zachow R. Developmental exposure to environmental endocrine disruptors: consequences within the ovary and on female reproductive function. Reprod Toxicol 2007; 23: 337 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A, Jolly P, Wei H. Genistein modulates neuroblastoma cell proliferation and differentiation through induction of apoptosis and regulation of tyrosine kinase activity and N-myc expression. Carcinogenesis 1998; 19: 991 997. [DOI] [PubMed] [Google Scholar]
- Gu Y, Zhu CF, Iwamoto H, Chen JS. Genistein inhibits invasive potential of human hepatocellular carcinoma by altering cell cycle, apoptosis, and angiogenesis. World J Gastroenterol 2005; 11: 6512 6517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han RM, Tian YX, Liu Y, Chen CH, Ai XC, Zhang JP, Skibsted LH. Comparison of flavonoids and isoflavonoids as antioxidants. J Agric Food Chem 2009; 57: 3780 3785. [DOI] [PubMed] [Google Scholar]
- Kouki T, Kishitake M, Okamoto M, Oosuka I, Takebe M, Yamanouchi K. Effects of neonatal treatment with phytoestrogens, genistein and daidzein, on sex difference in female rat brain function: estrous cycle and lordosis. Horm Behav 2003; 44: 140 145. [DOI] [PubMed] [Google Scholar]
- Nikaido Y, Yoshizawa K, Danbara N, Tsujita-Kyutoku M, Yuri T, Uehara N, Tsubura A. Effects of maternal xenoestrogen exposure on development of the reproductive tract and mammary gland in female CD-1 mouse offspring. Reprod Toxicol 2004; 18: 803 811. [DOI] [PubMed] [Google Scholar]
- Jefferson W, Newbold R, Padilla-Banks E, Pepling M. Neonatal genistein treatment alters ovarian differentiation in the mouse: inhibition of oocyte nest breakdown and increased oocyte survival. Biol Reprod 2006; 74: 161 168. [DOI] [PubMed] [Google Scholar]
- Jefferson W, Padilla-Banks E, Newbold RR. Disruption of the developing female reproductive system by phytoestogens: genistein as an example. Mol Nutr Food Res 2007; 51: 832 844. [DOI] [PubMed] [Google Scholar]
- Jefferson WN, Couse JF, Padilla-Banks E, Korach KS, Newbold RR. Neonatal exposure to genistein induces estrogen receptor (ER)alpha expression and multioocyte follicles in the maturing mouse ovary: evidence for ERbeta-mediated and nonestrogenic actions. Biol Reprod 2002; 67: 1285 1296. [DOI] [PubMed] [Google Scholar]
- Zhuang XL, Fu YC, Xu JJ, Kong XX, Chen ZG, Luo LL. Effects of genistein on ovarian follicular development and ovarian life span in rats. Fitoterapia 2010; 81: 998 1002. [DOI] [PubMed] [Google Scholar]
- Markey CM, Rubin BS, Soto AM, Sonnenschein C. Endocrine disruptors: from Wingspread to environmental developmental biology. J Steroid Biochem Mol Biol 2002; 83: 235 244. [DOI] [PubMed] [Google Scholar]
- Burridge E. Bisphenol A: product profile. Eur Chem News 2003; 17: 14 20. [Google Scholar]
- Ikezuki Y, Tsutsumi O, Takai Y, Kamei Y, Taketani Y. Determination of bisphenol A concentrations in human biological fluids reveals significant early prenatal exposure. Hum Reprod 2002; 17: 2839 2841. [DOI] [PubMed] [Google Scholar]
- Schonfelder G, Wittfoht W, Hopp H, Talsness CE, Paul M, Chahoud I. Parent bisphenol A accumulation in the human maternal-fetal-placental unit. Environ Health Perspect 2002; 110: A703 A707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calafat AM, Kuklenyik Z, Reidy JA, Caudill SP, Ekong J, Needham LL. Urinary concentrations of bisphenol A and 4-nonylphenol in a human reference population. Environ Health Perspect 2005; 113: 391 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter CA, Birnbaum LS, Farabollini F, Newbold RR, Rubin BS, Talsness CE, Vandenbergh JG, Walser-Kuntz DR, vom Saal FS. In vivo effects of bisphenol A in laboratory rodent studies. Reprod Toxicol 2007; 24: 199 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welshons WV, Nagel SC, vom Saal FS. Large effects from small exposures. III: endocrine mechanisms mediating effects of bisphenol A at levels of human exposure. Endocrinology 2006; 147: S56 S69. [DOI] [PubMed] [Google Scholar]
- Rodriguez HA, Santambrosio N, Santamaria CG, Munoz-de-Toro M, Luque EH. Neonatal exposure to bisphenol A reduces the pool of primordial follicles in the rat ovary. Reprod Toxicol 2010; 30: 550 557. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Sugihara A, Uchida K, Sato T, Ohta Y, Katsu Y, Watanabe H, Iguchi T. Developmental effects of perinatal exposure to bisphenol-A and diethylstilbestrol on reproductive organs in female mice. Reprod Toxicol 2002; 16: 107 116. [DOI] [PubMed] [Google Scholar]
- Hunt PA, Koehler KE, Susiarjo M, Hodges CA, Ilagan A, Voigt RC, Thomas S, Thomas BF, Hassold TJ. Bisphenol a exposure causes meiotic aneuploidy in the female mouse. Curr Biol 2003; 13: 546 553. [DOI] [PubMed] [Google Scholar]
- Susiarjo M, Hassold TJ, Freeman E, Hunt PA. Bisphenol A exposure in utero disrupts early oogenesis in the mouse. PLoS Genet 2007; 3: 66 70 (e5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newbold RR. Lessons learned from perinatal exposure to diethylstilbestrol. Toxicol Appl Pharmacol 2004; 199: 142 150. [DOI] [PubMed] [Google Scholar]
- Sheehan DM, Young M. Diethylstilbestrol and estradiol binding to serum albumin and pregnancy plasma of rat and human. Endocrinology 1979; 104: 1442 1446. [DOI] [PubMed] [Google Scholar]
- Newbold RR, Bullock BC, Mc Lachlan JA. Exposure to diethylstilbestrol during pregnancy permanently alters the ovary and oviduct. Biol Reprod 1983; 28: 735 744. [DOI] [PubMed] [Google Scholar]
- Iguchi T, Fukazawa Y, Uesugi Y, Takasugi N. Polyovular follicles in mouse ovaries exposed neonatally to diethylstilbestrol in vivo and in vitro. Biol Reprod 1990; 43: 478 484. [DOI] [PubMed] [Google Scholar]
- Iguchi T, Kamiya K, Uesugi Y, Sayama K, Takasugi N. In vitro fertilization of oocytes from polyovular follicles in mouse ovaries exposed neonatally to diethylstilbestrol. In Vivo 1991; 5: 359 363. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.