

Abstract
Streptococcus pneumoniae is a Gram-positive, extracellular bacterium that is responsible for significant mortality and morbidity worldwide. Pneumolysin (PLY), a cytolysin produced by all clinical isolates of the pneumococcus, is one of the most important virulence factors of this pathogen. We have previously reported that PLY is an essential factor for activation of caspase-1 and consequent secretion of IL-1β and IL-18 in macrophages infected with S. pneumoniae. However, the host molecular factors involved in caspase-1 activation are still unclear. To further elucidate the mechanism of caspase-1 activation in macrophages infected with S. pneumoniae, we examined the involvement of inflammasomes in inducing this cellular response. Our study revealed that apoptosis-associated speck like protein containing a caspase recruitment domain (ASC), an adaptor protein for inflammasome receptors such as NLR family, pyrin domain containing 3 (NLRP3) and absent in melanoma 2 (AIM2), is essentially required for the induction of caspase-1 activation by S. pneumoniae. Caspase-1 activation was partially impaired in NLRP3−/− macrophages, while knockdown and knockout of AIM2 resulted in a clear decrease in caspase-1 activation in response to S. pneumoniae. These results suggest that ASC inflammasomes, including AIM2 and NLRP3, are critical for caspase-1 activation induced by S. pneumoniae. Furthermore, ASC−/− mice were more susceptible than wild-type mice to S. pneumoniae, with impaired secretion of IL-1β and IL-18 into the bronchoalveolar lavage after intranasal infection, suggesting that ASC inflammasomes contribute to the protection of host from infection with PLY-producing S. pneumoniae.
Introduction
Streptococcus pneumoniae is a Gram-positive, extracellular bacterium that is responsible for significant mortality and morbidity worldwide, causing bacterial pneumonia, otitis media, meningitis and septicemia. Pneumolysin (PLY), a 53-kDa multifunctional protein toxin which is produced by virtually all clinical isolates of pneumococcus, is one of the key virulence factors of S. pneumoniae (1-4). PLY is a member of the cholesterol-dependent cytolysin family, which binds to cholesterol and form ring- or arc-shaped transmembrane pores on cholesterol-containing membranes, eventually causing cytolysis in various mammalian cells. As PLY lacks a signal sequence for secretion, it has been hypothesized that the cytolysin is released during the breakdown of the pneumococcal cell wall (5, 6). A number of reports have suggested various roles for PLY in the pathogenicity of S. pneumoniae, including disruption of tissue barriers, inhibition of ciliary beating on epithelial cells and bactericidal activity of neutrophils, induction of host cell apoptosis, and a possible subversion of complement-mediated opsonization, thereby facilitating colonization and survival of pneumococci in the host (6, 7). In addition to above-mentioned functions as the virulence factor, PLY is involved in the activation of innate immune responses of the host that generate inflammation and/or contribute to the host defense (6, 8, 9). PLY protein has been demonstrated to induce IL-1β secretion (9, 10), and it has been suggested that PLY is essential for the maturation and secretion, but not expression, of IL-1α, IL-1β and IL-18 upon infection of macrophages or dendritic cells (DCs) with S. pneumoniae (10-12). IL-1 cytokines, especially IL-1β, and their receptor play protective roles in pneumococcal pneumonia and meningitis in mice (13-16). IL-18 has been also reported to contribute to early host defense against pneumococcal pneumonia (17). Thus, it is likely that PLY-dependent induction of these cytokines contributes to the protection of the host from S. pneumoniae, although a host-detrimental role of IL-18 in a meningitis model has also been reported (18). Thus, the PLY-dependent induction of these inflammatory cytokines and their roles in vivo have been established, however, little is known about the mechanism by which these cytokine responses are induced in a PLY-dependent manner. Caspase-1 is a cysteine protease, which processes proforms of IL-1β and IL-18 into mature forms, and is important for secretion of these cytokines. We have also shown that caspase-1 activation is depending on PLY in macrophages infected with S. pneumoniae (10).
Recent studies have demonstrated that the inflammasome, a multiprotein complex, which typically consists of a nucleotide-binding oligomerization domain-like receptor (NLR) molecule and pro-caspase-1, mediates the activation of caspase-1 in response to a wide variety of stimuli (19-38). Several inflammasomes have been identified, and each has been designated according to the receptor within the protein complex. Of these, the NLR family, pyrin domain containing 3 (NLRP3) inflammasome has been shown to be activated by endogenous stimuli such as urate crystals (20), extracelluar ATP (21), cholesterol crystals (22), and also exogenous stimuli such as bacterial and viral pathogens, pore-forming toxins, asbestos, silica, and so on (23-30). It has been reported that the NLR family, caspase recruitment domain containing 4 (NLRC4) inflammasome is activated by flagellin, basal body rod proteins of the type III secretion systems and pilin, while cytosolic DNA activates the absent in melanoma 2 (AIM2) inflammasome (31-38). Apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) plays a critical role in the activation of inflammasomes as an adaptor protein that bridges pro-caspase-1 and inflammasome receptors like NLRP3 and AIM2 (19, 39). Recent reports have identified a distinct role for inflammasomes in host defense against several pathogens.
In this study, we sought to elucidate the induction mechanism of PLY-dependent activation of caspase-1 in macrophages infected with S. pneumoniae, by examining the involvement of inflammasomes in the cellular response. Our results suggest that the activation of caspase-1 and the subsequent maturation of IL-1β and IL-18 in response to PLY-producing S. pneumoniae are critically dependent on ASC inflammasomes, including AIM2 and NLRP3. We also demonstrated a significant role of ASC inflammasomes in host defense against pulmonary pneumococcal infection. Thus, this study has revealed a pathophysiological link between the virulence factor PLY and the inflammasome.
Materials and Methods
Mice
Female C3H/HeN, C3H/HeJ and C57BL/6 mice were purchased from Japan SLC (Hamamatsu, Japan). NLRP3−/− mice generated by Prof. Jürg Tschopp (University of Lausanne) (20), NLRC4−/− mice generated by Dr. Vishva Dixit (Genentech) (40), ASC−/− mice generated by Prof. Shun-ichiro Taniguchi (Shinshu University) (41), and caspase-1−/− mice generated by Dr. Keisuke Kuida (Millennium Pharmaceuticals) were kindly gifted. Mice were maintained in specific-pathogen-free conditions and used at 7 to 9 weeks of age. All of the experimental procedures performed on mice were approved by the Animal Ethics and Research Committee of Kyoto University Graduate School of Medicine, Kyoto, Japan.
Bacterial strains
Streptococcus pneumoniae D39 (serotype 2) was purchased from the National Collection of Type Cultures (NCTC 7466; Central Public Health Laboratory, London, United Kingdom). A deletion mutant of S. pneumoniae D39 for the PLY gene (Δply) was constructed by using homologous recombination method as described previously (10). Bacteria were grown on tryptic soy agar (Difco Laboratories, Detroit, MI) with 5% (vol/vol) defibrinated sheep blood (Nacalai Tesque, Kyoto, Japan) and in Todd-Hewitt broth (Difco) supplemented with 0.5% yeast extract (THY) at 37°C and 5% CO2 and subsequently stored at −80°C in THY plus 10% glycerol. For the preparation of bacterial stocks for macrophage stimulation, pneumococci were grown overnight on blood agar plates at 37°C and 5% CO2. Colonies were inoculated into the THY medium, grown until mid-logarithmic phase (optical density at 600 nm [OD600] =0.5), and centrifuged at 6,000 g for 15 min. The bacterial pellet was suspended in phosphate-buffered saline (PBS) and stocked at −80°C. The concentration was determined by viable cell counting on blood agar plates.
Collection of peritoneal macrophages and infection by S. pneumoniae in vitro
Mice were injected intraperitoneally (i.p.) with 3% thioglycollate broth (EIKEN CHEMICAL, Tokyo, Japan), and peritoneal exudate cells (PECs) were collected 3 days later. After washing with RPMI 1640 medium (Nacalai Tesque), PECs were suspended with RPMI 1640 supplemented with 10% FCS and incubated in 48-well microplates at a density of 2.5 × 105 cells/well at 37°C plus 5% CO2, and non-adherent cells were removed after two hours. Adherent macrophages were infected with S. pneumoniae at a multiplicity of infection (MOI) of 10 for 8 h, then 100 μg/ml of gentamicin (Wako Pure Chemical Industries, Osaka, Japan) was added to the cultures and incubated for an additional 16 h. In order to confirm the enrichment for macrophages in adherent PECs, the percentage of F4/80+ macrophages was determined by flow cytometry. Whole and adherent PECs collected by using Cell Dissociation Buffer Enzyme-Free PBS-based (Invitrogen, Carlsbad, CA) were incubated with anti-mouse CD16/32 (Biolegend, San Diego, CA) for 30 min at 4°C. Then the cells were stained with PE anti-mouse F4/80 (Biolegend) or isotype control (PE Rat IgG2a, Biolegend) for 30 min at 4°C and analyzed on a FACScalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ). The proportions of F4/80+ cells in whole PECs from WT, caspase-1−/−, ASC−/− and NLRP3−/− mice were 94.8%, 96.0%, 95.6% and 95.0%, respectively, and were increased to 98.2%, 98.5%, 98.3% and 95.9% in adherent PECs, respectively. Thus the adherent PECs were sufficiently enriched for F4/80+ macrophages.
Macrophage cell lines
Immortalized WT and AIM2−/− macrophage cell lines were generated by infecting primary bone marrow cells with J2 recombinant retrovirus (a kind gift from Dr. Howard Young) as described previously (42). The macrophage lines were infected with S. pneumoniae at a MOI of 10 for 8 h, then 100 μg/ml of gentamicin was added to the cultures and incubated for an additional 16 h.
ELISA
Levels of secreted cytokines in culture supernatants were determined by two-site sandwich enzyme-linked immunosorbent assay (ELISA). ELISA kits for IL-6 and IL-1β were purchased from eBioscience (San Diego, CA). For the titration of IL-18, biotin-labeled and unlabeled mAbs specific to IL-18 (Medical & Biological Laboratories, Nagoya, Japan) were used.
Western blot analysis
PECs were cultured on 12-well plates at a density of 5 × 105 cells/well in RPMI 1640 + FCS at 37°C for two hours. Then medium was replaced with Opti-MEM (Invitrogen), and adherent cells were infected with S. pneumoniae at a MOI of 10. Supernatants were collected 24h after infection, and the cells were lysed with Radio-Immunoprecipitation Assay (RIPA) buffer (Nacalai Tesque). The supernatants were concentrated 20-fold using 20% (w/v) trichloroacetate (TCA; Nacalai Tesque). The precipitates and cell lysates were subjected to SDS-PAGE and subsequently transferred to polyvinylidene difluoride membranes by electroblotting. The membranes were immunoblotted with anti-caspase-1 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-IL-1β antibody (R&D Systems, Minneapolis, MN) or anti-β-actin monoclonal antibody (Sigma-Aldrich, St. Louis, MO) (43).
Detection of lactate dehydrogenase (LDH) release
LDH activity was measured using an LDH detection kit (TaKaRa BIO, Otsu, Japan). The percentage of LDH release was calculated by using the following formula: percentage of release = 100 × (experimental LDH release − spontaneous LDH release)/(maximal LDH release − spontaneous LDH release). To determine the maximal LDH release, cells were treated with 1% Triton X-100. For spontaneous LDH release, adherent PECs were left uninfected.
RNA interference
RNA interference experiments were done using the method employed in our previous study (43). Briefly, PECs were transfected with Stealth siRNA duplexes (Invitrogen) at a final concentration of 20 nM using siPORT Amine (Ambion, Austin, TX). The transfected cells were incubated at 37°C for 48 h and used after washing with RPMI 1640 + FCS. The sense siRNA sequences were: AIM2-1, UUA UCU UCU GGA CUU UAA ACA GCC C and AIM2-2, UAA AGU CAU UGU CAC UGC GGG UGG C. Stealth RNAi Negative Control Medium GC Duplex #2 (Invitrogen) was used as a control siRNA.
RT-PCR analysis
Total cellular RNA of adherent PECs was extracted by using NucleoSpin RNA Clean-up (MACHEREY-NAGEL, Düren, Germany), and quantitative real time RT-PCR was carried out using the ABI PRISM 7000 (Applied Biosystems, Foster City, CA) and EXPRESS Two-Step SYBR GreenER (Invitrogen), according to the manufacturer’s instructions. Primers for quantitative real-time RT-PCR were as follows: Interferon (IFN)-β: Fw:5′-CTG GAG CAG CTG AAT GGA AAG-3′; Rv:5′-CTT GAA GTC CGC CCT GTA GGT-3′; β-Actin: Fw:5′-GCC CTG AGG CTC TTT TCC AG-3′; Rv:5′-TGC CAC AGG ATT CCA TAC CC-3′; AIM2: Fw:5′-TTG TAT CTA GGC TGA TCC TGG GAC-3′; Rv: 5′-ACC TGC ACT TTG AAT CAG GTG GTC-3′.
Extraction of S. pneumoniae genomic DNA
S. pneumoniae D39 was inoculated into THY broth and culture for overnight, then the bacteria was centrifuged and the pellet was suspended in the lysis buffer containing 10mM Tris buffer, 50 mM EDTA, 20 mg/ml lysozyme (Nacalai tesque) and 100 μg/ml RNaseA (Sigma-Aldrich), and then incubated at 37°C for 1h. Next, the lysate was mixed with 5 M NaCl and 10% CTAB/0.7 M NaCl solution, incubated at 65°C for 10 min. Finally, the genomic DNA was extracted by chloroform/isoamyl alcohol (24:1) and phenol/chloroform/isoamyl alcohol (25:24:1). To stimulate macrophages with purified S. pneumoniae DNA or poly (deoxyadenylic-thymidylic) acid (poly dA:dT, Sigma-Aldrich), macrophages were transfected with 400 ng of DNA by using Lipofectamine LTX (Invitrogen).
Quantitation of macrophage associated and phagocytosed S. pneumoniae
Adherent macrophages were infected with S. pneumoniae at MOI of 10 for 2, 4, 6, or 8 h. To enumerate S. pneumoniae associating with macrophages, cells were washed with chilled PBS three times to remove non-associating bacteria and lysed in PBS containing 0.1% Triton X-100. The cell lysates were diluted with PBS and plated on blood agar plates. CFU were counted after overnight incubation. To enumerate the phagocytosed bacteria, macrophages were infected with S. pneumoniae as above and additionally cultured for 30 min in the presence of 100 μg/ml gentamicin. The cells were then washed, and the bacterial numbers were counted. To monitor the survival of S. pneumoniae inside macrophages, the numbers of intracellular bacteria were determined every 0.5h until 3h after gentamicin addition. To inhibit the phagocytosis of bacteria by macrophages, cytochalasin B was added to the macrophage cultures at a final concentration of 10 μg/ml. Equal volume of DMSO was added to macrophages as a solvent control.
Intranasal infection of mice
Mice were anesthetized with pentobarbitone (Nacalai tesque) and inoculated with 5 × 107 bacteria in 25 μl PBS. Bronchoalveolar lavage fluid (BALF) were collected according to the method of Lauw et al. (17). BALF from non-infected mice were collected as negative control. Survival of infected mice was monitored every day until two weeks after infection and analyzed using the Kaplan-Meier method. Lungs were excised 48h after infection and homogenized in 1ml PBS, serial diluted in PBS and plated onto blood agar, CFUs were counted after overnight culture.
Statistical analysis
For comparisons between two groups, Student’s t test was used. Multigroup comparisons of mean values were made according to the ANOVA and the Bonferroni post hoc test. Statistical analysis for survival curves was performed using the log-rank test. Statistical significance was determined as p < 0.05.
Results
Caspase-1 activation in macrophages induced by S. pneumoniae is independent of TLR4
We have previously reported that PLY plays an important role in caspase-1 activation and the subsequent maturation and secretion of caspase-1-dependent cytokines in macrophages infected with S. pneumoniae (10). However, the host receptors involved in this process remains still unclear. In our previous report, we also showed that secretion of IL-1β and IL-18 induced following the stimulation of macrophages with recombinant protein of PLY is critically dependent on Toll-like receptor 4 (TLR4). We therefore first tested whether PLY-producing S. pneumoniae activates caspase-1 through TLR4. Macrophages from C3H/HeN and C3H/HeJ (tlr4Lps-d) mice were infected with S. pneumoniae D39 at a MOI of 10 for 24 h, then the culture supernatants were assayed for the p10 fragment of mature caspase-1 by Western blotting. There was no significant difference between the two mouse strains in caspase-1 activation in response to S. pneumoniae (Fig. 1A). Furthermore, HeN and HeJ macrophages secreted comparable levels of mature IL-1β after infection with S. pneumoniae (Fig. 1A and 1B). The level of IL-1β in HeJ macrophages was almost the same with that in HeN macrophages following a treatment with Pam3CSK4 (a TLR2 ligand) plus nigericin, whereas HeJ macrophages never responded to the treatment with LPS (a TLR4 ligand) plus nigericin (Fig. 1C and 1D). These results suggested that S. pneumoniae induces caspase-1 activation in a TLR4-independent manner.
FIGURE 1.
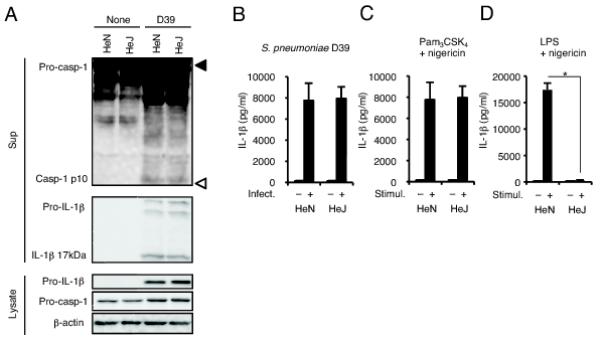
Caspase-1 activation in macrophages induced by S. pneumoniae is independent of TLR4. A and B: Adherent PECs from C3H/HeN or C3H/HeJ mice were left uninfected or infected with S. pneumoniae D39 at a MOI of 10, 8 h later gentamicin was added to cultures (final concentration 100 μg/ml) and culture supernatants were collected after an additional 16 h incubation (24 h after infection). A: Culture supernatants were concentrated by TCA precipitation. The precipitates and cell lysates were subjected to Western blot analysis using antibodies specific for caspase-1, IL-1β or β-actin. Filled and open triangles indicate MW markers of 47 kDa and 9 kDa, respectively. B: Levels of IL-1β in the culture supernatants were determined by ELISA. C and D: Adherent PECs were left unstimulated or stimulated with Pam3CSK4 (100 ng/ml) or LPS (10 ng/ml) for 3 h and sequentially with nigericin (5 μM). After 21 h, culture supernatants were collected to assess the levels of IL-1β by ELISA. All of the experiments were repeated three times. The ELISA data are presented as the mean and SD of triplicate assays. Statistical significance was determined by one-way ANOVA followed by the Bonferroni’s test. *, p < 0.05.
ASC plays a critical role in caspase-1 activation in macrophages infected with S. pneumoniae
Caspase-1 was essential for S. pneumoniae-induced secretion of IL-1β and IL-18, as macrophages from caspase-1−/− mice were incapable of secreting these cytokines after infection with S. pneumoniae D39 (Fig. 2A and 2B). LDH release occurred following infection of normal macrophages with S. pneumoniae D39 but not the Δply strain (Fig. 2C), and D39-induced LDH release was not observed in caspase-1−/− macrophages. It appeared that this bacterium induces macrophage pyroptosis in a PLY-dependent manner. NLRP3 is a well-studied NLR receptor and has been shown to induce caspase-1 activation in response to cholesterol-dependent cytolysins (24-26). Therefore, we investigated whether NLRP3 and its adaptor protein ASC are involved in caspase-1 activation upon S. pneumoniae infection. Caspase-1 activation, secretion of IL-1β and IL-18, and LDH release induced after infection with S. pneumoniae D39 were completely abolished in ASC−/− macrophages, whereas these cellular responses in NLRP3−/− macrophages were only partially reduced as compared with those in WT macrophages (Fig. 2A-C). Deficiency of NLRC4 did not affect or only slightly reduced the secretion of IL-1β and IL-18 and LDH release induced by S. pneumoniae D39 infection (Fig. 2B and 2C). Taken together, these results suggested that ASC is essential for the caspase-1 response to S. pneumoniae and additional receptor(s), other than NLRP3, is involved in ASC-dependent caspase-1 activation induced by this pathogen.
FIGURE 2.
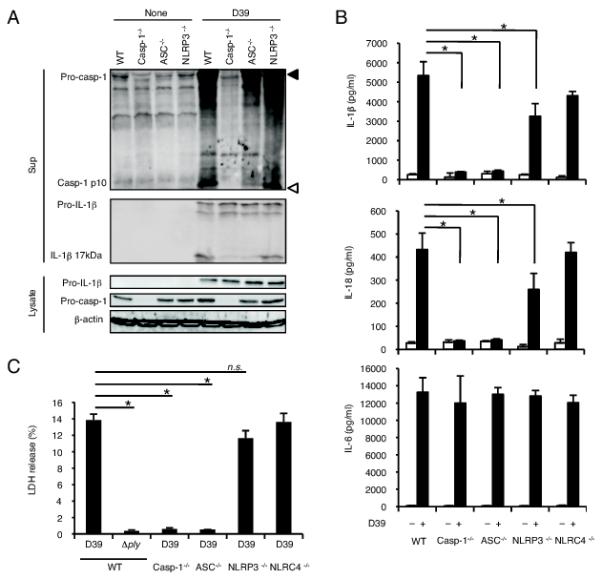
ASC is essentially required for caspase-1 activation in S. pneumoniae infected macropahges. Adherent PECs from C57BL/6 WT, caspase-1−/−, ASC−/−, NLRP3−/− or NLRC4−/− mice were left uninfected or infected with S. pneumoniae D39 or the Δply strain at a MOI of 10 for 24 h as described in Fig. 1, and culture supernatants and cell lysates were then collected. A: Culture supernatants and cell lysates were subjected to Western blot analysis as described in Fig. 1. Filled and open triangles indicate MW markers of 47 kDa and 9 kDa, respectively. B: Levels of IL-1β, IL-18 and IL-6 in the culture supernatants were determined by ELISA. C: LDH release from D39- or Δply-infected macrophages derived from each mouse strain was determined by the LDH activity in the culture supernatants, and the data are expressed as percent LDH release. All of the experiments were repeated more than three times. The results are presented as the mean and SD of triplicate assays. Tests for statistical significance were performed by using one-way ANOVA followed by the Bonferroni’s test. n.s., not significant. *, p < 0.05.
AIM2 inflammasome is involved in caspase-1 activation induced by S. pneumoniae
AIM2 has been shown to recognize cytoplasmic DNA and to be responsible for ASC-dependent caspase-1 activation in response to DNA (35-38). We tested whether the AIM2 inflammasome is involved in caspase-1 activation in macrophages infected with S. pneumoniae. Knockdown of AIM2 was carried out in macrophages using two siRNAs, AIM2-1 and AIM2-2, which target independent sequences in the AIM2 transcript, as described previously (43). The efficiency of the knockdown was assessed by real time RT-PCR, and the expression of AIM2 was lowered by both AIM2-targeting siRNAs (Fig. 3A). Knockdown of AIM2 resulted in a reduction of IL-18 secretion induced by poly dA:dT transfection (Fig. 3B) without affecting the secretion of IL-1β in response to LPS plus nigericin (Fig. 3C). A significant reduction in S. pneumoniae-induced caspase-1 activation was observed in AIM2 knockdown macrophages (Fig. 3D). Moreover, secretions of IL-1β, IL-18 and LDH release in response to S. pneumoniae were decreased by knockdown of AIM2, whereas IL-6 response was not affected (Fig. 3D-3F). These findings suggest that the AIM2 inflammasome plays an important role in caspase-1 activation induced by S. pneumoniae. To confirm the involvement of AIM2, similar experiments were conducted with immortalized macrophages established from WT or AIM2−/− mice and the results showed that D39-induced caspase-1 activation, secretion of caspase-1-dependent cytokines and LDH release were significantly decreased in the absence of AIM2 (Fig. 3G-3J), though the NLRP3 inflammasome was intact in both macrophage lines (Fig. 3K), further suggesting the involvement of AIM2 in caspase-1 activation upon S. pneumoniae infection. However, a question may be raised here as to how S. pneumoniae, an extracellular bacterium, is recognized by AIM2, a sensor molecule of cytosolic DNA. Since PLY is important for caspase-1 activation in response to S. pneumoniae, we hypothesized that PLY may play a role in the delivery of bacterial DNA into the macrophage cytoplasm. If this is the case, a type I IFN response could also be induced by S. pneumoniae in a PLY-dependent manner, as cytosolic DNA induces not only activation of AIM2 inflammasome but also expression of type I IFNs (44, 45). Consistent with this hypothesis, we found that the expression of IFNβ was induced in macrophages infected with S. pneumoniae D39 but not with the Δply strain (Fig. 3L).
FIGURE 3.
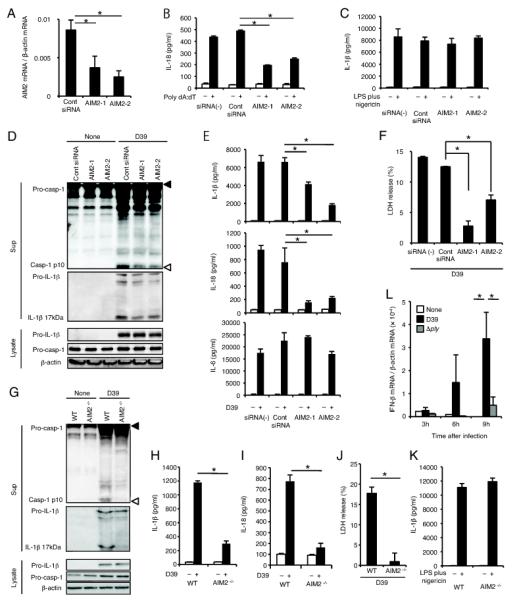
AIM2 is involved in caspase-1 activation upon S. pneumoniae infection. A-F: Adherent PECs were transfected with siRNAs (AIM2-1, AIM2-2 or control siRNA) and cultivated for 2 days. G-K: Immortalized macrophages from WT or AIM2−/− mice were employed. A: AIM2 expression levels were analyzed by real time RT-PCR. B: Cells were transfected or not with poly dA:dT and incubated for 24 h. Culture supernatants were then collected, and levels of IL-18 in the culture supernatants were determined by ELISA. C and K: Cells were left unstimulated or stimulated with LPS (10 ng/ml) for 3 h and sequentially with nigericin (5 μM). After 21 h, culture supernatants were collected, and levels of IL-1β in the culture supernatants were determined by ELISA. D-F and G-J: Cells were left uninfected or infected with S. pneumoniae D39 at a MOI of 10 for 24 h as described in Fig. 1, and culture supernatants and cell lysates were then collected. Levels of IL-1β (E and H), IL-18 (E and I), and IL-6 (E) in the culture supernatants were determined by ELISA. Culture supernatants and cell lysates were subjected to Western blot analysis as described in Fig. 1 (D and G). Filled and open triangles indicate MW markers of 47 kDa and 9 kDa, respectively. LDH release from D39-infected macrophages was determined by the LDH activity in the culture supernatants, and the data are expressed as percent LDH release (F and J). L: Adherent PECs were infected with S. pneumoniae D39 or Δply at a MOI of 10 for 3, 6, or 9 h, and the expression of IFN-β was quantified by real time RT-PCR. White bars, untreated macrophages; black bars, macrophages infected with S. pneumoniae D39; and gray bars, macrophages infected with the Δply strain. All of the experiments were repeated more than three times. The results are presented as the mean and SD of triplicate assays. Tests for statistical significance were performed by using one-way ANOVA followed by the Bonferroni’s test (A, B, E, F, H, I and L) or the Student’s t test (J). *, p < 0.05.
S. pneumoniae genomic DNA activates the AIM2 inflammasome when introduced into macrophages
We next checked whether S. pneumoniae genomic DNA can be a ligand for AIM2. We purified genomic DNA from liquid cultures of S. pneumoniae and the DNA sample was split into one sample treated with DNase I and another left untreated (Fig. 4A). The DNA was then transfected into macrophages and the activation of caspase-1 and the secretion of IL-18 were determined. S. pneumoniae genomic DNA without treatment could induce the activation of caspase-1 and the secretion of IL-18 while DNase I-treated genomic DNA did not retain such an activity (Fig. 4B and 4C). Moreover, knockdown of AIM2 in macrophages resulted in a decrease of secretion of IL-18 induced by S. pneumoniae genomic DNA (Fig. 4D). IL-1β was not secreted from macrophages transfected with S. pneumoniae genomic DNA (data not shown), probably due to the absence of pro-IL-1β expression (Fig. 4B). Taken together, these results indicated that S. pneumoniae genomic DNA can be recognized by AIM2, and that this then induces caspase-1 activation and the secretion of a caspase-1-dependent cytokine.
FIGURE 4.
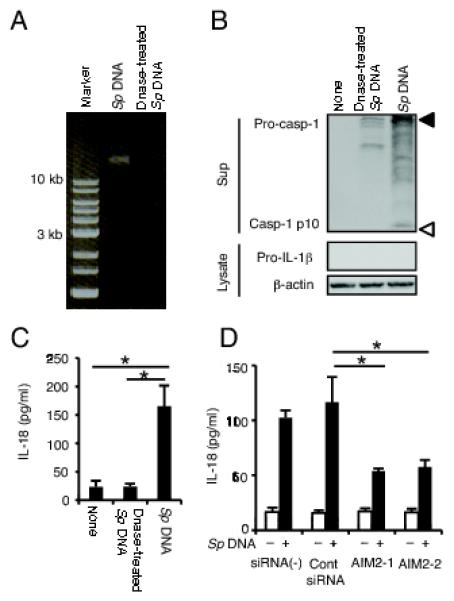
S. pneumoniae genomic DNA activates the AIM2 inflammasome. A: DNase I untreated and treated S. pneumoniae (Sp) genomic DNA was electrophoresed on a 0.8% agarose gel. B and C: Adherent PECs were transfected or not with Sp genomic DNA or DNase-treated Sp genomic DNA and incubated for 24h. B: Culture supernatants and cell lysates were subjected to Western blot analysis as described in Fig. 1. Filled and open triangles indicate MW markers of 47 kDa and 9 kDa, respectively. C: IL-18 in the supernatants was determined by ELISA. D: AIM2 knockdown or control macrophages were left untreated or transfected with Sp genomic DNA for 24 h and IL-18 in the supernatants was determined by ELISA. All of the experiments were repeated two times. The results are presented as the mean and SD of triplicate assays. Tests for statistical significance were performed by using one-way ANOVA followed by the Bonferroni’s test. *, p<0.05.
Phagocytosis of S. pneumoniae by macrophages is important for caspase-1 dependent cytokine production
Though S. pneumoniae is an extracellular bacterium that is not capable of invading the macrophage cytoplasm, the present findings indicated the recognition of S. pneumoniae by AIM2 that functions in the cytosolic space. A possible interpretation is the recognition of bacterial DNA possibly released from the phagosome after phagocytosis of bacteria by macrophages. We therefore tested whether phagocytosis of bacteria is a prerequisite for caspase-1 activation induced by S. pneumoniae. Before the examination, we determined the fate of S. pneumoniae D39 in macrophages after infection, because the bacterial strain has a capsule, which is known to affect phagocytic processes (1-4). The numbers of bacteria associated with macrophages or phagocytosed by macrophages were counted at different time points after infection, showing that the numbers of both macrophage-associated bacteria and phagocytosed bacteria reached their peaks at 6 h after infection (Fig. 5A and 5B). We observed a rapid decrease of intracellular bacteria after addition of gentamicin 4 h after infection (Fig. 5C). These results suggested that S. pneumoniae D39, though the strain is encapsulated, is phagocytosed by macrophages for several hours after infection and the phagocytosed bacteria undergo rapid death, possibly due to killing by host macrophages or pneumococcal autolysis within the phagosome. The fate of the Δply strain after infection of macrophages was almost the same as that of wild-type D39 (Fig. 5A-C). Moreover, caspase-1−/− and ASC−/− macrophages phagocytosed and killed S. pneumoniae D39 as well as WT macrophages (Fig. 5D), suggesting that the observed pneumococcal death was not the consequence of pyroptotic macrophage cell death. To elucidate whether phagocytosis is required for S. pneumoniae-induced caspase-1 activation, macrophages were pretreated with cytochalasin B to inhibit phagocytic activity. Phagocytosis of S. pneumoniae by macrophages was successfully inhibited by cytochalasin B (Fig. 5E), and this impaired engulfment of bacteria resulted in a significant decrease in the activation of caspase-1 (Fig. 5F). Maturation, but not expression, of IL-1β in response to S. pneumoniae was abolished by cytochalasin B pretreatment, suggesting that phagocytosis is, while dispensable for pro-IL-1β induction, indispensable for IL-1β processing and release through the activation of caspase-1 (Fig. 5F). To rule out the possibility that cytochalasin B directly affects the caspase-1 activation process besides inhibiting phagocytosis, cytochalasin B-treated macrophages were transfected with DNA or infected with S. pneumoniae, and levels of IL-18 and IL-1β secreted into the culture supernatants were determined. DNA-induced IL-18 secretion was only slightly affected by pretreatment with cytochalasin B, whereas that induced by S. pneumoniae was significantly decreased (Fig. 5G). Also IL-1β secretion from S. pneumoniae-infected macrophages was significantly decreased by cytochalasin B (Fig. 5H). Thus, it appears that phagocytosis is important for the caspase-1 activation in macrophages infected with S. pneumoniae.
FIGURE 5.
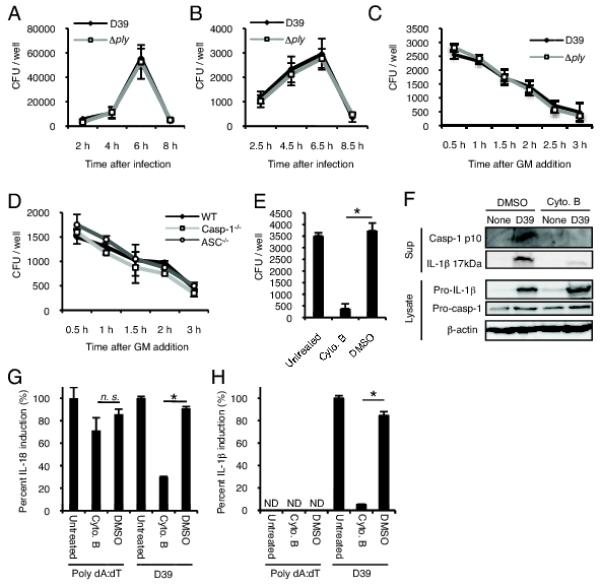
Phagocytosis of S. pneumoniae by macrophages is important for caspase-1 activation. A-D: Adherent PECs were infected with S. pneumoniae D39 (A-D) or Δply (A-C) at a MOI of 10. A: Bacteria associating with macrophages were counted. Infected cells were washed with chilled PBS three times and lysed at the indicated time points. The lysates were plated on blood agar plates after serial dilutions and CFU were counted after overnight incubation. B: Phagocytosed bacteria were counted. Non-cell-associated bacteria were removed by washing at 2, 4, 6, and 8 h after infection, and macrophages were cultured for an additional 30 min in the presence of 100 μg/ml gentamicin. The numbers of bacteria in cell lysates were then counted. C and D: The viability of phagocytosed bacteria was determined. WT macrophages were infected with D39 or the Δply mutant (C). WT, ASC−/− or caspase-1 macrophages were infected with D39 (D). Gentamicin was added to the cultures 4 h after infection and the cells were additionally incubated for the indicated times. The numbers of bacteria in cell lysates were then counted. E: WT macrophages were left untreated or pretreated with cytochalasin B or DMSO and then infected with S. pneumoniae D39. The phagocytosed bacteria were counted 6.5 h after infection as described in Fig. 5B. F: Cytochalasin-pretreated or untreated PECs were infected by S. pneumoniae for 24 h, and culture supernatants and cell lysates were subjected to Western blot analysis as described in Fig. 1. G and H: Cytochalasin-pretreated or untreated macrophages were transfected with DNA or infected by S. pneumoniae for 24 h, levels of IL-18 and IL-1β in the supernatants were determined by ELISA, and the data are expressed as percent IL-18 induction (G) and percent IL-1β induction (H) with respect to untreated controls. All the experiments were repeated three times. Tests for statistical significance were performed by using one-way ANOVA followed by the Bonferroni’s test. ND, not detected. n.s., not significant. *, p<0.05.
ASC inflammasomes protect the host from S. pneumoniae infection in vivo
In order to determine whether ASC-dependent inflammasome formation plays a role in vivo, we have employed an intranasal infection of S. pneumoniae in WT, ASC−/− and NLRP3−/− mice. Consistent with the in vitro findings using cultured macrophages, a significant level of IL-1β was detected in the BALF of WT mice infected with S. pneumoniae D39 but not the Δply strain (Fig. 6A). To assess whether ASC inflammasomes are required for the secretion of IL-1β and IL-18 upon infection with S. pneumoniae D39 in vivo, the levels of these cytokines in the BALF were compared among WT, ASC−/− or NLRP3−/− mice. Compared with WT mice, ASC−/− mice secreted significantly lower levels of IL-1β and IL-18 after S. pneumoniae infection (Fig. 6B and 6C). On the other hand, NLRP3 was partially required for secretion of these cytokines (Fig. 6B and 6C). IL-6 secretion in response to S. pneumoniae infection, which is not depending on inflammasome, was not affected at all in ASC−/− and NLRP3−/− mice (Fig. 6D). These results are consistent with the in vitro findings that multiple ASC inflammasomes are necessary for inducing caspase-1 activation and maturation of IL-1β and IL-18 in S. pneumoniae-infected macrophages. Furthermore, the protection of mice as determined by survival curves and bacterial numbers in the lungs was significantly impaired in ASC−/− and NLRP3−/− mice (Fig. 6E and 6F). The Δply mutant was cleared more rapidly from the lungs of WT mice than the D39 parental strain (n = 4 in each group, data not shown), and when WT or ASC−/− mice were infected with the mutant strain, the bacterial numbers were comparable in the lungs of both mice at 48 h after infection (n = 3 in each group, data not shown). These results have clearly indicated that ASC-inflammasomes are essential for the secretion of caspase-1-dependent cytokines and eventual protection in mice infected with the PLY-producing strain of S. pneumoniae.
FIGURE 6.
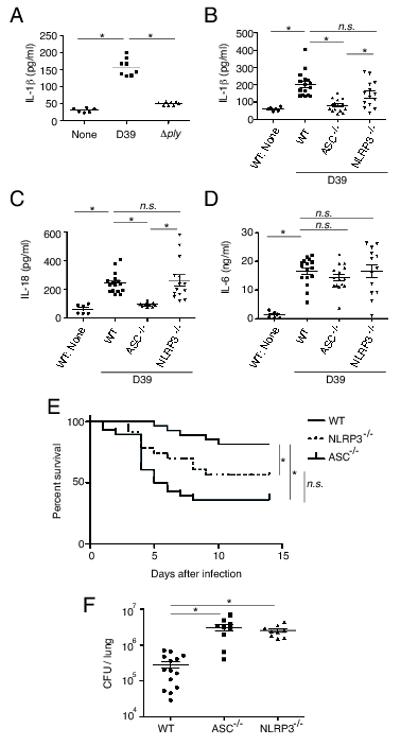
ASC inflammasomes are important for host protection against PLY-producing S. pneumonie infection in vivo. A: WT mice were left uninfected or intranasally challenged with S. pneumoniae D39 or Δply, BALF were collected from the mice 12 h after infection, and levels of IL-1β were measured by ELISA. Data are from two independent experiments (n = 6-8 in each group). B-D: WT, NLRP3−/− or ASC−/− mice were intranasally infected or not by S. pneumoniae D39, BALF were collected 12 h after infection, and levels of IL-1β (B), IL-18 (C) and IL-6 (D) were measured by ELISA. Data are pooled from three individual experiments (n = 6 for control WT mice, n=16 for D39-infected WT mice, n = 14 for D39-infected NLRP3−/− mice and n = 15 for D39-infected ASC−/− mice). E: WT (n = 27), NLRP3−/− (n = 23), or ASC−/− (n = 28) mice were intranasally infected by S. pneumoniae D39, and survival was monitored for two weeks. Data are pooled from two individual experiments with consistent results. F: WT (n = 14), NLRP3−/− (n = 9), or ASC−/− (n = 9) mice were intranasally infected by S. pneumoniae D39, and the numbers of bacteria in the lungs were determined 48 h after infection. Data are pooled from two individual experiments. The lines indicate the means ± SEM of each group (A-D, and F). Tests for statistical significance were performed by using one-way ANOVA followed by the Bonferroni’s test (A-D, and F) or the log-rank test (E). n.s., not significant.*, p<0.05.
Discussion
Innate immune responses play important roles in host defense against S. pneumoniae infection. In this study, we elucidated the mechanism of caspase-1 activation in macrophages infected with PLY-producing S. pneumoniae. We found that ASC inflammasomes, including both AIM2 and NLRP3, but not TLR4, are indispensable for inducing the activation of caspase-1, and the maturation and secretion of IL-1β and IL-18, and for pyroptosis. In addition, our results clearly demonstrate the essential requirement for ASC for the secretion of IL-1β and IL-18 into the BALF in a mouse model of pneumococcal pneumonia. We also showed that the absence of ASC results in a significant increase in the susceptibility to S. pneumoniae infection in vivo. Thus, this study revealed a novel role of ASC inflammasomes in mediating host resistance to pneumococcal pneumonia, most likely through the induction of the protective cytokines, IL-1β and IL-18.
In contrast to our results, PLY has been shown to inhibit the production of pro-inflammatory cytokines including IL-1β by human DCs infected with the non-encapsulated mutant of S. pneumoniae TIGR4 (46), suggesting that the role of PLY in inducing host inflammatory responses may vary in different situations. On the other hand, findings similar to ours were presented in recent reports demonstrating that PLY expression and the NLRP3 inflammasome are involved in the secretion of IL-1β from mouse DCs or macrophages infected with S. pneumoniae D39 (11, 12). These studies also suggested that NLRP3 is important for host defense against S. pneumoniae. All these results are in agreement with ours, as in our present study NLRP3−/− macrophages exhibited a decreased secretion of IL-1β and IL-18 in response to S. pneumoniae compared with WT macrophages, and we found NLRP3−/− mice to be susceptible to pneumococcal pneumonia than WT mice. However, our data are indicative of the involvement of other inflammasome receptors in addition to NLRP3, judging from the observations that ASC seemed to be more essential for the caspase-1 response and host resistance to S. pneumoniae than NLRP3. Indeed, we identified AIM2 as a receptor involved in the activation of caspase-1 in S. pneumoniae-infected macrophages. Thus, we suggest that not only NLRP3 but also other receptors, such as AIM2, are responsible for ASC-dependent secretion of IL-1β and IL-18 and consequent early defense against pneumococcal pneumonia.
We and other groups have demonstrated the ability of PLY to activate the TLR4 signaling, and we have previously shown that TLR4−/− macrophages secrete a lower level of IL-18 after infection with S. pneumoniae D39 than WT macrophages (8, 10, 47, 48). In addition, it has been shown that in some cases lipopolysaccharide, a canonical TLR4 ligand, induces the activation of the NLRP3 inflammasome dependent on TLR4 and an TLR adaptor protein, toll-interleukin-1 receptor domain-containing adapter protein inducing interferon beta (49). For these reasons, we assumed that PLY activates the NLRP3 inflammasome through recognition by TLR4. However, there was no significant difference in caspase-1 activation in response to S. pneumoniae between HeN macrophages and HeJ macrophages (Fig. 1). Moreover, McNeela et al. recently showed that secretion of IL-1β induced by rPLY plus other TLR stimuli, including heat-killed S. pneumoniae, does not require TLR4 (11). Thus, it appears that TLR4 is dispensable for the caspase-1 response to PLY-producing S. pneumoniae, and that the decrease in S. pneumoniae-induced secretion of IL-18 in the absence of TLR4 that we previously published might be due to reasons other than caspase-1 activation. As experiments demonstrating the ability of PLY to activate TLR4 signaling were usually performed using large amounts of PLY protein, the interaction between PLY and TLR4 may be not always be accurately reflected in innate immune responses to PLY-producing S. pneumoniae.
AIM2 is a cytoplasmic receptor that recognizes dsDNA in the cytoplasm, and we have verified that S. pneumoniae genomic DNA can be a ligand for AIM2. In the present study, knockdown or the deficiency of AIM2 caused significant decreases in caspase-1 activation, secretion of IL-1β and IL-18, and pyroptosis in response to S. pneumoniae. These results suggest that the AIM2 inflammasome is involved in caspase-1 activation in S. pneumoniae-infected macrophages. However, how AIM2 detects DNA upon infection with the extracellular pathogen remains unclear. In our study, inhibiting of bacterial uptake significantly reduced the activation of caspase-1 in S. pneumoniae-infected macrophages, suggesting that phagocytosis of bacteria by macrophages is an important process leading to the activation of AIM2 inflammasome. Although S. pneumoniae engulfed by macrophages was shown to undergo rapid death, which might cause the release of bacterial DNA from the bacterial cell, DNA in the phagosome would be hardly diffused across the phagosomal membrane, degraded by lysosomal hydrolases, such as DNase II, and therefore not be detected by AIM2. Importantly, PLY, a pore-forming toxin secreted upon the breakdown of the pneumococcal cell wall, is required for the activation of caspase-1 in response to S. pneumoniae. It is thus conceivable that PLY released from autolyzed or killed bacteria in the phagosome might cause destabilization of the phagosomal membrane, leading to the leakage of bacterial DNA into the cytoplasm. AIM2 has been reported to play a role in caspase-1 activation in infection with F. tularensis, L. monocytogenes, and DNA viruses that are all characterized as intracellular parasitic pathogens (50-54). By contrast, S. pneumoniae is generally regarded as an extracellular bacterium. Therefore, activation of AIM2 inflammasome is not a host response restricted to infection by intracellular parasitic microbes, and other pathogens may also activate the AIM2 inflammasome regardless of their niches if they possess the ability to deliver DNA into the cytoplasm.
Our data suggest that NLRP3 is also involved in the activation of caspase-1 in S. pneumoniae-infected macrophages. The NLRP3 inflammasome is activated by a wide variety of stimuli. In particular, pore-forming toxins, including PLY, have been reported to activate the NLRP3 inflammasome, owing to their pore-forming activities by causing K+ efflux (11, 21, 23-26). Since cell membrane permeation by PLY leads to K+ efflux (55) and PLY-induced activation of the NLRP3 inflammasome is inhibited by adding KCl to the cultures (11), it is conceivable that NLRP3 inflammasome activation induced after infection with S. pneumoniae depends on K+ efflux caused by PLY. Consistent with the assumption, addition of 20 mM KCl to cell cultures resulted in a significant decrease in the secretion of IL-1β and IL-18 from WT macrophages infected with S. pneumoniae (data not shown). However, it was difficult to judge whether the effect of KCl was due to specific inhibition of the NLRP3 inflammasome, as the same concentration of KCl also decreased the cytokine responses of NLRP3−/− macrophages to S. pneumoniae (data not shown). Since the presence of the cathepsin B inhibitor CA-074-Me reduces the secretion of IL-1β from PLY-stimulated DC (11), cathepsin B may also participate in the activation of the NLRP3 inflammasome in response to S. pneumoniae. If it is assumed that PLY released from phagocytosed S. pneumoniae damages the phagosomal membrane, S. pneumoniae-containing phagosomes may provide cathepsin B for the NLRP3 pathway.
Based on our experimental results, we have developed a mechanistic model of caspase-1 activation in macrophages infected by PLY-producing S. pneumoniae. PLY is an important virulence factor of S. pneumoniae that promotes inflammation, bacterial survival in the host, and ultimately pathogenicity of the pathogen. On the other hand, PLY is required for the activation of ASC imflammasomes that are critical for host defense against pneumococcal pneumonia by means of inducing the secretion of protective cytokines. The findings of this study have revealed a pathophysiological link between a virulence factor PLY and the inflammasome.
Acknowledgments
We thank Prof. Jürg Tschopp, University of Lausanne, for permission to use the NLRP3−/− mice; Dr. Vishva Dixit, Genentech, for permission to use the NLRC4−/− mice; Prof. Shun-ichiro Taniguchi, Shinshu University, for permission to use the ASC−/− mice; and Dr. Keisuke Kuida, Millennium Pharmaceuticals, for permission to use the caspase-1−/− mice. We thank Prof. Hiroko Tsutsui, Hyogo College of Medicine, for providing the NLRP3−/− mice, NLRC4−/− mice, ASC−/− mice, and caspase-1−/− mice.
This study was supported by a Grant-in-Aid for Scientific Research on Priority Areas from the Ministry of Education, Science, Culture, and Sports of Japan; Grants-in-Aid for Scientific Research (B) and (C); Grant-in-Aid for Young Scientists (B) from Japan Society for Promotion of Science; and grants from the National Institute of Health (NIH AG14357 and AR055398 to E.S.A.).
Footnotes
Disclosures The authors have no financial conflicts of interest.
References
- 1.Balakrishnan I, Crook P, Morris R, Gillespie SH. Early predictors of mortality in pneumococcal bacteraemia. J Infect. 2000;40:256–261. doi: 10.1053/jinf.2000.0653. [DOI] [PubMed] [Google Scholar]
- 2.Tuomanen EI, Austrian R, Masure HR. Pathogenesis of pneumococcal infection. N Engl J Med. 1995;332:1280–1284. doi: 10.1056/NEJM199505113321907. [DOI] [PubMed] [Google Scholar]
- 3.Mitchell TJ, Alexander JE, Morgan PJ, Andrew PW. Molecular analysis of virulence factors of Streptococcus pneumoniae. Soc Appl Bacteriol Symp Ser. 1997;26:62S–71S. [PubMed] [Google Scholar]
- 4.Gillespie SH, Balakrishnan I. Pathogenesis of pneumococcal infection. J Med Microbiol. 2000;49:1057–1067. doi: 10.1099/0022-1317-49-12-1057. [DOI] [PubMed] [Google Scholar]
- 5.Alouf JE. Cholesterol-binding cytolytic protein toxins. Int J Med Microbiol. 2000;290:351–356. doi: 10.1016/S1438-4221(00)80039-9. [DOI] [PubMed] [Google Scholar]
- 6.Marriott HM, Mitchell TJ, Dockrell DH. Pneumolysin: a double-edged sword during the host-pathogen interaction. Curr Mol Med. 2008;8:497–509. doi: 10.2174/156652408785747924. [DOI] [PubMed] [Google Scholar]
- 7.Paton JC, Rowan-Kelly B, Ferrante A. Activation of human complement by the pneumococcal toxin pneumolysin. Infect Immun. 1984;43:1085–1087. doi: 10.1128/iai.43.3.1085-1087.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malley R, Henneke P, Morse SC, Cieslewicz MJ, Lipsitch M, Thompson CM, Kurt-Jones E, Paton JC, Wessels MR, Golenbock DT. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc Natl Acad Sci U S A. 2003;100:1966–1971. doi: 10.1073/pnas.0435928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Houldsworth S, Andrew PW, Mitchell TJ. Pneumolysin stimulates production of tumor necrosis factor alpha and interleukin-1 beta by human mononuclear phagocytes. Infect Immun. 1994;62:1501–1503. doi: 10.1128/iai.62.4.1501-1503.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shoma S, Tsuchiya K, Kawamura I, Nomura T, Hara H, Uchiyama R, Daim S, Mitsuyama M. Critical involvement of pneumolysin in production of interleukin-1alpha and caspase-1-dependent cytokines in infection with Streptococcus pneumoniae in vitro: a novel function of pneumolysin in caspase-1 activation. Infect Immun. 2008;76:1547–1557. doi: 10.1128/IAI.01269-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McNeela EA, Burke A, Neill DR, Baxter C, Fernandes VE, Ferreira D, Smeaton S, El-Rachkidy R, McLoughlin RM, Mori A, Moran B, Fitzgerald KA, Tschopp J, Petrilli V, Andrew PW, Kadioglu A, Lavelle EC. Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog. 2010;6:e1001191. doi: 10.1371/journal.ppat.1001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Witzenrath M, Pache F, Lorenz D, Koppe U, Gutbier B, Tabeling C, Reppe K, Meixenberger K, Dorhoi A, Ma J, Holmes A, Trendelenburg G, Heimesaat MM, Bereswill S, van der Linden M, Tschopp J, Mitchell TJ, Suttorp N, Opitz B. The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J Immunol. 2010;187:434–440. doi: 10.4049/jimmunol.1003143. [DOI] [PubMed] [Google Scholar]
- 13.Kafka D, Ling E, Feldman G, Benharroch D, Voronov E, Givon-Lavi N, Iwakura Y, Dagan R, Apte RN, Mizrachi-Nebenzahl Y. Contribution of IL-1 to resistance to Streptococcus pneumoniae infection. Int Immunol. 2008;20:1139–1146. doi: 10.1093/intimm/dxn071. [DOI] [PubMed] [Google Scholar]
- 14.Jones MR, Simms BT, Lupa MM, Kogan MS, Mizgerd JP. Lung NF-kappaB activation and neutrophil recruitment require IL-1 and TNF receptor signaling during pneumococcal pneumonia. J Immunol. 2005;175:7530–7535. doi: 10.4049/jimmunol.175.11.7530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zwijnenburg PJ, van der Poll T, Florquin S, Roord JJ, Van Furth AM. IL-1 receptor type 1 gene-deficient mice demonstrate an impaired host defense against pneumococcal meningitis. J Immunol. 2003;170:4724–4730. doi: 10.4049/jimmunol.170.9.4724. [DOI] [PubMed] [Google Scholar]
- 16.Rijneveld AW, Florquin S, Branger J, Speelman P, Van Deventer SJ, van der Poll T. TNF-alpha compensates for the impaired host defense of IL-1 type I receptor-deficient mice during pneumococcal pneumonia. J Immunol. 2001;167:5240–5246. doi: 10.4049/jimmunol.167.9.5240. [DOI] [PubMed] [Google Scholar]
- 17.Lauw FN, Branger J, Florquin S, Speelman P, van Deventer SJ, Akira S, van der Poll T. IL-18 improves the early antimicrobial host response to pneumococcal pneumonia. J Immunol. 2002;168:372–378. doi: 10.4049/jimmunol.168.1.372. [DOI] [PubMed] [Google Scholar]
- 18.Zwijnenburg PJ, van der Poll T, Florquin S, Akira S, Takeda K, Roord JJ, van Furth AM. Interleukin-18 gene-deficient mice show enhanced defense and reduced inflammation during pneumococcal meningitis. J Neuroimmunol. 2003;138:31–37. doi: 10.1016/s0165-5728(03)00088-2. [DOI] [PubMed] [Google Scholar]
- 19.Lamkanfi M, Dixit VM. The inflammasomes. PLoS Pathog. 2009;5:e1000510. doi: 10.1371/journal.ppat.1000510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 21.Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 22.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCoy AJ, Koizumi Y, Toma C, Higa N, Dixit V, Taniguchi S, Tschopp J, Suzuki T. Cytotoxins of the human pathogen Aeromonas hydrophila trigger, via the NLRP3 inflammasome, caspase-1 activation in macrophages. Eur J Immunol. 2010;40:2797–2803. doi: 10.1002/eji.201040490. [DOI] [PubMed] [Google Scholar]
- 24.Chu J, Thomas LM, Watkins SC, Franchi L, Nunez G, Salter RD. Cholesterol-dependent cytolysins induce rapid release of mature IL-1beta from murine macrophages in a NLRP3 inflammasome and cathepsin B-dependent manner. J Leukoc Biol. 2009;86:1227–1238. doi: 10.1189/jlb.0309164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harder J, Franchi L, Munoz-Planillo R, Park JH, Reimer T, Nunez G. Activation of the Nlrp3 inflammasome by Streptococcus pyogenes requires streptolysin O and NF-kappa B activation but proceeds independently of TLR signaling and P2X7 receptor. J Immunol. 2009;183:5823–5829. doi: 10.4049/jimmunol.0900444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meixenberger K, Pache F, Eitel J, Schmeck B, Hippenstiel S, Slevogt H, N’Guessan P, Witzenrath M, Netea MG, Chakraborty T, Suttorp N, Opitz B. Listeria monocytogenes-infected human peripheral blood mononuclear cells produce IL-1beta, depending on listeriolysin O and NLRP3. J Immunol. 2010;184:922–930. doi: 10.4049/jimmunol.0901346. [DOI] [PubMed] [Google Scholar]
- 27.Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med. 2009;206:79–87. doi: 10.1084/jem.20081667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hise AG, Tomalka J, Ganesan S, Patel K, Hall BA, Brown GD, Fitzgerald KA. An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe. 2009;5:487–497. doi: 10.1016/j.chom.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, Aderem A. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol. 2006;7:569–575. doi: 10.1038/ni1344. [DOI] [PubMed] [Google Scholar]
- 32.Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozoren N, Jagirdar R, Inohara N, Vandenabeele P, Bertin J, Coyle A, Grant EP, Nunez G. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat Immunol. 2006;7:576–582. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- 33.Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, Leaf IA, Aderem A. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci U S A. 2010;107:3076–3080. doi: 10.1073/pnas.0913087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arlehamn CS, Evans TJ. Pseudomonas aeruginosa pilin activates the inflammasome. Cell Microbiol. 2011;13:388–401. doi: 10.1111/j.1462-5822.2010.01541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, Hardy LL, Garceau V, Sweet MJ, Ross IL, Hume DA, Stacey KJ. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science. 2009;323:1057–1060. doi: 10.1126/science.1169841. [DOI] [PubMed] [Google Scholar]
- 36.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burckstummer T, Baumann C, Bluml S, Dixit E, Durnberger G, Jahn H, Planyavsky M, Bilban M, Colinge J, Bennett KL, Superti-Furga G. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol. 2009;10:266–272. doi: 10.1038/ni.1702. [DOI] [PubMed] [Google Scholar]
- 39.Srinivasula SM, Poyet JL, Razmara M, Datta P, Zhang Z, Alnemri ES. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J Biol Chem. 2002;277:21119–21122. doi: 10.1074/jbc.C200179200. [DOI] [PubMed] [Google Scholar]
- 40.Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, Dixit VM. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430:213–218. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- 41.Yamamoto M, Yaginuma K, Tsutsui H, Sagara J, Guan X, Seki E, Yasuda K, Yamamoto M, Akira S, Nakanishi K, Noda T, Taniguchi S. ASC is essential for LPS-induced activation of procaspase-1 independently of TLR-associated signal adaptor molecules. Genes Cells. 2004;9:1055–1067. doi: 10.1111/j.1365-2443.2004.00789.x. [DOI] [PubMed] [Google Scholar]
- 42.Wu J, Fernandes-Alnemri T, Alnemri ES. Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by Listeria monocytogenes. J Clin Immunol. 2010 doi: 10.1007/s10875-010-9425-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsuchiya K, Hara H, Kawamura I, Nomura T, Yamamoto T, Daim S, Dewamitta SR, Shen Y, Fang R, Mitsuyama M. Involvement of absent in melanoma 2 in inflammasome activation in macrophages infected with Listeria monocytogenes. J Immunol. 2010;185:1186–1195. doi: 10.4049/jimmunol.1001058. [DOI] [PubMed] [Google Scholar]
- 44.Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24:93–103. doi: 10.1016/j.immuni.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 45.Ishii KJ, Coban C, Kato H, Takahashi K, Torii Y, Takeshita F, Ludwig H, Sutter G, Suzuki K, Hemmi H, Sato S, Yamamoto M, Uematsu S, Kawai T, Takeuchi O, Akira S. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat Immunol. 2006;7:40–48. doi: 10.1038/ni1282. [DOI] [PubMed] [Google Scholar]
- 46.Littmann M, Albiger B, Frentzen A, Normark S, Henriques-Normark B, Plant L. Streptococcus pneumoniae evades human dendritic cell surveillance by pneumolysin expression. EMBO Mol Med. 2009;1:211–222. doi: 10.1002/emmm.200900025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dessing MC, Hirst RA, de Vos AF, van der Poll T. Role of Toll-like receptors 2 and 4 in pulmonary inflammation and injury induced by pneumolysin in mice. PLoS One. 2009;4:e7993. doi: 10.1371/journal.pone.0007993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Srivastava A, Henneke P, Visintin A, Morse SC, Martin V, Watkins C, Paton JC, Wessels MR, Golenbock DT, Malley R. The apoptotic response to pneumolysin is Toll-like receptor 4 dependent and protects against pneumococcal disease. Infect Immun. 2005;73:6479–6487. doi: 10.1128/IAI.73.10.6479-6487.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Imamura M, Tsutsui H, Yasuda K, Uchiyama R, Yumikura-Futatsugi S, Mitani K, Hayashi S, Akira S, Taniguchi S, Van Rooijen N, Tschopp J, Yamamoto T, Fujimoto J, Nakanishi K. Contribution of TIR domain-containing adapter inducing IFN-beta-mediated IL-18 release to LPS-induced liver injury in mice. J Hepatol. 2009;51:333–341. doi: 10.1016/j.jhep.2009.03.027. [DOI] [PubMed] [Google Scholar]
- 50.Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J, Datta P, McCormick M, Huang L, McDermott E, Eisenlohr L, Landel CP, Alnemri ES. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol. 2010;11:385–393. doi: 10.1038/ni.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, Hornung V, Vogel SN, Szomolanyi-Tsuda E, Fitzgerald KA. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010;11:395–402. doi: 10.1038/ni.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim S, Bauernfeind F, Ablasser A, Hartmann G, Fitzgerald KA, Latz E, Hornung V. Listeria monocytogenes is sensed by the NLRP3 and AIM2 inflammasome. Eur J Immunol. 2010;40:1545–1551. doi: 10.1002/eji.201040425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sauer JD, Witte CE, Zemansky J, Hanson B, Lauer P, Portnoy DA. Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe. 2010;7:412–419. doi: 10.1016/j.chom.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Warren SE, Armstrong A, Hamilton MK, Mao DP, Leaf IA, Miao EA, Aderem A. Cutting edge: Cytosolic bacterial DNA activates the inflammasome via Aim2. J Immunol. 2010;185:818–821. doi: 10.4049/jimmunol.1000724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cockeran R, Theron AJ, Steel HC, Matlola NM, Mitchell TJ, Feldman C, Anderson R. Proinflammatory interactions of pneumolysin with human neutrophils. J Infect Dis. 2001;183:604–611. doi: 10.1086/318536. [DOI] [PubMed] [Google Scholar]