

Abstract
Advances in genotyping and sequencing technologies have revolutionized the genetics of complex disease by locating rare and common variants that influence an individual’s risk for diseases, such as diabetes, cancers, and psychiatric disorders. However, to capitalize on this data for prevention and therapies requires the identification of causal alleles and a mechanistic understanding for how these variants contribute to the disease. After discussing the strategies currently used to map variants for complex diseases, this Primer explores how variants may be prioritized for follow-up functional studies and the challenges and approaches for assessing the contributions of rare and common variants to disease phenotypes.
Most common diseases are complex: many genetic and environmental factors mediate the risk for developing the disease and each individual factor explains only a small proportion of population risk (Cardon and Abecasis, 2003). Genome-wide genotyping with high-throughput approaches has led to the identification of >2,600 associated common risk alleles, with convincing associations in >350 different complex traits (most with modest effect size of odds ratio <1.5) (Hindorff et al., 2009). More recently, low cost, high throughput sequencing of exomes and whole genomes are giving investigators access to the spectrum of rare inherited variants and de novo mutations. Once an associated allele is discovered, a critical step to characterizing pathogenesis is the definition of the causal allele, that is the functional allele that influences disease susceptibility and explains the observed association. However for the vast majority of associated alleles, the identities of causal genes and variants, as well as the function of these variants, remain uncertain. This Primer discusses the population genetics features of rare and common alleles, strategies for connecting these alleles to disease, and strategies to prioritize them for functional follow up studies.
POPULATION GENETICS OF RARE AND COMMON ALLELES
Geneticists have long debated the extent to which rare and common alleles contribute to complex disease (Pritchard, 2001; Pritchard and Cox, 2002; Reich and Lander, 2001). While there is evidence of susceptibility alleles across the frequency spectrum in many complex diseases, it is important to realize that rare alleles and common alleles have different population characteristics that are relevant to medical genetics.
The exact distinction between rare and common alleles is to an extent an arbitrary one. We define common alleles as those with frequencies >1%; these alleles are frequent enough that they can be queried by genotyping in standard marker panels. Rare alleles are polymorphic alleles with <1% frequency that might now be effectively studied with sequencing technologies. The rarest alleles are seen in only a handful of individuals or are private to a single individual –and require sequencing for discovery.
The Origin of Polymorphic Alleles
De novo mutations occurring spontaneously in individuals are constantly and rapidly introduced into any population. These mutations are initially “private” to the individual that they occurred in, but might then be passed on to progeny. Most of these mutations are quickly filtered out or lost by genetic drift and will never achieve appreciable allele frequencies. I illustrate this concept by a simulation in which de novo neutral mutations (conferring no effect on fitness) are introduced into a population of 2,000 diploid individuals. In 31 generations 95% of these mutations disappear from the general population, and not one of these mutations achieves an allele frequency of >1% in 200 generations (see Figure S1). Mutations that are deleterious are even more rapidly purged from the populations. While any de novo mutation is very unlikely to become a common allele, even a somewhat deleterious mutation may persist for a few subsequent generations as a rare allele before disappearing.
Thus populations harbor many rare alleles, most of which have been derived recently, but relatively few common ones. In fact, there is only about one common variant on average per ~500 bp in European populations (The 1000 Genomes Project Consortium, 2010). On the other hand, recent and rapid expansion of human populations has resulted in the presence of many rare alleles. At the extreme of the allele frequency spectrum are de novo mutations; each individual harbors ~40 de novo point mutations that may not be present in any other individuals (Conrad et al., 2011).
Common alleles tend to be more ancient than rare ones as it takes many generations for a rare allele to rise to a reasonable allele frequency. There are important exceptions to these generalizations. An ancient allele may be rare because it is being depleted from the population. A common allele may be recent, if it confers a critical survival advantage or has emerged after a rapid population expansion from a small founder population.
Linkage Disequilibrium and Haplotypes
Genetic linkage is the tendency of alleles at nearby loci to be transmitted together; two nearby loci are in linkage disequilibrium (LD) when recombination events occur between them very infrequently. Two common metrics quantify pairwise LD between bi-allelic markers (see Figure 1A). The R-squared (r2) between two markers is their correlation across chromosomes within a population. If two markers have r2=1, then alleles are always in phase (or in cis) with each other; in a genetic study their association statistics will be identical. The D-prime (D’) between two markers is inversely related to the fraction of chromosomes that have had historical recombination between them. If D’=1, two bi-allelic variants constitute only two or three haplotypes, while if D’<1, all four possible haplotypes are present in the population. If D’=0 or r2=0 then the two markers are unlinked and statistically independent of each other.
Figure 1. Linkage disequilibrium (LD) metrics.


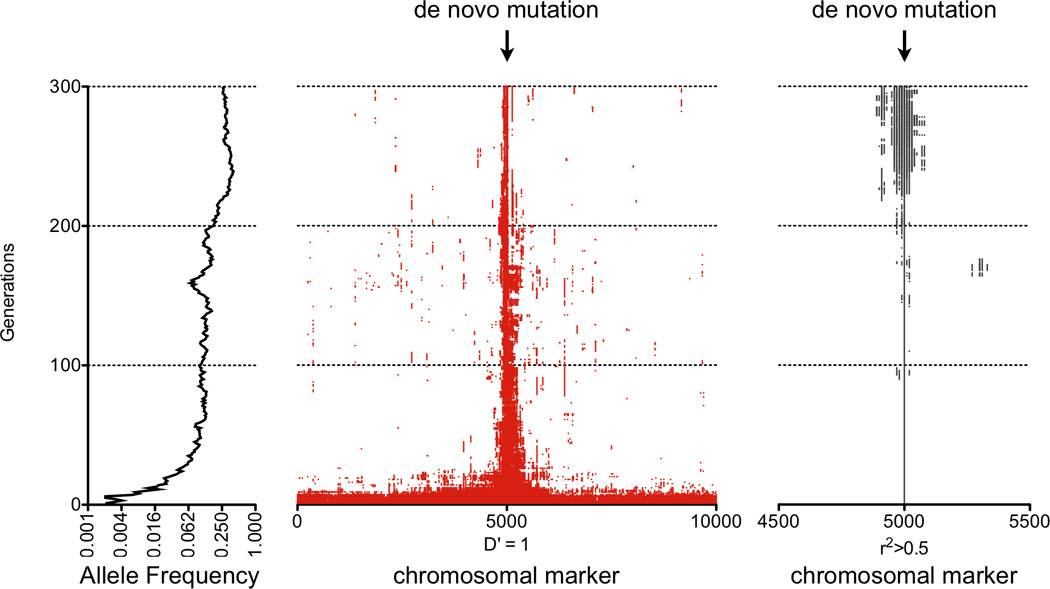
A. Haplotype frequency with changes in LD. Left: For two markers that are random with respect to each other, each with a 0.5 allele frequency, there is no linkage between them; each resulting haplotype has a frequency of 0.25. Middle left: Here the two markers are not entirely random, and alleles at one marker correlate partially with alleles at the other marker. The A allele on the left is observed more frequently with the C allele on the right, and the T allele on the left is observed more frequently with the G allele on the right. Middle right: Here the two alleles are more tightly linked or have tighter LD than in the previous case. In this instance, the presence of the T allele on the left predicts with certainty the G allele on the right. This could be the case if the T allele arose de novo on a haplotype with the G allele in the right marker. Right: For instances of tight LD, an allele at one marker predicts perfectly the allele at the other marker; in this case, these two markers form only two haplotypes. B. Changing LD properties as a de novo mutation propagates. A de novo event (circle), when it first occurs on a chromosome (bottom), is on one haplotypic background defined by the chromosomal markers on which it forms (red). As generations pass (moving upward), the event propagates through the population. Recombination events (Xs) occur, reducing the common haplotype (red) on which variant is present and decoupling it from distal markers (blue). C. Simulating LD structure of a de novo event as it becomes a common variant. Here a computer simulation depicts a chromosome with 10,000 common markers with 1,000 randomly assigned hotspots. Random mating is allowed with an average of one recombination event per generation. A single rare variant is introduced in the middle of the chromosome on one individual (bottom) and allowed to propagate through the population. The left panel depicts the allele frequency as it increases through the generations (upwards). In the middle panel, all markers in LD with that variant (with D’=1) are indicated with a red dot. Initially that variant is in LD with every common marker that it is in phase with on that chromosome, revealed by the red band stretching across the bottom of the plot. As random recombination events occur and the allele becomes more frequent, the number of markers in phase decreases, revealed by the shrinking red band in the middle. On the right panel, a grey dot indicates markers for which the genotypes correlate with the rare variant (r2>0.5). For the first few generations, there are no other variants that correlate with the de novo mutation as it becomes a rare allele. As time progresses and the allele becomes more common, it begins to develop genotypic correlations with nearby variants that remain on the same haplotype.
Recombination events break down pairwise linkage between markers over time, and reduce the length of haplotypes in a population. Recombination events are much more likely to occur in hotspot regions in the genome than other regions (Myers et al., 2005). As a result, markers without a recombination hotspot between them are often linked over long periods of time and have high pairwise D’. Those markers can often be grouped into a set of limited number of common haplotypes (see Figure 2). Phasing algorithms can be applied to determine markers in cis and to define the most likely haplotypes.
Figure 2.


A. Common variants. This image illustrates the structure of common variants and linkage disequilibrium blocks. The top lists a reference genome spanning ~10 kb and the reference genotypes of the variants. The haplotype structure is broken up into two blocks by a recombination hotspot. Each block contains a set of markers in tight LD, which can be phased into a small number of haplotypes. Below that, a limited number of genotypes are depicted for a hypothetical individual because a commercial array would assay only a limited collection of all of the common variants in a region. The bottom row demonstrates how data for those genotypes can be phased using reference population data and how missing genotypes can be imputed if the haplotype can be inferred accurately. In some instances, imputed genotypes may not be uncertain. B. Fine mapping with conditional haplotype analysis. This figure illustrates the basic concept of conditional haplotype mapping. The left-hand side lists genotypes at ten variant sites (numbered) that define 7 common haplotypes. Each row represents a haplotype, and genotypes at variant sites are listed in each column. Assuming that a common variant association is observed at marker 1, identical associations will be observed at the markers 2, 3, and 5 because their genotypes are correlated across haplotypes. In the first step, haplotypes are grouped by marker 1. The result is that the seven haplotypes form two subgroups (indicated by purple and red bars on the right). One group demonstrates association with disease (right). Including marker 7 breaks the groups up further into four haplotypes (indicated by purple, green, blue, and red bars on the far right). By adding marker 7, differential risk association between haplotypes is apparent. Whereas the T/G haplotype confers risk, the T/T haplotype confers even more risk.
Rare alleles generally sit on long haplotypes while common alleles sit on shorter ones. When a mutation first occurs de novo on a chromosome, it occurs on the background of a single rare haplotype defined by all markers on that chromosome (see Figure 1B). Since the de novo mutation appeared as a random event, it initially has no correlation with other markers on that chromosome (r2=0). In initial generations, prior to a recombination event, the mutation has D’=1 with other markers across the chromosome. But, if the mutation survives generations and becomes a common allele, repeated recombination events fragment that haplotype and reduce its length. The allele retains high D’ to only proximate markers that are not separated from it by a recombination hotspot. As the variant becomes more frequent, so does the haplotype that it occurs on; over time the emerging variant develops correlation (r2≫0) with the markers on that short haplotype (see Figure 1C).
Finding Pathogenic Variants, Both Rare and Common
Common variant associations to phenotype are often facile to find. Their high frequencies allow case-control studies to be adequately powered to detect even modest effects. Their high r2 to other proximate common variants allows for association signals to be discovered by genotyping the marker directly, or other nearby correlated markers. But mapping those associated variants to the specific variant that functionally influence disease risk can be challenging since the statistical signals invoked by inter-correlated variants are difficult to disentangle.
On the other hand, individual rare variant associations are challenging to find. Their low frequency renders current cohorts underpowered to detect all but the strongest effects, and lack of correlation to other markers often prevents them from being picked up by a standard genotyping marker panels. But, once a rare associated variant is identified, mapping the causal rare variants is relatively facile since recent ancestry is likely to limit the number of inter-correlated markers.
Functional properties of pathogenic variants, both rare and common
Since common alleles tend to be ancient, they have weathered the influences of purifying negative selection. Therefore, common variants that influence disease risk are likely to have functionally modest effects that are compatible with their high population frequency. There are two possibilities outlined by Kruyokov et al that might allow for this (Kryukov et al., 2007). First, common variants that are medically detrimental act subtly or specifically to confer disease without altering evolutionary fitness. As an example consider a variant that confers risk of addiction to tobacco (Thorgeirsson et al., 2008). Such a variant might have little impact on survival historically, but might have specific neuropsychiatric effects that mediate the risk of 21st century diseases such as lung cancer or coronary artery disease that play a role later in life after reproduction. Second, forces that select specifically for these common variants counteract their medically detrimental qualities; the variant while causing disease also offers evolutionary benefit simultaneously. For example common ApoL1 variants that confer high risk of chronic kidney disease in African Americans, but protects from Trypanosoma brucei rhodesiense infection at the same time (Genovese et al., 2010).
Since rare alleles are more recent, they have not been subjected to the same negative selective pressures yet and may include among them more relatively deleterious mutations. Rare alleles therefore often include those more likely to have more dramatic functional consequences. This is supported by data indicating that rare deletions are more likely than more common deletions to remove entire genes, exons, promotors, or stop codons (Conrad et al., 2009). Similarly, rare variants are twice as likely as common ones to be to be non-synonymous (The 1000 Genomes Project Consortium, 2010). Since rare variants are relatively unrestricted in terms of their functional impact in general, a subset of rare pathogenic variants with large effect might offer more obvious insight about disease mechanism.
COMMON VARIANTS
Detecting common variants with high throughput SNP arrays
High throughput genotyping of standard marker panels of common SNPs has become possible with microarrays (Gunderson et al., 2005). Their application to large case-control sample collections has facilitated detection of even the most modest risk alleles, with odds ratios of 1.1 or less. There are a finite number of common variants present in the general population, i.e. <6 million are estimated in European populations (The 1000 Genomes Project Consortium, 2010). But nearby common SNPs are in LD with one another and define a limited number of haplotypes (see Figure 2A); so the effective number of independent variants is much fewer. Thus genotyping a limited number of common variants genome-wide has the effect of covering many more common variants. In European populations, the Affymetrix 5.0 array with 440K SNPs has r2>0.8 for 57% of common variants, and the Affymetrix 6.0 array with roughly double the number of SNPs (900K) has r2>0.8 for 66% of common variants (Bhangale et al., 2008).
Genome-wide genotyping also allows investigators to use imputation to estimate genotypes of markers not directly genotyped; in doing so it becomes possible to combine samples genotyped on different platforms. Probabilistic multipoint imputation algorithms, using a limited number of genotyped common variants, can determine the genotypes of ungenotyped common variants by comparing to a reference panel of comprehensively genotyped individuals, (see Figure 2A). Most of these methods currently use probabilistic Hidden Markov Model approaches to infer the local LD structure (Browning, 2008; de Bakker et al., 2008).
Selecting populations for study
Initial efforts to map complex traits emphasized selected isolated populations, for example the Finish populations (Peltonen et al., 2000). These populations can offer the advantage of increased inbreeding, more uniform genetic and environmental backgrounds, detailed genealogical records, availability of intact extended families, and longer LD intervals. Populations that have undergone rapid population expansion may be of particular use since LD intervals are longer. The most successful validation of this approach is represented by the deCODE genetics and their study of a wide-range of complex diseases in Iceland.
Now, investigators are increasingly focused on the Inclusion of individuals from multiple ethnic backgrounds in order to enhance the ability of studies to discover risk alleles with variable allele frequencies across different backgrounds (Rosenberg et al., 2010). Different ethnic backgrounds might highlight different mechanisms of disease pathogenesis, including differences in environmental exposures, as well as reflect different degrees of genetic diversity and linkage disequilibrium patterns. A striking example of this is the discovery of an IL18B variant that predicts response to hepatitis C treatment with equivalent effect in European, African, and Hispanic American patients; allele frequency differences of the variant explains about half of the differences in treatment response across populations (Ge et al., 2009).
Genome-wide association studies
In a case-control genome-wide association study (GWAS) samples are genotyped for a set of 100,000 – 2,000,000 markers; case and control allele frequencies are compared directly to each other. Statistical significance is assessed with a simple 2×2 chi-square test, or with logistic regression when genotypes are probabilistic (e.g. from imputation).
Critical to the success of GWASs has been the application of stringent statistical significance thresholds that result in reproducible associations that account for the large number of simultaneous tests (Risch and Merikangas, 1996). Testing for common variant associations throughout the genome represents about ~1 million independent tests (Hoggart et al., 2008). Thus investigators routinely use a genome-wide significance threshold representing a Bonferoni correction for multiple tests (p=0.05/106 = 5×10−8).
Since effect sizes for most common variants are modest, large sample sizes and careful adjustment for subtle technical artifacts that can easily obscure results or produce false positive associations are of paramount importance (Balding, 2006; Clayton et al., 2005; McCarthy et al., 2008). The genomic inflation factor is an important metric that indicates the extent of inflation due to stratification and other technical confounders. Fortuitously, the strength of genome-wide genotyping goes beyond simply measuring case-control allele frequency differences throughout the genome. It also allows investigators to look at patterns in the genotyping data to identify key technical confounders. For instance, patterns of excessive “missing” genotype data for an individual indicates that intensity data could not be clustered into genotype, likely as a function of low DNA quality or concentration. Another key confounder is population stratification, that is the presence of the systematic allele frequency differences observed in a population as a consequence of ancestry rather than case-control status. As a dramatic example, Campbell et al, showed that even in studies using only European populations that not carefully adjusting for an individual’s country of origin results in a highly statistically significant false-positive association for height at a lactase SNP (Campbell et al., 2005). Genome-wide genotype data allow investigators to identify and correct for case-control population stratification.
Once markers are identified with having statistically significant allele frequency differences in cases and controls, they are ideally replicated in independent populations. The value of replicating in an independent population not only adds statistical confidence to the results, but also adds confidence that the results of the initial study is not the consequence of technical confounding or stratification.
Identifying an associated marker rarely clarifies if the marker itself is the functional allele that causes altered disease susceptibility. The observed association at a marker might be the result of an underlying causal allele with high r2 with the associated variant, a rare functional allele on a shared haplotypic background as the associated variant, or multiple functional alleles that cause an apparent association. Nevertheless, the causal alleles must closely correlate and be in LD with associated variants.
Fine-mapping common variant loci
Dense genotyping of markers in the region, followed by fine-mapping can identify the causal allele, or at least reduce the number of potential candidates. The underlying assumption is that the causal allele will most parsimoniously explain the entirety of the evidence of association. In many instances, however, fine mapping is complicated if the association is not being driven by a marker that has been genotyped; in those instances it might be possible to identify a risk haplotype defined by genotyped markers, and to then sequence selected individuals to identify the causal allele. Thus in order to fine map effectively, dense genotyping to include all known markers in the region is key. Fine-Additionally, in many instances there might be multiple causal alleles, and in order to be powered to detect multiple effects it is often necessary to densely genotype a large number of samples, perhaps more than those used to discover the association.
After densely genotyping a large number of samples, there are two major statistical tools utilized in fine-mapping common variants. The first is conditional regression. If a single lead marker (or another marker in perfect LD with it) is causal, then applying conditional regression adjusting for that lead marker should obviate all other association in the region. The second statistical tool is conditional haplotype analysis. With conditional haplotype analyses, investigators start with data from a subset of the genotyped markers and phase genotypes to define haplotypes. If the selected markers are causal then the defined haplotypes should parsimoniously explain the risk at that locus. That is the addition of additional markers (and thus creation of more haplotypes) should not explain risk better, and removal of any marker (and thus removal of haplotypes) should reduce the explained risk. With both approaches, if the causal allele is in perfect LD (r2=1) with other markers, then identifying statistically indistinguishable associations may not be possible.
One striking example of fine-mapping was an effort by Pereyra et al where with a GWAS they demonstrated that multiple HLA-B classical alleles are associated with long-term viral load control in HIV infected individuals (Pereyra et al., 2010). Then, with conditional haplotype analysis, they were demonstrated that allelic risk was best defined by amino acid variation at a few sites along the binding groove of HLA-B.
Data from multiple ethnic populations may be particularly useful to fine-map associations (Rosenberg et al., 2010). Ideally a single allele might explain risk across multiple ethnic groups. This approach is effective only if the same causal allele is present with a high allele frequency in both, and there are ethnic differences in local LD structure. The inclusion of African populations might be particularly useful since LD patterns are generally shorter. This approach might be complicated if multiple different alleles in populations influence disease susceptibility within the same locus. Adrianto et al looked at SNPs associated with systemic lupus erythematosus (SLE) spanning the TNFAIP3 gene (Adrianto et al., 2011). When they looked at markers associated in Asian and European populations they were able to fine-map the associated region from a span of ~100 kb to ~50 kb. Subsequent sequencing identified a novel AA>T single base pair deletion polymorphism that acts to disrupt an NF-κb binding site. This single variant explained the associated risk of the locus.
RARE VARIANTS
It is possible that associated rare variants for complex diseases will be more facile to fine-map and to evaluate for functional impact. The discovery of a rare variant near a common variant might be particularly informative. A rare variant is clearly impacting one of the multiple nearby candidate genes in LD with the common variant might clarify the pathogenic gene and offer clues about mechanism of the common variant. There have been several examples of this reported in the literature already. Five genes with confirmed type II diabetes common risk alleles, PPARg, HNF1A, KCNJ11, WFS1 and HNF1B, also have known familial forms as a consequence of nearby rare mutations (Voight et al., 2010). Similarly, 18 of the 95 known common variants associated with serum lipid levels are near genes that have been implicated in monogenic lipid disorders (Teslovich et al., 2010). Indeed studies to find rare coding variants near common risk loci have already shown success in type I diabetes (Nejentsev et al., 2009), age-related macular degeneration (Raychaudhuri et al., In Press), and Crohn’s Disease (Momozawa et al., 2011).
The extent to which rare variants explain complex disease susceptibility in general remains an open question. It has been speculated that the gap between the heritability explained by known common variants and that which might be predicted from family studies might be explained by rare variants (Bansal et al., 2010), and that even many observed common variant associations might be the consequence of functional undiscovered are variants (Anderson et al., 2011; Dickson et al., 2010). Other investigators have suggested that undiscovered common variants themselves might explain much of that missing heritability (Purcell et al., 2009; Yang et al., 2010).
Identifying rare variants with high throughput sequencing
Advances in DNA capture and sequencing technology has greatly facilitated targeted, exome, and whole genome sequencing (Maxmen, 2011; Ng et al., 2010), and has in the process enhanced the search for rare variants. While the cost of sequencing is rapidly dropping, the computational and statistical challenges to rapidly aligning sequences to reference sequences, separating variant calls (SNPs, indels, and structural variants) from sequencing artifact, data storage, and establishing associations are mounting (McKenna et al., 2010).
Second-generation sequencing technologies have now come online, and are distinct from prior approaches in that they do not use Sanger chemistry, and are characterized by high sequencing yield with shorter reads (Shendure and Ji, 2008). The Illumina HiSeq 2000 system for example generates >1 billion 100 bp paired-end useable reads per run. Efficiently mapping a large volume of short reads to the reference genome accurately has been an important area of methodological progress (Li and Homer, 2010). Look-up (or hash-table) based methods map reads quickly, but are not as accurate as less-efficient alignment-based methods. Accurate alignment is especially important in regions with short insertions or deletions (indels); poor alignment in such regions can result in false positive SNP calls and false negative indel calls. Repetitive genomic regions and regions with homology can be challenging to map, and in some instances may not be possible to query effectively. Paired-end sequencing generates two sequence reads generated from opposite ends of the same contiguous genomic fragment, and helps overcome some of these alignment issues.
To sensitively and accurately call a heterozygote non-reference base, a minimum of ~20x coverage is necessary to overcome the uncertainty resulting from sampling short sequence reads across a diploid genome. Additional coverage may be necessary to compensate for random and non-random sequencing error, which may vary across technologies. Even with a high-coverage sequencing experiment, the coverage is typically non-uniform across the targeted region. Non-uniform coverage can be related to biases in DNA capture technologies, in unequal pooling of amplicon products from different genomic regions or individuals, and intrinsic sequence properties (Harismendy et al., 2009). Careful experimental technique and sample normalization can minimize some biases in coverage. Average coverage of an experiment is thus not as useful of a metric as is the percentage of target genomic region achieving more than a prespecified coverage threshold. A set of independently genotyped SNPs to verify sequence-based genotype calls and assess the accuracy of sequencing studies is useful to confirm accuracy.
Sequencing can be applied to a set of samples to discover variants or genotype variants. For variant discovery, sequence data can be pooled across multiple samples to boost power to detect a non-reference base. After application of sequencing to discover rare variants, confirming the presence of the variant in discovery samples with TaqMan or capillary electrophoresis sequencing is useful before exploring in independent samples to establish disease association.
Power considerations and significance testing
One of the challenges to convincingly establishing a common variant association in human populations and families is the rarity of SNP itself. Genetic studies are more poorly powered to detect a rare SNP association than they are to detect more common association with the same effect size (see Figure 3). Thus to detect associations at the same statistical threshold, larger sample collections might be necessary than ones currently used. Establishing association of a de novo or private mutations may not be possible since they may be seen only once in an entire study.
Figure 3.


A. Power to find rare variants. Here is a plot of 80% power to discover rare associated alleles at p<10−7 and p<10−11 for cohorts of both 500 and 5000 cases and controls. The control allele frequency and odd ratio (OR) are plotted along the x-axis and the y-axis, respectively. Diagonal lines indicate corresponding case allele frequencies. B. Burden testing. Here data from sequenced cases (top) and controls (bottom) are depicted around a gene of interest. Each horizontal line represents an individual. Variants are shown as red Xs. Certain variants are rare (i.e., seen once), and others are more common (vertical line). In this example, the case variants within the candidate gene (arrow at bottom, and blue shading) are seen more frequently than in controls. If common variants are excluded, there are five case chromosomes with a rare variant compared to one control chromosome. This pattern of enrichment is not evident outside the gene. A burden test of association for rare variants within the gene might be statistically significant.
For rare variant associations, the field has not yet defined accepted standards for statistical significance that account for the burden of multiple hypothesis testing. Since there are many more rare variants than common ones, and they are not typically inter-correlated with each other, a more stringent threshold may be necessary than applied for common variants. One conservative approach is to correct for the total number of bases genome-wide, ie p=0.05/3000000000 ~ 10−11 as a significance threshold. Most recent studies have limited themselves to exomes or to a subset of targeted genes; in these instances the multiple-hypothesis testing burden might be significantly less. But with spectre of genetic studies with genome-sequencing in the very near future this conservative threshold may ultimately turn out to be appropriate.
Despite limitations in power and the need for achieving greater significance, rare variant associations with strong effects might be imminently detectable. For instance Holm et al was able to identify a rare variant for sick sinus syndrome as part of a genome-wide study (Holm et al., 2011).; the coding variant that explained the association was highly statistically significant in a modestly sized cohort since it had such a large effect size (OR>12). One strategy to further enhance the prospects of discovery is to identify those individuals most likely to have highly penetrant rare mutations. For example, individuals with younger onset or more severe disease, familial forms of disease, or those individuals that have disease despite a lack of other clinical or genetic risk factors might be promising candidates for rare variant association studies.
Burden testing
If a genomic region is critical to disease pathogenesis rare mutations may modulate disease susceptibility. Then many affected individuals may have rare mutations more frequently in that region, though the mutations may be different from and unrelated to one another. This concept has sparked interest in the genetics community, and workers in statistical genetics have devised strategies to examine rare variants in aggregate across a target region (Bansal et al., 2010). These “burden” tests assess if rare variants within a specific region are distributed in a non-random way, suggesting that they might be playing a roll in disease pathogenesis (see Figure 3B). For example a simple burden test might assess whether cases are enriched for rare variants compared to controls.
More sophisticated tests account for the possibility that the region contains both protective and risk conferring mutations. The target region might be a specific sub-region of a gene, an entire gene transcript, or the entire genome.
This approach is an important alternative to the challenging task of establishing the association of individual rare variants; using these approaches to test multiple variants simultaneously might enhance power over testing individual variants. For instance, a burden test might be able to identify non-random distributions even of private mutations.
In an early application of rare variant burden testing, Cohen et al examined individuals from the general population with high and low HDL levels, and assessed the burden of rare variation in three candidate genes known to harbor Mendelian mutations that cause familial low serum high density lipoprotein (HDL) levels (Cohen et al., 2004). They found that individuals with the low HDL individuals were significantly more likely to contain rare nonsynoymous mutations than those with high HDL levels; of the low HDL individuals 16% had at least one rare mutation compared to 2% of high HDL individuals. This suggested strongly that for individuals with low HDL levels ~14% of them may have mutations in these three genes mediating phenotype. The idea of comparing the proportion of case individuals with a rare alleles to control individuals with rare alleles was formalized into a statistical test, the “Cohort Allelic Sums Test” (CAST) (Morgenthaler and Thilly, 2007). Subsequently, more sophisticated tests have been proposed, that allow investigators to combine association testing of rare and common alleles by either testing for association together in multivariate tests (Li and Leal, 2008) or by combining rare and common alleles weighted inversely to their allele frequency (Madsen and Browning, 2009).
One very powerful way of enhancing burden testing is to filter variants that are more likely to be causal from those that are likely not to be causal. For example, investigators may to focus their study on nonsynonymous alleles. Alternative approaches might include filtering variants based on sequence conservation properties or other bioinformatics approaches (Adzhubei et al., 2010; Ng and Henikoff, 2003).
A successful test, where statistical significance is obtained, can be used to argue that (1) the tested rare variants play a role in a specific disease and (2) that the target region tested plays an important role in disease pathology. But, it fails to implicate specific variants, and ambiguity about the causal variants might remain. For example if rare variants are enriched in a gene two-fold in cases compared to controls, then roughly half the variants seen in cases might be pathogenic, but the other half are part of the background distribution of rare variation in that gene, and may not influence disease risk.
Structural variants
Rare structural variants have gained recent interest; the frequency and size of structural variants have repeatedly showed enrichment in schizophrenia and other neuropscychiatric disease (International Schizophrenia Consortium, 2008; Sebat et al., 2007; Walsh et al., 2008). However, except for a few specific regions such as 22q11 and 16p11, most of rare events have uncertain pathogenecity. For instance, while the rate of >100 kb deletion events are significantly increased in cases compared to controls –there is great uncertainty as to which individual events are pathogenic and which ones are non-pathogenic events that might occur in the general healthy population. This is analogous to the circumstance that might occur with a statistically significant burden test for point mutations described above.
Extended haplotypes
As previously discussed, many rare variants are recent and occur on extended haplotypes that can be identified using common variant markers. Thus GWAS data sets may be used to identify long-range haplotypes based on common markers, and to then assess if they are associated with phenotype. If this is the case, the phenotypic association might be driven by a highly penetrant rare variant. We used this approach to find an extended haplotype in the CFH gene that conferred high risk of age-related macular degeneration; subsequent sequencing identified the causal mutation to be an argenine to cysteine change in the C-terminus of the protein (Raychaudhuri et al., In Press).
This approach might be most effective in isolated populations where reduced genetic diversity and founder effects make it possible to identify long-range haplotypes (Kong et al., 2008). One recently published method to identify long and rare haplotypes, and to then to test for association to phenotype has been successfully applied to multiple phenotypes in out-bred populations (Gusev et al., 2011).
FROM VARIANTS TO FUNCTION
Translating rare and common variants to function can be challenging. In many instances the presence of an association does not clarify which variants are functionally causing disease susceptibility. For common variants, fine-mapping might be stymied by local LD. For rare variants, burden testing might be able to identify a genomic region enriched for rare variants, but may not be able to specifically distinguish the individual causal rare variants from spurious non-pathogenic variants. Here we describe broad approaches that might be pursued to clarify pathogenic functions and causality, in the absence of genetic mapping that has clearly identified a single causal variant.
Evaluating Nonsynonymous Coding variants
About 1% of the genome consists of protein coding sequences. Variants in this portion of the genome are potentially the most amenable to follow-up by biochemical characterization of the protein product in vitro, characterization in cell lines, or evaluation in transgenic model organisms. Only a minority of associated common variants can be explained by a non-synonymous coding variant (~10%) (Hindorff et al., 2009). Currently, most studies of rare variation emphasize nonsynomous coding variants; in many cases non-coding variants are altogether ignored even if they are sequenced. An important challenge in the field is to prioritize discovered coding variants for potentially time-consuming functional follow-up.
Computational approaches can be effective at assessing the degree to which a specific amino acid substitution in a protein, induced by a variant, might disrupt function. The functional impact of a substitution can often be estimated by using information about sequence conservation at the mutated site from comparative sequence analysis of a gene with orthologs and paralogs. If an amino acid position in a protein sequence is functionally critical, then most de novo mutations are deleterious and are subject to purifying selection; these sites then are expected to show little variation. Thus, non-synonymous sites in highly conserved regions are likely to be deleterious. Sequence conservation in organisms more closely related to human is particularly informative since more distantly related organisms may have divergent biology and protein function. A variety of software tools using this these principles to assess coding variants have now been devised (Cooper and Shendure, 2011). One example of such a program is Polymorphism Phenotyping 2 (or PolyPhen 2) (Adzhubei et al., 2010). The most predictive features in this method are the estimated likelihood that the mutant allele fits the substitution pattern observed in the multiple-sequence alignment; the evolutionary distance to the organism with a protein harboring a similar nonsynonymous substitution; and whether the mutant allele occurs at a site that is hypermutable. The method uses these features and others, including information from the three-dimensional protein structure, to define a statistical model that defines the probability of disease based on a catalog of known pathogenic Mendielian mutations. The functional importance of an amino acid replacement is predicted from these features based on a naive Bayes classifier. Polyphen 2 and other related methods demonstrate similar performance in their ability to predict pathogenic mutations achiving an area under the curve (AUC) of 75–80% (Hicks et al., 2011).
Experimental approaches to individually interrogate rare variants with functional assays to can also be very powerful. But, for an approach to be effective, it is critical that the functional assay is high throughput, and that it assayed function that is relevant to the phenotype. Otherwise, mutations that affect the assayed gene function might not in fact be pathogenic. In one application of this approach, Davis et al used this approach to look at individual mutations with the TTC21B gene, and to show that they cause human ciliopathies (Davis et al., 2011). First they demonstrated that a translation-blocking morpholino specific for TTC21B resulted in gastrulation defects in zebrefish that were consistent with cilliary dysfunction. Then, when they re-sequenced TTC21B in a large, clinically diverse ciliopathy cohort and matched controls they observed a similar frequency of rare variants. But, when they tested those rare alleles that caused gastrulation defects in zebrefish, they observed a significant enrichment of functional alleles in cases compared to controls.
Evaluating non-coding variants
Non-coding variants pose a particular challenge to the field at the moment. The non-coding genome represents 99% of the genome and at present is poorly annotated (Alexander et al., 2010). About 10% of the non-coding genome is under-purifying selection, suggesting that they harbor critical processes that if disrupted could be pathogenic (Davydov et al., 2010). Many common variants, if they contribute to disease likely act by impacting the non-coding genome. As one example, an associated Crohn’s disease SNP in LD with polymorphic deletion overlapping the IRGM gene promotor and modulates gene expression (McCarroll et al., 2008). In the last several years, however, several promising approaches have emerged to evaluate non-coding variants that might point the way to causality, such as analyzing sequence conservation, gene expression and chromatin state.
Sequence Conservation
A computational approach to prioritizing non-coding variants is to identify those at sites with a high degree of sequence conservation across mammalian organisms, and are thus under purifying negative selection (Cooper et al., 2005; Miller et al., 2007). These approaches differ from those approaches used to prioritize coding substitutions, since they can only use nucleotide sequence similarity. Indeed, investigators have argued that the conservation information from nucleotide sequences is as predictive as the information gained by peptide sequence similarity and protein structural features (Cooper et al., 2010). The value of assessing common variants with sequence conservation approaches is uncertain, since common variants are presumably not under purifying negative selection. But, rare non-coding rare variants that have dramatic effects on disease susceptibility might be effectively prioritized with this approach.
eQTL data can suggest causal genes and mechanism
Expression quantitative loci (eQTL) are genetic variants that correlate with the transcript level of a gene (Jansen and Nap, 2001). To date, most reported eQTLs are cis-effects, acting on nearby genes by encoding variants that modulate promotor activity, enhancer activity, or mRNA stability. Expression QTLs acting in trans have been largely unexplored thus far. While, most recently discovered eQTLs have been common variants, there is evidence of rare eQTLs also (Montgomery et al., 2011). Identifying rare eQTLs might be challenging given the limited power of currently sized cohorts. In the future, burden tests previously described might be able to effectively identify small genomic regions where rare variants dramatically impact transcript levels.
It has been shown that common trait-associated variants have a significant overlap with eQTLs, suggesting the possibility that many common disease variants act by altering transcript levels (Nicolae et al., 2010). Thus, it might be insightful to assess whether a specific disease associated common variant is itself an eQTL. If it is then the gene whose transcript is influenced by the risk allele might be the causal gene. Furthermore, if the risk allele is increasing the transcript level, then the gene may increase disease risk by magnifying gene function; alternatively if the risk allele reduces transcript level, then the gene may cause disease by mitigating gene function. A convincing eQTL effect can be fine-mapped by transfecting constructs with risk haplotype fragments, as was done to to identify the causal variant in the SORT1 lipid locus (Musunuru et al., 2010). Another compelling example of a eQTL that influences disease susceptibility is a type II diabetes associated variant upstream of the KLF14 transcription factor. Investigators showed that this variant not only acts as a cis-eQTL influencing KLF14 levels in adipose tissue, but also as a trans-eQTL for many genes regulated by KLF14 that are important in metabolic traits (Small et al., 2011).
There are a few important caveats about this seemingly straightforward approach. First, eQTL since eQTLs are spread throughout the genome, spurious overlap between disease associated variants and eQTLs is possible (Nica et al., 2010). If a risk variant confers risk by modulating transcript levels, and it is itself causal (or in LD with the causal variant), then it should also be consistent with the strongest eQTL effect in the region. Checking to ensure that the disease-associated variant is consistent with the strongest eQTL effect itself mitigates the risk of spurious overlap. However, it is still possible that the causal allele and the strongest eQTL effect are strongly correlated by chance, and that eQTL association is unrelated to disease risk.
Second, while many eQTLs act generically, most are tissue specific (Dimas et al., 2009; Price et al., 2011). In fact, certain eQTLs may not be detectible unless the cell has responded to a specific stimulus or stress. In order to understand the transcriptional impact of disease alleles most effectively, identifying eQTLs in the pathogenic tissues is key. Current eQTL databases are based on a small number of resting cell-types, for example lymphoblastoid cell-lines (Stranger et al., 2007). Many important pathogenic tissues are not easily accessible for eQTL studies. In the near future the catalog of available tissues profiled will expand dramatically with the NIH sponsored Genotype Tissue Expression (GTEx) project, aiming to profile >60 separate tissues (https://commonfund.nih.gov/GTEx/).
Finally, while eQTL data can offer potential in identifying the likely causal gene and provide hints about mechanism for common variants, it may not clarify ambiguity about the causal variant if there are multiple variants in LD. Certain variants may seem more promising, for example structural variant or SNP overlapping a regulatory variant. As with disease associated common variants, eQTL datasets often face challenges in fine-mapping signals.
Chromatin modifications
Identifying regions of the genome that act as regulatory elements can offer important complementary information to eQTL data in evaluating non-coding variants. Specific functional regulatory elements can be identified from genome-wide profiles of key histone modifications: H3K4me3 marks active promoters; H3K4me1 marks enhancers; H3K4me2 and most histone acetylation marks are enriched at both promoters and enhancers (Barski et al., 2007; Heintzman et al., 2007; Wang et al., 2008). Similarly, DNase I hypersensitive sites also flag open chromatin regions harboring promoters and enhancers (Sabo et al., 2006). With the advancement of high throughput sequencing technologies and development of techniques such as ChIP-seq (Park, 2009) and DNase-seq (John et al., 2011), there are mounting public data on genome-wide chromatin profiles. For instance, histone mark ChIP-seq and DNase-seq data on over 100 cell lines and tissues has now been generated through the ENCODE and Roadmap Epigenomics projects (Bernstein et al., 2010; Birney et al., 2007).
While computational approaches to identify putative binding sites based on sequence data alone is non-specific, recent reports suggest that the prediction of active regulatory sites within assayed tissues is possible by including ChIP-seq and DNase-seq data (Ernst and Kellis, 2010; He et al., 2010; Pique-Regi et al., 2011; Song et al., 2011). One potential approach then to prioritize non-coding variants for followup is to identify those that are in regions that have been predicted to be regulatory elements. These variants might, for example, disrupt or enhance a transcription factor binding at an enhancer or a promoter. Particularly promising variants might be those that have eQTL activity in the same cell-type. Histone marks locations and DNase hypersensitive sites have been shown to be enriched near associated variants (Ernst et al., 2011; McDaniell et al., 2010). A key limitation of this approach is, that like eQTL data, it requires genome-wide chromatin data from the same or similar cell types as those that are pathogenic.
Identifying causal processes with integrative analyses
In many instances where the specific gene of a locus cannot be specifically identified, examination of the genes implicated may still help to suggest the key underlying functional networks and pathways that might be active in a disease. For instance age-related macular degeneration associations have implicated the complement pathway without necessarily identifying causal variants. This task can be challenging in general since for any given associated allele 20 or more genes might be implicated by LD, and any of them may harbor the causal mutation
But despite that, statistically significant connectivity between genes in different associated loci can often be identified. We and others have devised strategies to look for functional connections or similarity between genes across implicated loci. These networks can predict novel gene loci and offer insight about disease mechanism. Gene Relationships Across Implicated Loci (GRAIL) uses >400,000 published scientific PubMed text to assess pairwise gene similarity between genes across loci (Raychaudhuri et al., 2009a). In addition to repeatedly showing highly statistically significant connectivity between genes across loci in multiple diseases, GRAIL has been used to prospectively predict and prioritize associated variants (Raychaudhuri et al., 2009b) and prioritize disease genes within a locus (Beroukhim et al., 2010). Investigators used a similar approach, Disease Association Protein-Protein Link Evaluator (DAPPLE) algorithm to demonstrate that protein-protein interactions are enriched among genes within disease loci more than by chance alone (Rossin et al., 2011). They demonstrated enrichment most convincingly in autoimmune diseases, and furthermore demonstrated that the enrichment of interactions was often between genes within the same immune cell types. These networks offer insight as to how protein products of genes across many loci might be interacting together to initiate disease. We note importantly that pathway analyses can be easily confounded, in particular in neuropsychiatric diseases since there is a correlation between the size of transcripts and the likelihood that they will have brain function (Raychaudhuri et al., 2010).
Conclusions
The advances in genotyping and sequencing technologies over the last few years have revolutionized the genetics of complex diseases. Only 5 years ago, researchers were still tackling the challenges of gene mapping and discovery. Now we face an embarrassment of riches in which the ability to map loci has become quick and reproducible. The next important challenge is streamlining functional validation, which in most cases, is still a critical bottleneck. Rare variant discovery has the potential to yield more obviously functional variants with larger effect sizes because they are less constrained by purifying selection. The discovery of rare variant associations might shed light on those loci discovered by common variant mapping. However, strategies to prioritize functional follow up studies will be key at those loci where common variants cannot be effectively fine mapped or individual rare variants (beyond the presence of case enrichment) cannot be identified. Strategies to use regulatory variants, chromatin state data, and sequence conservation offer a potential path forward to prioritize candidate variants
Supplementary Material
ACKNOWLEDGEMENTS
The author would like to acknowledge help discussions and feedback from colleagues including Drs. Mark Daly, Paul I.W. de Bakker, X Shirley Liu, Cynthia Sandor, Eli A. Stahl, Barbara E. Stranger, and Shamil Sunyaev.
Glossary
- Associated Allele
An allele that, in a genetic study, is observed to have differential allele frequencies in cases compared to controls. The presence of an association suggests that it, or some other variant in LD, is influencing disease susceptibility.
- Causal Allele
The functional allele that influences disease susceptibility and explains the observed associated allele.
- Common Alleles
Alleles with a high population frequency, typically defined as >1%. Standard marker panels can often be used to identify common allele associations.
- Rare Alleles
Alleles with a lower allele frequency of <1%. These alleles can be polymorphic in the population being seen in multiple distantly related individuals; alternately they might be alleles that are private to an individual or seen in a small number of closely related individuals.
- De novo Mutations
A mutation that has occurred in an individual and that was not inherited from a parent. These mutations are private. If a de novo mutation is passed on and persists through generations, it can become a polymorphic allele.
- Linkage disequilibrium (LD)
Two polymorphic loci are in LD when they are collocated on the genome, and alleles at those loci are distributed nonrandomly with respect to each other on chromosomes in the population. Linkage disequilibrium is present when recombination events occur between two loci occur infrequently. Two metrics for LD are r2 and D’.
- Recombination hotspots
Individual regions within the genome that have frequent recombination events.
- Negative Selection
Selection acting to remove new deleterious mutations that reduces evolutionary fitness of an individual. Also known as purifying selection.
- Positive Selection
Selection acting to propagate new advantageous mutations that increase evolutionary fitness of an individual.
- Balancing Selection
Selection acting to increase allelic variability at a locus.
- Genotype Imputation
A statistical technique to infer missing genotypes in a set of individuals using a reference panel of genotyped individuals. Invariably, imputation exploits linkage disequilibrium between genotyped and ungenotyped variants.
- Genome-wide significance
A level of statistical significance typically used to establish association for a common variant in a genome-wide association studies (p=5×10−8), which assumes that there are ~1,000,000 million effective number of independent tests genome-wide.
- Stratification
A genetic confounder if there are differences in the ancestral origin of cases and controls. The resulting systematic allele frequency differences can result in false positive associations.
- Genomic Inflation Factor (λ)
The ratio of the median of the observed chi-square statistics for an association study and the expected median chi-square statistic. If there is stratification the test statistic is inflated, causing the genomic inflation factor to be substantially greater than 1, causing inappropriately significant p-values.
- Fine-mapping
The use of dense genotyping data around an associated allele to identify the causal allele(s) to account for the observed statistical signal in the region.
- Second Generation Sequencing
Recent sequencing technologies not using Sanger chemistry, that characteristically generates many short read sequences.
- Targeted Region
The region of the genome selected for a sequencing experiment.
- Whole-Genome-Sequencing
A sequencing experiment where the full ~3 GBp of whole genome is sequenced. Does not require DNA capture. For most medical genetic studies the sequencing data is not reassembled, but mapped and compared to a reference genome sequence.
- Whole Exome Sequencing
A sequencing experiment where the protein coding sequences of all known genes are targeted, captured, and sequenced (~30Mbp).
- Coverage
In a sequencing experiment, coverage at a genomic position is the total number of reads mapped to that position.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adrianto I, Wen F, Templeton A, Wiley G, King JB, Lessard CJ, Bates JS, Hu Y, Kelly JA, Kaufman KM, et al. Association of a functional variant downstream of TNFAIP3 with systemic lupus erythematosus. Nature genetics. 2011;43:253–258. doi: 10.1038/ng.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander RP, Fang G, Rozowsky J, Snyder M, Gerstein MB. Annotating non-coding regions of the genome. Nature reviews. 2010;11:559–571. doi: 10.1038/nrg2814. [DOI] [PubMed] [Google Scholar]
- Anderson CA, Soranzo N, Zeggini E, Barrett JC. Synthetic associations are unlikely to account for many common disease genome-wide association signals. PLoS Biol. 2011;9:e1000580. doi: 10.1371/journal.pbio.1000580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balding DJ. A tutorial on statistical methods for population association studies. Nature reviews. 2006;7:781–791. doi: 10.1038/nrg1916. [DOI] [PubMed] [Google Scholar]
- Bansal V, Libiger O, Torkamani A, Schork NJ. Statistical analysis strategies for association studies involving rare variants. Nature reviews. 2010;11:773–785. doi: 10.1038/nrg2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhangale TR, Rieder MJ, Nickerson DA. Estimating coverage and power for genetic association studies using near-complete variation data. Nature genetics. 2008;40:841–843. doi: 10.1038/ng.180. [DOI] [PubMed] [Google Scholar]
- Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning SR. Missing data imputation and haplotype phase inference for genome-wide association studies. Hum Genet. 2008;124:439–450. doi: 10.1007/s00439-008-0568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell CD, Ogburn EL, Lunetta KL, Lyon HN, Freedman ML, Groop LC, Altshuler D, Ardlie KG, Hirschhorn JN. Demonstrating stratification in a European American population. Nature genetics. 2005;37:868–872. doi: 10.1038/ng1607. [DOI] [PubMed] [Google Scholar]
- Cardon LR, Abecasis GR. Using haplotype blocks to map human complex trait loci. Trends Genet. 2003;19:135–140. doi: 10.1016/S0168-9525(03)00022-2. [DOI] [PubMed] [Google Scholar]
- Clayton DG, Walker NM, Smyth DJ, Pask R, Cooper JD, Maier LM, Smink LJ, Lam AC, Ovington NR, Stevens HE, et al. Population structure, differential bias and genomic control in a large-scale, case-control association study. Nature genetics. 2005;37:1243–1246. doi: 10.1038/ng1653. [DOI] [PubMed] [Google Scholar]
- Cohen JC, Kiss RS, Pertsemlidis A, Marcel YL, McPherson R, Hobbs HH. Science. Vol. 305. New York, NY: 2004. Multiple rare alleles contribute to low plasma levels of HDL cholesterol; pp. 869–872. [DOI] [PubMed] [Google Scholar]
- Conrad DF, Keebler JE, Depristo MA, Lindsay SJ, Zhang Y, Casals F, Idaghdour Y, Hartl CL, Torroja C, Garimella KV, et al. Variation in genome-wide mutation rates within and between human families. Nature genetics. 2011;43:712–714. doi: 10.1038/ng.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2009;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Goode DL, Ng SB, Sidow A, Bamshad MJ, Shendure J, Nickerson DA. Single-nucleotide evolutionary constraint scores highlight disease-causing mutations. Nat Methods. 2010;7:250–251. doi: 10.1038/nmeth0410-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Shendure J. Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nature reviews. 2011;12:628–640. doi: 10.1038/nrg3046. [DOI] [PubMed] [Google Scholar]
- Cooper GM, Stone EA, Asimenos G, Green ED, Batzoglou S, Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome research. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis EE, Zhang Q, Liu Q, Diplas BH, Davey LM, Hartley J, Stoetzel C, Szymanska K, Ramaswami G, Logan CV, et al. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nature genetics. 2011;43:189–196. doi: 10.1038/ng.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++ PLoS Comput Biol. 2010;6:e1001025. doi: 10.1371/journal.pcbi.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bakker PI, Ferreira MA, Jia X, Neale BM, Raychaudhuri S, Voight BF. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Human molecular genetics. 2008;17:R122–R128. doi: 10.1093/hmg/ddn288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010;8:e1000294. doi: 10.1371/journal.pbio.1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimas AS, Deutsch S, Stranger BE, Montgomery SB, Borel C, Attar-Cohen H, Ingle C, Beazley C, Gutierrez Arcelus M, Sekowska M, et al. Science. Vol. 325. New York, NY: 2009. Common regulatory variation impacts gene expression in a cell type-dependent manner; pp. 1246–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat Biotechnol. 2010;28:817–825. doi: 10.1038/nbt.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, Heinzen EL, Qiu P, Bertelsen AH, Muir AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, et al. Science. Vol. 329. New York, NY: 2010. Association of trypanolytic ApoL1 variants with kidney disease in African Americans; pp. 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunderson KL, Steemers FJ, Lee G, Mendoza LG, Chee MS. A genome-wide scalable SNP genotyping assay using microarray technology. Nature genetics. 2005;37:549–554. doi: 10.1038/ng1547. [DOI] [PubMed] [Google Scholar]
- Gusev A, Kenny EE, Lowe JK, Salit J, Saxena R, Kathiresan S, Altshuler DM, Friedman JM, Breslow JL, Pe'er I. DASH: a method for identical-by-descent haplotype mapping uncovers association with recent variation. American journal of human genetics. 2011;88:706–717. doi: 10.1016/j.ajhg.2011.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harismendy O, Ng PC, Strausberg RL, Wang X, Stockwell TB, Beeson KY, Schork NJ, Murray SS, Topol EJ, Levy S, et al. Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol. 2009;10:R32. doi: 10.1186/gb-2009-10-3-r32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He HH, Meyer CA, Shin H, Bailey ST, Wei G, Wang Q, Zhang Y, Xu K, Ni M, Lupien M, et al. Nucleosome dynamics define transcriptional enhancers. Nature genetics. 2010;42:343–347. doi: 10.1038/ng.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nature genetics. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- Hicks S, Wheeler DA, Plon SE, Kimmel M. Prediction of missense mutation functionality depends on both the algorithm and sequence alignment employed. Hum Mutat. 2011;32:661–668. doi: 10.1002/humu.21490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoggart CJ, Clark TG, De Iorio M, Whittaker JC, Balding DJ. Genome-wide significance for dense SNP and resequencing data. Genetic epidemiology. 2008;32:179–185. doi: 10.1002/gepi.20292. [DOI] [PubMed] [Google Scholar]
- Holm H, Gudbjartsson DF, Sulem P, Masson G, Helgadottir HT, Zanon C, Magnusson OT, Helgason A, Saemundsdottir J, Gylfason A, et al. A rare variant in MYH6 is associated with high risk of sick sinus syndrome. Nature genetics. 2011;43:316–320. doi: 10.1038/ng.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen RC, Nap JP. Genetical genomics: the added value from segregation. Trends Genet. 2001;17:388–391. doi: 10.1016/s0168-9525(01)02310-1. [DOI] [PubMed] [Google Scholar]
- John S, Sabo PJ, Thurman RE, Sung MH, Biddie SC, Johnson TA, Hager GL, Stamatoyannopoulos JA. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nature genetics. 2011;43:264–268. doi: 10.1038/ng.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A, Masson G, Frigge ML, Gylfason A, Zusmanovich P, Thorleifsson G, Olason PI, Ingason A, Steinberg S, Rafnar T, et al. Detection of sharing by descent, long-range phasing and haplotype imputation. Nature genetics. 2008;40:1068–1075. doi: 10.1038/ng.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryukov GV, Pennacchio LA, Sunyaev SR. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. American journal of human genetics. 2007;80:727–739. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. American journal of human genetics. 2008;83:311–321. doi: 10.1016/j.ajhg.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Homer N. A survey of sequence alignment algorithms for next-generation sequencing. Brief Bioinform. 2010;11:473–483. doi: 10.1093/bib/bbq015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen BE, Browning SR. A groupwise association test for rare mutations using a weighted sum statistic. PLoS genetics. 2009;5:e1000384. doi: 10.1371/journal.pgen.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxmen A. Exome sequencing deciphers rare diseases. Cell. 2011;144:635–637. doi: 10.1016/j.cell.2011.02.033. [DOI] [PubMed] [Google Scholar]
- McCarroll SA, Huett A, Kuballa P, Chilewski SD, Landry A, Goyette P, Zody MC, Hall JL, Brant SR, Cho JH, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn's disease. Nature genetics. 2008;40:1107–1112. doi: 10.1038/ng.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JP, Hirschhorn JN. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nature reviews. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- McDaniell R, Lee BK, Song L, Liu Z, Boyle AP, Erdos MR, Scott LJ, Morken MA, Kucera KS, Battenhouse A, et al. Science. Vol. 328. New York, NY: 2010. Heritable individual-specific and allele-specific chromatin signatures in humans; pp. 235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller W, Rosenbloom K, Hardison RC, Hou M, Taylor J, Raney B, Burhans R, King DC, Baertsch R, Blankenberg D, et al. 28-way vertebrate alignment and conservation track in the UCSC Genome Browser. Genome research. 2007;17:1797–1808. doi: 10.1101/gr.6761107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momozawa Y, Mni M, Nakamura K, Coppieters W, Almer S, Amininejad L, Cleynen I, Colombel JF, de Rijk P, Dewit O, et al. Resequencing of positional candidates identifies low frequency IL23R coding variants protecting against inflammatory bowel disease. Nature genetics. 2011;43:43–47. doi: 10.1038/ng.733. [DOI] [PubMed] [Google Scholar]
- Montgomery SB, Lappalainen T, Gutierrez-Arcelus M, Dermitzakis ET. Rare and common regulatory variation in population-scale sequenced human genomes. PLoS genetics. 2011;7:e1002144. doi: 10.1371/journal.pgen.1002144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenthaler S, Thilly WG. A strategy to discover genes that carry multi-allelic or mono-allelic risk for common diseases: a cohort allelic sums test (CAST) Mutat Res. 2007;615:28–56. doi: 10.1016/j.mrfmmm.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Musunuru K, Strong A, Frank-Kamenetsky M, Lee NE, Ahfeldt T, Sachs KV, Li X, Li H, Kuperwasser N, Ruda VM, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466:714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers S, Bottolo L, Freeman C, McVean G, Donnelly P. Science. Vol. 310. New York, NY: 2005. A fine-scale map of recombination rates and hotspots across the human genome; pp. 321–324. [DOI] [PubMed] [Google Scholar]
- Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Science. Vol. 324. New York, NY: 2009. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes; pp. 387–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic acids research. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, et al. Exome sequencing identifies the cause of a mendelian disorder. Nature genetics. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nica AC, Montgomery SB, Dimas AS, Stranger BE, Beazley C, Barroso I, Dermitzakis ET. Candidate causal regulatory effects by integration of expression QTLs with complex trait genetic associations. PLoS genetics. 2010;6:e1000895. doi: 10.1371/journal.pgen.1000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, Cox NJ. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS genetics. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nature reviews. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltonen L, Palotie A, Lange K. Use of population isolates for mapping complex traits. Nature reviews. 2000;1:182–190. doi: 10.1038/35042049. [DOI] [PubMed] [Google Scholar]
- Pereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PI, Walker BD, Ripke S, Brumme CJ, Pulit SL, Carrington M, et al. Science. Vol. 330. New York, NY: 2010. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation; pp. 1551–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pique-Regi R, Degner JF, Pai AA, Gaffney DJ, Gilad Y, Pritchard JK. Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome research. 2011;21:447–455. doi: 10.1101/gr.112623.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Helgason A, Thorleifsson G, McCarroll SA, Kong A, Stefansson K. Single-tissue and cross-tissue heritability of gene expression via identity-by-descent in related or unrelated individuals. PLoS genetics. 2011;7:e1001317. doi: 10.1371/journal.pgen.1001317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK. Are rare variants responsible for susceptibility to complex diseases? American journal of human genetics. 2001;69:124–137. doi: 10.1086/321272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Cox NJ. The allelic architecture of human disease genes: common disease-common variant…or not? Human molecular genetics. 2002;11:2417–2423. doi: 10.1093/hmg/11.20.2417. [DOI] [PubMed] [Google Scholar]
- Purcell SM, Wray NR, Stone JL, Visscher PM, O'Donovan MC, Sullivan PF, Sklar P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychaudhuri S, Iartchouk O, Chin K, Tan PL, Tai A, Ripke S, Gowrisankar S, Vemuri S, Montgomery K, Yu Y, et al. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nature genetics. doi: 10.1038/ng.976. (In Press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychaudhuri S, Korn JM, McCarroll S, International Schizophrenia Consortium. Altshuler D, Sklar P, Purcell S, Daly MJ. Accurately assessing the risk of schizophrenia conferred by rare copy-number variation affecting genes with brain function. PLoS genetics. 2010;6:e1001097. doi: 10.1371/journal.pgen.1001097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychaudhuri S, Plenge RM, Rossin EJ, Ng ACY, International Schizophrenia Consortium. Purcell SM, Sklar P, Scolnick EM, Xavier RJ, Altshuler D, et al. Identifying Relationships Among Genomic Disease Regions: Predicting Genes at Pathogenic SNP Associations and Rare Deletions. PLoS genetics. 2009a;5:e1000534. doi: 10.1371/journal.pgen.1000534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychaudhuri S, Thomson BP, Remmers EF, Eyre S, Hinks A, Guiducci C, Catanese JJ, Xie G, Stahl EA, Chen R, et al. Genetic variants at CD28, PRDM1 and CD2/CD58 are associated with rheumatoid arthritis risk. Nature genetics. 2009b;41:1313–1318. doi: 10.1038/ng.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich DE, Lander ES. On the allelic spectrum of human disease. Trends Genet. 2001;17:502–510. doi: 10.1016/s0168-9525(01)02410-6. [DOI] [PubMed] [Google Scholar]
- Risch N, Merikangas K. Science. Vol. 273. New York, NY: 1996. The future of genetic studies of complex human diseases; pp. 1516–1517. [DOI] [PubMed] [Google Scholar]
- Rosenberg NA, Huang L, Jewett EM, Szpiech ZA, Jankovic I, Boehnke M. Genome-wide association studies in diverse populations. Nature reviews. 2010;11:356–366. doi: 10.1038/nrg2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossin EJ, Lage K, Raychaudhuri S, Xavier RJ, Tartar D, Benita Y, Consortium IIG, Cotsapas C, Daly MJ. Proteins encoded in genomic regions associated to immune-mediated disease physically interact and suggest underlying biology. PLoS genetics. 2011;7:e1001273. doi: 10.1371/journal.pgen.1001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo PJ, Kuehn MS, Thurman R, Johnson BE, Johnson EM, Cao H, Yu M, Rosenzweig E, Goldy J, Haydock A, et al. Genome-scale mapping of DNase I sensitivity in vivo using tiling DNA microarrays. Nat Methods. 2006;3:511–518. doi: 10.1038/nmeth890. [DOI] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, et al. Science. Vol. 316. New York, NY: 2007. Strong association of de novo copy number mutations with autism; pp. 445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- Small KS, Hedman AK, Grundberg E, Nica AC, Thorleifsson G, Kong A, Thorsteindottir U, Shin SY, Richards HB, Soranzo N, et al. Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nature genetics. 2011;43:561–564. doi: 10.1038/ng.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Zhang Z, Grasfeder LL, Boyle AP, Giresi PG, Lee BK, Sheffield NC, Graf S, Huss M, Keefe D, et al. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome research. 2011 doi: 10.1101/gr.121541.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Nica AC, Forrest MS, Dimas A, Bird CP, Beazley C, Ingle CE, Dunning M, Flicek P, Koller D, et al. Population genomics of human gene expression. Nature genetics. 2007;39:1217–1224. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorgeirsson TE, Geller F, Sulem P, Rafnar T, Wiste A, Magnusson KP, Manolescu A, Thorleifsson G, Stefansson H, Ingason A, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452:638–642. doi: 10.1038/nature06846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, Zeggini E, Huth C, Aulchenko YS, Thorleifsson G, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nature genetics. 2010;42:579–589. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM, Nord AS, Kusenda M, Malhotra D, Bhandari A, et al. Science. Vol. 320. New York, NY: 2008. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia; pp. 539–543. [DOI] [PubMed] [Google Scholar]
- Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nature genetics. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, Madden PA, Heath AC, Martin NG, Montgomery GW, et al. Common SNPs explain a large proportion of the heritability for human height. Nature genetics. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.