

Abstract
OBJECTIVE
To assess the potential role of FoxP3-expressing regulatory T cells (Tregs) in reversing obesity-linked insulin resistance and diabetic nephropathy in rodent models and humans.
RESEARCH DESIGN AND METHODS
To characterize the role of Tregs in insulin resistance, human visceral adipose tissue was first evaluated for Treg infiltration and second, the db/db mouse model was evaluated.
RESULTS
Obese patients with insulin resistance displayed significantly decreased natural Tregs but an increase in adaptive Tregs in their visceral adipose tissue as compared with lean control subjects. To further evaluate the pathogenic role of Tregs in insulin resistance, the db/db mouse model was used. Treg depletion using an anti-CD25 monoclonal antibody enhanced insulin resistance as shown by increased fasting blood glucose levels as well as an impaired insulin sensitivity. Moreover, Treg-depleted db/db mice developed increased signs of diabetic nephropathy, such as albuminuria and glomerular hyperfiltration. This was paralleled by a proinflammatory milieu in both murine visceral adipose tissue and the kidney. Conversely, adoptive transfer of CD4+FoxP3+ Tregs significantly improved insulin sensitivity and diabetic nephropathy. Accordingly, there was increased mRNA expression of FoxP3 as well as less abundant proinflammatory CD8+CD69+ T cells in visceral adipose tissue and kidneys of Treg-treated animals.
CONCLUSIONS
Data suggest a potential therapeutic value of Tregs to improve insulin resistance and end organ damage in type 2 diabetes by limiting the proinflammatory milieu.
CD4+CD25+FoxP3+ natural regulatory T cells (Tregs) have attracted attention as a potent immunosuppressive population in inflammatory disorders. According to the current paradigm, they counteract proinflammatory cell populations, among which TH1 and TH17 cells are most important (1). Treg transfer has proven to be beneficial in various animal models of inflammation and autoimmunity (2–4). Moreover, patients suffering from autoimmune diseases such as rheumatoid arthritis, multiple sclerosis, or Goodpasture disease display a numerical and/or functional deficit in the Treg compartment (5–8). It is generally accepted that Tregs inhibit the respective target cells in a direct cell-to-cell contact manner and that their immunomodulatory effects are primarily conveyed by membrane-bound transforming growth factor-β (9). Moreover, soluble factors such as interleukin (IL)-10 have also been implicated in Treg-induced immunomodulation (10,11). Emerging data provide evidence for a functional heterogeneity and lineage plasticity within the Treg compartment since Tregs derive on one hand in the thymus and on the other hand develop in the periphery upon inflammatory stimuli. Depending on their origin, they are classified as natural Tregs and adaptive Tregs, respectively (12). Helios, an Ikaros-family transcription factor, has recently shown to be selectively expressed by natural Tregs (13). Patients with type 1 diabetes have been shown to have increasing numbers of adaptive but diminished numbers of natural Tregs in their peripheral blood as compared with healthy control subjects (14).
There is overwhelming evidence from human and preclinical studies that insulin sensitivity deteriorates as a result of subclinical inflammation. Recently, T cells have been found to play a key role in the pathogenesis of insulin resistance, since blocking of T cells by a CD3-depleting antibody protected mice from the development of insulin resistance (15). T-cell depletion tipped the balance from a pro- toward an anti-inflammatory milieu by limiting the TH1 response and favoring a dominance of Tregs (15,16). The anti-inflammatory effects were mirrored by a decreased macrophage infiltration and tumor necrosis factor (TNF)-α expression in murine visceral adipose tissue (mVAT). The importance of Tregs in the pathogenesis of insulin resistance is further supported by data from Feuerer et al. (17) who found significantly decreased Treg numbers in mVAT of obese mice as compared with lean control animals. In humans, conflicting data exist on the abundance of Tregs in human visceral adipose tissue (hVAT) of obese patients with or without insulin resistance when compared with lean control subjects (17–19).
Type 2 diabetes, including its end organ damages, such as diabetic nephropathy, is a major health burden that requires the development of novel and innovative therapeutic strategies. Db/db mice, which lack signaling of the leptin receptor, are an excellent model of type 2 diabetes because these animals develop hyperphagia, obesity, and overt hyperglycemia (20). When uninephrectomized at the age of 5 weeks, they develop an early phase of diabetic nephropathy (21).
Using this model, we provide first evidence that Tregs are critically involved in the pathogenesis of type 2 diabetes and of diabetic nephropathy. Our study might set the stage for future testing of strategies to increase Treg numbers in vivo (e.g., by adoptive Treg transfer), which limits inflammation in visceral fat tissue and thereby restores insulin sensitivity and prevents the development of end organ damage.
RESEARCH DESIGN AND METHODS
Human samples.
The study included 79 obese patients undergoing weight-reducing surgery and 14 lean patients who underwent elective cholecystectomy. Lean patients with elevated C-reactive protein were excluded from the study. Participants were included if they had fasting blood glucose levels <7.0 mmol/L, no history of diabetes or use of glucose- or lipid-lowering drugs, and no weight changes >3% during the previous 2 months. All study participants provided informed consent, and study protocols were approved by the local ethics committee. BMI, fasting blood glucose, and insulin were determined as described. Adipose tissue biopsies were collected in RNAlater (Ambion, Austin, TX) and stored at −80°C until RNA isolation (22).
Animal studies.
Five-week-old male db/db leptin receptor–deficient mice (BKS.Cg-Dock7m +/+ Lepr db/J) were obtained from Charles River (Sulzfeld, Germany) and maintained in a virus/antibody-free central animal facility of the Innsbruck Medical University. At 6 weeks of age, the animals underwent surgical uninephrectomy. Body weight, fasting glucose levels, and urinary albumin excretion were measured regularly once a week, and 24-h urine samples were collected in metabolic cages. Mice were treated with either 200 μg of an anti-CD25 monoclonal antibody (mAb; clone PC61; BD Biosciences, San Diego, CA) or an isotype control antibody administered intraperitoneally the day after uninephrectomy and 28 days thereafter. In a second set of experiments, db/db mice were treated intravenously with either 0.5–2 × 106 CD4+FoxP3+GFP+ or CD4+GFP− cells on days 4, 18, 32, and 46 after uninephrectomy. Adoptively transferred cells were isolated from pooled spleen and lymph node suspensions (minced and passed through a 70-μm mesh) of B6.Cg-Foxp3tm2Tch/J reporter mice (Jackson Laboratories, Bar Harbor, ME). After staining for CD4 (BD Biosciences), CD4+FoxP3+GFP+ Tregs were purified by fluorescence-activated cell sorter (FACS) using a BD FACSAria cell sorter (BD Biosciences). A purity of 99.14 ± 0.43% was achieved.
All animal experiments were approved by Austrian veterinary authorities and corresponded to the European Commission Directive 86/609/EEC.
Flow cytometry.
Single cell suspensions from liver, spleen, mVAT, and murine subcutaneous adipose tissue (mSAT) were costained for surface CD4, CD8, and CD69 (all BD Biosciences) and intracellular FoxP3+ (eBiosciences, San Diego, CA) strictly adhering to the manufacturer’s instructions. Data collection and analysis were done on a FACSCalibur (BD Biosciences).
Metabolic studies.
Blood glucose levels were measured using OneTouch Ultra2 (LifeScan, Vienna, Austria), urine creatinine was quantified spectrophotometrically using a Picric acid–based method (Sigma, St. Louis, MO), and urinary albumin was determined by a double-sandwich ELISA (Abcam, Cambridge, MA) as reported previously (23). The insulin tolerance test was performed on day 56 after uninephrectomy. In brief, we injected 0.75 IU/kg body wt (in the Treg-depletion experiments) and 1.5 IU/kg body wt (in the Treg-treatment experiments) of human insulin aspartat (Novo Nordisk, Sorgenfri, Denmark) and measured the blood glucose level prior to and 15, 30, 45, 60, 75, and 90 min after insulin application. On the same day, we also measured fasting insulin levels (Crystal Chem, Downers Grove, IL) by ELISA. The homeostasis model assessment of insulin resistance (HOMA-IR) was calculated by using the glucose-insulin product divided by the constant 22.5. Furthermore, we measured the fat cell diameter in mSAT and mVAT (epididymal) and the glomerular diameter at a magnification of 100 using a ProgRes C12 camera and the ProgRes Capture Pro 2.5 software with the straight line tool (Jenoptik GmbH, Jena, Germany). At least 200 adipocytes from both mSAT and mVAT or 50 glomeruli were evaluated for each mouse in a blinded fashion.
Renal stainings.
Formalin-fixed renal tissue was embedded in paraffin, cut in 4-μm sections, and stained with hematoxylin-eosin and Periodic acid-Schiff using standard staining techniques. The three-layer immunoperoxidase technique was used for the detection of macrophages and T cells in frozen kidney sections. Macrophages were stained using rat anti-mouse F4/80 and CD68 antibodies (Serotec, Oxford, U.K.). A semiquantitative scoring system was performed as follows: 0, 0 to 4 cells; 1+, 5 to 10 cells; 2+, 10 to 50 cells; 3+, 50 to 200 cells; and 4+, >200 cells stained positive per low-power field. For the detection of CD4+ and CD8+ T cells, rat anti-mouse CD4 and CD8-α mAb (Serotec) were used. Biotin-conjugated goat anti-rat IgG antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) was used as a secondary antibody, followed by incubation with a streptavidin-biotin complex and subsequent development with 0.4% 3-amino-9-ethylcarbazole for 6 min and counterstaining with Gill's Hematoxylin No. 3 (Polysciences, Warrington, PA). Quantification of T cells was done by counting the number of cells in six adjacent high-power fields of renal cortex and medulla. Scoring of cells was performed by a blinded observer.
Real-time RT-PCR.
Human and murine tissue was stored at −80°C in RNAlater. Total RNA was isolated from the respective tissue using Trizol (Sigma) according to a standard protocol. The integrity of RNA was detected by RNA-gel electrophoresis in stratified random RNA samples. Thereafter, 2 μg of total RNA was reverse transcribed using Superscript III Transcription Kit (Nitrogen, Carlsbad, CA) and random primers (Roche, Basel, Switzerland). Real-time PCR was performed on a StepOnePlus real-time instrument (Applied Biosystems, Foster City, CA) or a CFX96 Real-Time System (Biorad, Hercules, CA). For amplification of murine FoxP3 and TNF-α, SYBR Green Master Mix (Nitrogen) and the following primers were used: Foxp3 forward 5′-TCT TGC CAA GCT GGA AGA CT-3′, reverse 5′-AGC TGA TGC ATG AAG TGT GG-3′; and TNF-α forward 5′-GAA CTG GCA GAA GAG GCA CT-3′, reverse 5′-AGG GTC TGG GCC ATA GAA CT-3′. The chosen primer pairs span the exon-intron border. The Foxp3 primer pair detects all murine Foxp3 splicing variants. For quantification of human FOXP3, Helios and TNF-α Taqman Master mix (Applied Biosystems) and the gene expression assays Hs01085834_m1, Hs00212361_m1, and Hs00174128_m1 (Applied Biosystems) were used. The human FOXP3 gene expression assay detects all human FOXP3 splicing variants. For quantification of murine IL-6, interferon-γ (IFN-γ), Gata-3, IL-10, the gene expression assays Mm00446190_m1, Mm00901778_m1, Mm00484683_m1, and Mm00439616_m1 (Applied Biosystems) were used. In murine samples, 18S mRNA served as reference. For the human studies, we evaluated the mean expression of three different housekeeping genes, namely, 18S, β-actin, and HPRT-1, which was then set as reference. We tested for intra- and interassay variance in our real-time PCR experiments. The coefficient of variation for the intra-assay variance was <0.0005 and for the interassay variance, <0.001.
Detection of GFP+ cells by PCR.
Enhanced green fluorescent protein–positive cells were detected using the following primers: forward 5′-AAG TTC ATC TGC ACC ACC G-3′, reverse 5′-TCC TTG AAG AAG ATG GTG CG-3′. Nested PCR was performed with the following primer pair: 5′-GTG TCC GGC GAG GGC GAG G-3′, reverse 5′-CGT GCT GCT TCA TGT GGT CG-3′. cDNA isolated from spleens of B6.Cg-Foxp3tm2Tch/J served as positive control. RT-PCR was carried out using HotStarTaq Polymerase (Qiagen, Hilden, Germany).
Statistical analysis.
Human data were analyzed for normal distribution using Kolmogorov-Smirnov test with Lilliefors correction. Since the data were not normally distributed, the bivariate associations between Helios/FOXP3 mRNA levels, BMI, fasting glucose levels, and HOMA-IR were calculated using the Spearman correlation coefficient. In a case of comparing three groups, the significance level was lowered to P < 0.025.
Results from animal experiments are presented as mean ± SEM. Normal distribution of data were assessed by the Kolmogorov-Smirnov test with Lilliefors correction. Both groups were compared by either nonparametric Mann-Whitney U test or unpaired t test as appropriate, depending on the distribution of the tested variable. A two-tailed P < 0.05 was considered statistically significant. For the insulin-sensitivity assay in Treg-depleted mice versus controls, the Fisher exact test was performed. All statistical analyses were done with SPSS 15.0 for Windows (SPSS, Chicago, IL).
RESULTS
Obese patients with insulin resistance display decreased Helios but increased FOXP3 mRNA expression in their visceral adipose tissue.
To test whether Tregs accumulate in visceral obesity, we analyzed hVAT from 14 lean control subjects, 39 obese patients without insulin resistance (HOMA-IR <2.5), and 39 obese patients with insulin resistance (HOMA-IR >5). We evaluated the mRNA expression of the thymic Treg marker Helios as well as of the master regulator of Tregs FOXP3 in hVAT. We found the mRNA expression of Helios to be downregulated in both obese patient groups, whereas FOXP3 was significantly downregulated only in obese patients without insulin resistance. In obese patients with insulin resistance, mRNA expression of FOXP3 was similar to lean control subjects (Fig. 1A). The Helios-to-FOXP3 ratio correlated inversely with BMI (Fig. 1B), HOMA-IR (Fig. 1C), and fasting glucose levels (Fig. 1D). Significance was reached in the correlation with fasting blood glucose level and HOMA-IR.
FIG. 1.
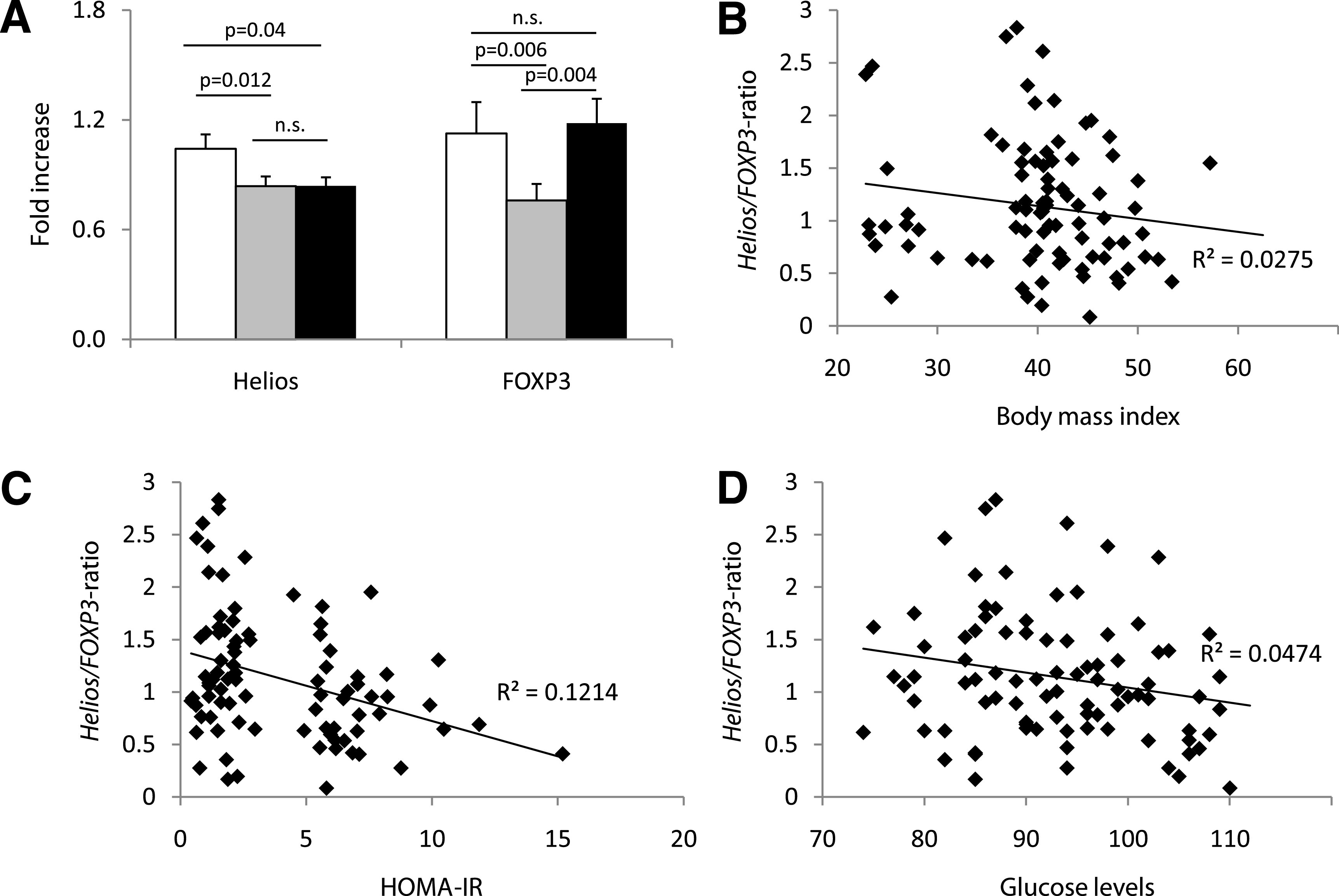
Obese patients with insulin resistance display significantly decreased Helios but increased FOXP3 mRNA expression in their hVAT. A: hVAT of lean control subjects (white bar, n = 14), obese patients with normal insulin sensitivity (HOMA <2.5, gray bar, n = 39), and obese patients with impaired insulin sensitivity (HOMA >5, black bar, n = 39) was analyzed for Helios and FOXP3 mRNA expression. P < 0.025 was considered significant; n.s., not significant. Correlation of the Helios-to-FOXP3 mRNA ratio in hVAT (n = 92) with BMI (P = 0.121) (B), HOMA-IR (P = 0.003) (C), and fasting blood glucose levels (P = 0.045) (D) was analyzed using Spearman ρ. P < 0.05 was considered significant.
Treg depletion aggravates insulin resistance.
Since Treg lineage plasticity is influenced by insulin resistance, we next studied their pathogenic role in a murine model of type 2 diabetes. We therefore used a well-established model of murine type 2 diabetes (i.e., the leptin-deficient db/db mice). Tregs were eliminated in these mice using the Treg-depleting anti-CD25 antibody, which was injected intraperitoneally on days 1 and 28 after uninephrectomy. We chose this application schedule since we could not detect any CD4+CD25+ T cells in the inguinal lymph nodes 3 weeks after application of a single dose of the antibody (data not shown). Treg-depleted mice and the respective controls had a comparable body weight throughout the observation period of 56 days (Fig. 2A); however, fasting blood glucose levels were significantly increased as early as 21 days after uninephrectomy in Treg-depleted db/db mice when compared with vehicle-treated db/db mice (Fig. 2B). On day 56, insulin-sensitivity testing revealed that insulin lowered blood glucose levels more efficiently in vehicle-treated db/db mice compared with Treg-depleted db/db mice (Table 1). In addition, Treg-depleted db/db mice displayed a significantly increased HOMA-IR on day 56 as compared with control animals (Fig. 2C). To analyze whether these metabolic differences were also reflected by changes in the inflammatory profile within the mVAT and mSAT compartment, real-time PCR for mRNA expression of IFN-γ, IL-6, TNF-α, Gata-3, and IL-10 was performed. We could detect a prominent increase of the Th1 cytokines IFN-γ, IL-6, and TNF-α in mVAT of Treg-depleted db/db mice as compared with control animals (Fig. 3A). No difference in the mRNA expression of Gata-3 and IL-10 was detected in mVAT (Fig. 3A). Furthermore, we did not detect any difference in the expression of the respective transcripts in mSAT (Fig. 3B). In parallel, flow cytometry revealed increased numbers of activated CD4+CD69+ T cells in the mVAT of Treg-depleted mice (Fig. 3C and D), while no difference in the abundance of this cell population was found in mSAT (Fig. 3C). No difference in the percentage of CD8+ T cells and CD11c+ dendritic cells in mVAT was detected (data not shown).
FIG. 2.
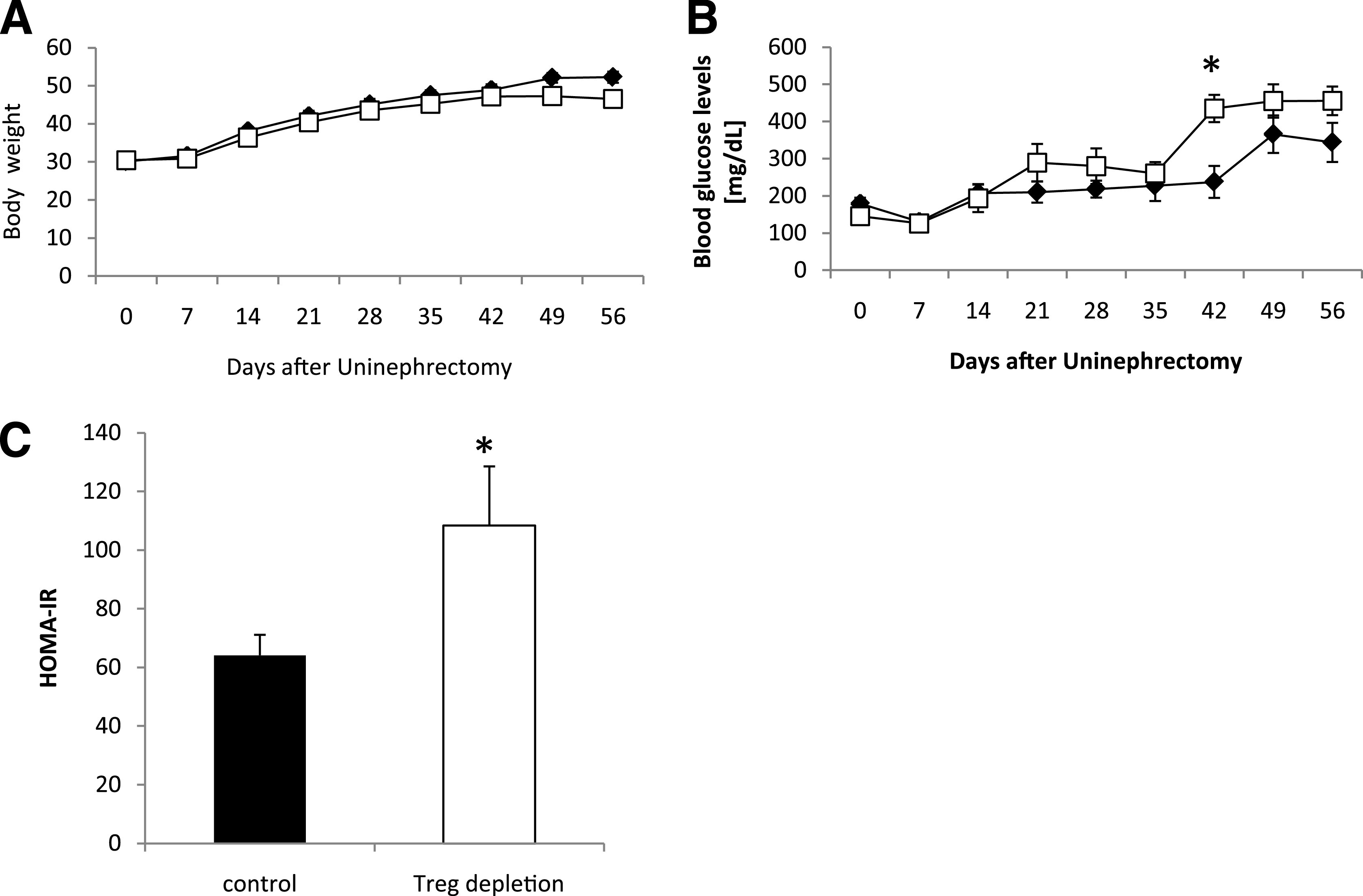
Treg-depletion aggravates insulin resistance. Db/db mice were uninephrectomized at the age of 6 weeks and followed for 56 days. One group intraperitoneally received an anti-CD25 antibody starting the day of and 4 weeks after uninephrectomy (□ and white bar, n = 8). Control animals received an isotype control antibody (♦ and black bar, n = 8). Body weight (A) and blood glucose levels (B) were measured weekly. C: On day 56, the HOMA-IR index was calculated. Pooled data of two independent experiments are shown. *P < 0.05.
TABLE 1.
Treg depletion aggravates insulin resistance
| Minutes |
|||||||
|---|---|---|---|---|---|---|---|
| 0 | 15 | 30 | 45 | 60 | 75 | 90 | |
| Control | 0/8 | 4/8 | 3/8 | 2/8 | 1/8 | 1/8 | 1/8 |
| Treg depleted | 2/8 | 8/8 | 6/8 | 5/8 | 6/8* | 7/8* | 7/8* |
Db/db mice were uninephrectomized at the age of 6 weeks and followed for 56 days. One group received an anti-CD25 antibody intraperitoneally starting the day of and 4 weeks after uninephrectomy (n = 8). Control animals received an isotype control antibody (n = 8). Insulin-sensitivity testing was performed on day 56. The number of mice with blood glucose levels >600 mg/dL at the indicated time points are shown. Pooled data of two independent experiments are shown.
*P < 0.05 as evaluated by Fisher exact test.
FIG. 3.
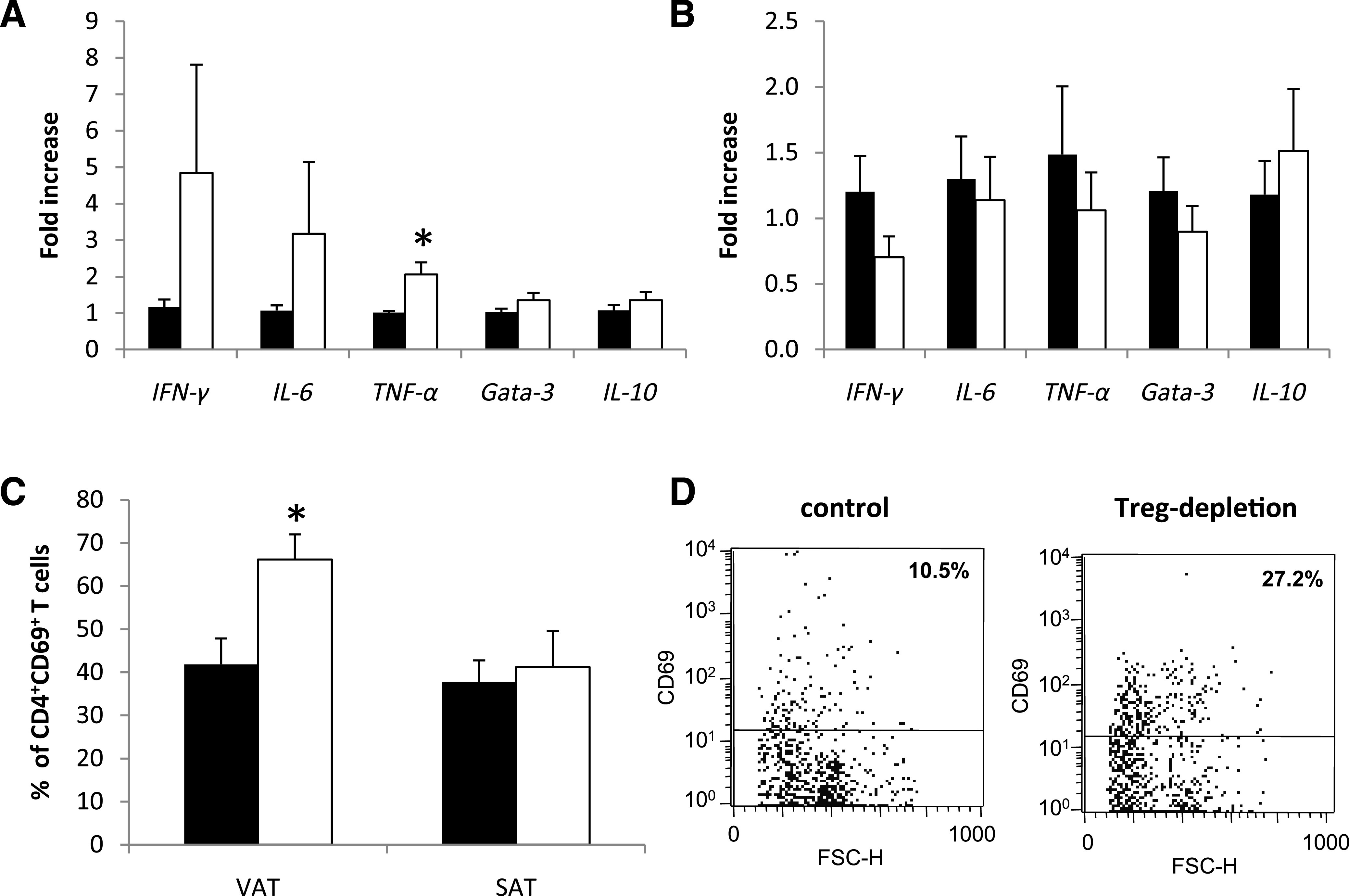
Treg depletion induces increased inflammation in visceral adipose tissue. Db/db mice were uninephrectomized at the age of 6 weeks and followed for 56 days. One group intraperitoneally received an anti-CD25 mAb the day of and 4 weeks after uninephrectomy (white bar, n = 8); the control group received an isotype control antibody (black bar, n = 8). The cytokine response in VAT (A) and SAT (B) was evaluated by real-time PCR. The fold increase to the mean expression of controls is provided. C: The percentage of CD4+CD69+ T cells was evaluated in VAT and SAT by flow cytometry. Pooled data of two independent experiments are shown. *P < 0.05. D: Representative dot plots from gated CD4+ T cells in VAT. FSC-H, forward scatter height.
Of note, Treg depletion by anti-CD25 antibody did not compromise endogenous insulin production since mice that received the CD25-depleting antibody had even slightly higher insulin levels as compared with control mice (control: 3.63 ± 0.62 ng/mL; Treg depleted: 3.93 ± 0.67 ng/mL).
Treg depletion deteriorates diabetic nephropathy.
The urinary albumin-to-creatinine ratio as a marker of renal impairment was increased in Treg-depleted versus nondepleted db/db mice starting on day 28 and reached statistical significance on day 42 after uninephrectomy (Fig. 4A). Accordingly, the glomerular diameter was found to be significantly increased in Treg-depleted mice on day 56 after uninephrectomy (Fig. 4B). Semiquantitative assessment of renal inflammatory cell infiltration revealed a profound increase in CD4+ and CD8+ T cells in the kidneys of Treg-depleted db/db mice (Fig. 4C), whereas F4/80+ cells and CD68-expressing macrophages were not different (Fig. 4D). Real-time analysis revealed a significantly increased mRNA expression of the Th1 cytokine TNF-α in the kidneys of Treg-depleted db/db mice (Fig. 4E). IFN-γ and IL-10 tended to be increased in Treg-depleted mice, whereas IL-6 and Gata-3 were not differentially regulated (Fig. 4E).
FIG. 4.

Treg depletion fosters organ damage in diabetic nephropathy. Db/db mice were uninephrectomized at the age of 6 weeks and followed for 56 days. One group intraperitoneally received an anti-CD25 antibody on the day of and 4 weeks after uninephrectomy (white bar, n = 8). Control animals received an isotype control antibody (black bar, n = 8). A: The albumin-to-creatinine ratio was evaluated in the urine every 14 days. B: After 56 days, kidneys were harvested and the glomerular diameter was measured. Kidney sections were stained for the infiltration of CD4+ and CD8+ T cells (C) as well as F4/80+ cells and CD68+ macrophages (D). Hpf, high-power field. E: The cytokine response in the kidney was evaluated by real-time PCR. The fold increase to the mean expression of control animals is provided. Pooled data of two independent experiments are shown. *P < 0.05.
Adoptive Treg transfer improves insulin sensitivity.
Db/db mice underwent uninephrectomy at the age of 6 weeks. On days 4, 18, 32, and 46 after uninephrectomy, CD4+FoxP3+GFP+ (Tregs) or CD4+FoxP3−GFP− (control) T cells isolated from healthy donor animals were adoptively transferred into db/db mice. No difference in the body weight was observed between the two groups (Fig. 5A). The blood glucose levels were significantly decreased only in the Treg-treated mice on day 28, whereas on the other evaluated time points, no difference between the groups was detectable (Fig. 5B). Moreover, significantly improved insulin sensitivity was observed in db/db mice treated with Tregs compared with db/db mice injected with control T cells (Fig. 5C). A trend toward an improved HOMA-IR in Treg-treated obese animals was detected, but no statistical significance was reached (Fig. 5D). In addition, Treg-treated db/db mice displayed a significantly decreased mVAT cell diameter as compared with controls (Fig. 6A), whereas we could not detect any difference in the mSAT cell diameters (data not shown). Treg transfer increased the expression of the Treg marker FoxP3 and the Th1 cytokine IFN-γ in mVAT, whereas IL-6, TNF-α, Gata-3, and IL-10 mRNA were not differentially regulated (Fig. 6B). It is interesting that the percentage of proinflammatory mVAT-infiltrating CD8+CD69+ effector T cells was significantly decreased in Treg-treated db/db mice (Fig. 6C and D), whereas no difference of these cells could be detected in mSAT (Fig. 6C). Of note, no difference in the mVAT and mSAT infiltration of CD4+CD69+ T cells was observed (Fig. 6C).
FIG. 5.
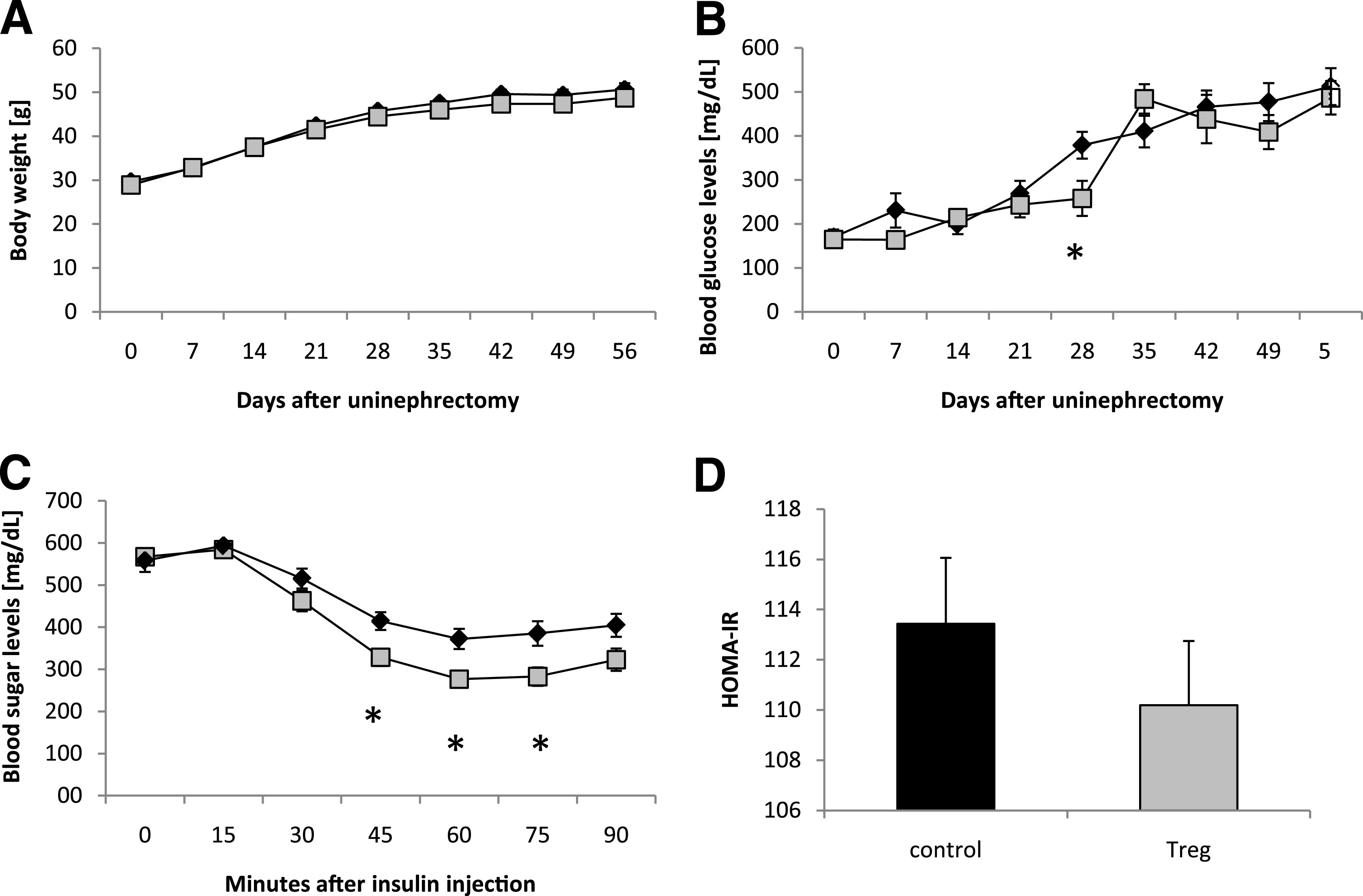
Adoptive Treg transfer restores insulin sensitivity in type 2 diabetes. Db/db mice were uninephrectomized at the age of 6 weeks and followed for 56 days. One group intravenously received FACS-sorted CD4+FoxP3+ Tregs every 14 days (gray cubes and bar, n = 8). Control animals received CD4+FoxP3− cells (black diamonds and bar, n = 8). Body weight (A) and blood glucose levels (B) were measured weekly. On day 56, the insulin-sensitivity testing was performed (C) and the HOMA-IR index was calculated (D). Pooled data of two independent experiments are shown. *P < 0.05.
FIG. 6.
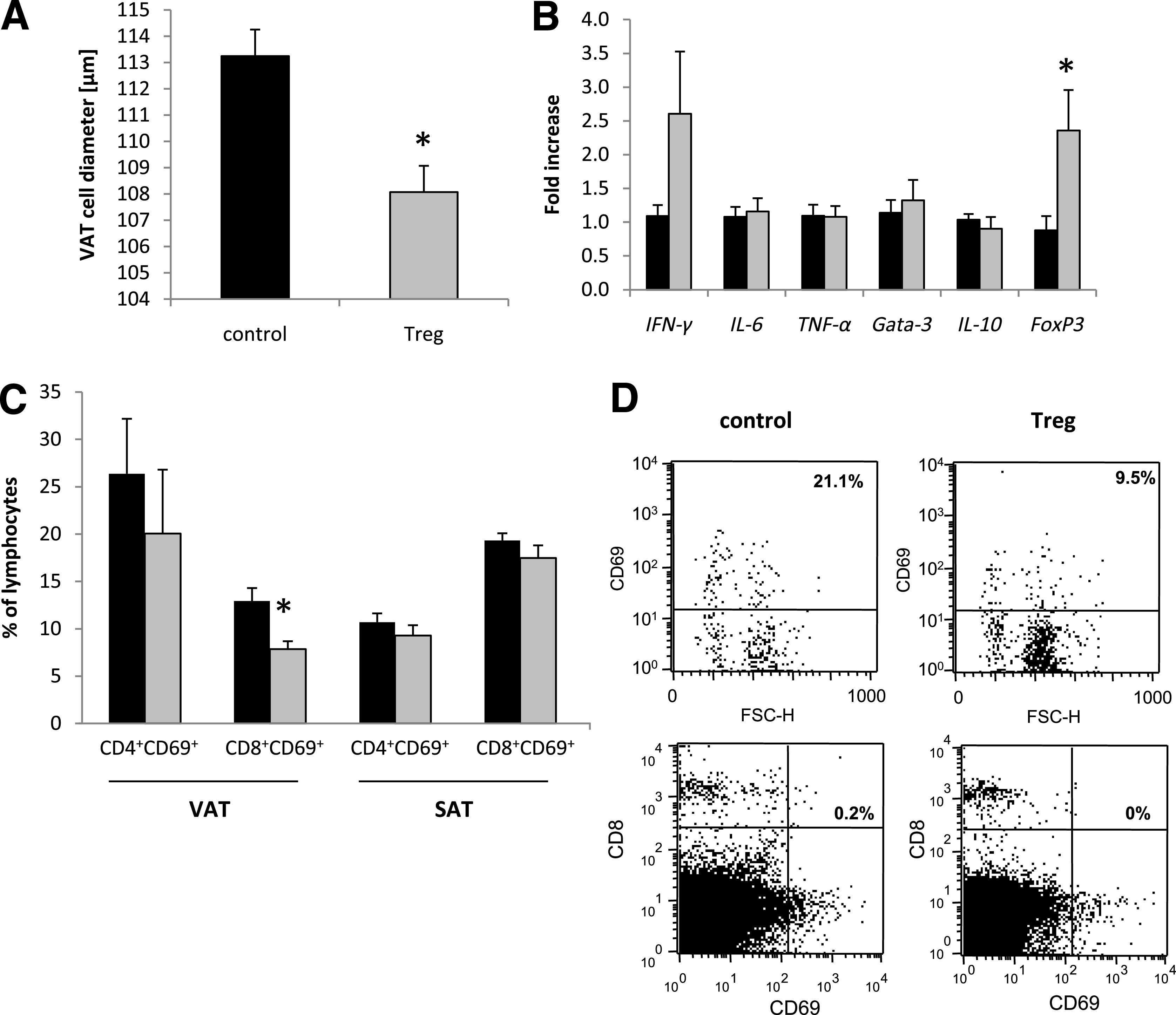
Treg treatment induces a shift toward an anti-inflammatory milieu in VAT. Db/db mice were uninephrectomized at the age of 6 weeks and followed for 56 days. One group intravenously received FACS-sorted CD4+FoxP3+ Tregs every 14 days (gray bar), whereas the control group was injected with CD4+FoxP3− control T cells (black bar). A: The VAT cell diameter was determined by morphological evaluation (n = 4 per group). B: The cytokine response in VAT was quantified by real-time PCR (n = 8 per group). The fold increase to the mean expression of controls is provided. C: The percentage of CD4+CD69+ and CD8+CD69+ T cells was evaluated in VAT and SAT by flow cytometry (n = 4 per group). Pooled or representative data of two independent experiments are shown. *P < 0.05. D: Representative dot plots from gated CD8+ T cells and from CD8/CD69 double staining in VAT. FSC-H, forward scatter height.
Treg treatment prevents the development of diabetic nephropathy.
Adoptive transfer of Treg significantly decreased urinary albumin-to-creatinine ratio in db/db mice when compared with respective controls on days 42 and 56 after uninephrectomy (Fig. 7A). In addition, the glomerular diameter, which reflects hyperfiltration, was also significantly decreased in kidneys of Treg-treated mice (Fig. 7B). In line with data from mVAT T-cell infiltration, CD8+ T cells were significantly less abundant in the kidneys of Treg-injected db/db mice (Fig. 7C), whereas the infiltration of CD4+ T cells and macrophages was not affected (data not shown). Despite a trend toward higher expression of the proinflammatory Th1 markers IFN-γ, IL-6, and TNF-α in kidneys of Treg-treated db/db mice, there was a predominant and statistically significant effect on the expression of the anti-inflammatory markers Gata-3, IL-10, and FoxP3 (Fig. 7D). Of note, we were not able to detect adoptively transferred GFP+ Tregs in harvested spleens, livers, kidneys, mSAT, and mVAT of uninephrectomized db/db mice on day 56 (i.e., 14 days after the final Treg-transfer) using either flow cytometry or a highly sensitive PCR for GFP (data not shown).
FIG. 7.

Treg treatment improves diabetic nephropathy. Db/db mice were uninephrectomized at the age of 6 weeks and followed for 56 days. One group intravenously received FACS-sorted CD4+FoxP3+ Tregs every 14 days (gray bar), whereas control animals received CD4+FoxP3− T cells (black bar). A: The albumin-to-creatinine ratio was evaluated in the urine every 14 days. B and C: After 56 days, kidneys were harvested and the glomerular diameter was morphologically determined (n = 4 per group) (B), and kidney sections were stained for the infiltration of CD8+ T cells (n = 8 per group) (C). Hpf, high-power field. D: The cytokine response in the kidney was evaluated by real-time PCR (n = 8 per group). The fold increase to the mean expression of controls is provided. Pooled data of two independent experiments are shown. *P < 0.05.
DISCUSSION
The pandemic of type 2 diabetes is one of the major health burdens in the industrial world today, and diabetic nephropathy due to type 2 diabetes has become the leading cause of end stage renal disease. Thus, new therapeutic options are eagerly awaited. Insulin resistance and type 2 diabetes are driven by chronic inflammation (24) where not only macrophages but also T cells are critically involved (24). Here, we present evidence that Tregs are key regulatory cells in the pathogenesis of insulin resistance and that they have the capacity to improve insulin resistance and diabetic nephropathy when transferred in vivo.
It is long known that macrophages accumulate in the mVAT of obese mice (25), but recently Winer et al. (15) found that T cells drive macrophage infiltration. By blocking T cells, they detected significantly improved insulin resistance in their murine type 2 diabetes model. In a similar approach, Ilan et al. (16) showed that the ratio between TH1 effector cells and Tregs changed toward Tregs as a result of oral administration of anti-CD3 and β-glucosylceramide. The importance of T cells and Tregs in insulin resistance was suggested by observational human studies. Nevertheless, published human data on Tregs in hVAT have been, thus far, conflicting (17–19). While Zeyda et al. (19) found T cells together with Tregs to accumulate in hVAT of obese patients, others detected decreased infiltrating Treg numbers (17,18). Our patient population consisted of lean, obese insulin-sensitive, and obese insulin-resistant patients. We detected decreased FOXP3 mRNA signals only in obese insulin-sensitive patients as compared with lean control subjects. Obese patients with insulin resistance had similar FOXP3 mRNA expression as lean control subjects. Most interesting, Helios—a new marker for thymic-derived natural Tregs (13)—differed significantly from our FOXP3 data, since Helios expression was significantly lowered in hVAT of all obese patients irrespective of their insulin resistance. Our data might thus explain the differences observed in the previous human studies. Obese patients seem to have decreased thymic-derived Tregs in their hVAT. Moreover, peripherally induced adaptive FoxP3+ Tregs seem to accumulate in the inflammatory microenvironment of hVAT in obese patients with overt insulin resistance. Limitations for these suggestions come from the technique used throughout our studies since we had no access to fresh hVAT to perform FoxP3/Helios double staining. Since FoxP3+ cell populations have also been shown to produce IL-17 or IFN-γ (26,27), we cannot exclude that our FOXP3 mRNA expression reflects other CD4+ T cells infiltrating hVAT of obese patients with insulin resistance. To gain further insights in the functional role of Treg in insulin resistance, we used db/db mice, which lack signaling capacity of the leptin receptor and therefore develop hyperphagia, diet-induced obesity, and type 2 diabetes (20). After treatment with a CD25-depletion antibody, which has been used for Treg depletion in various disease models (28–30), db/db mice displayed augmented insulin resistance and a more severe diabetic nephropathy. In line with recent reports (15,17), we detected significantly increased TH1 cytokines and activated T cells in the mVAT of Treg-depleted db/db mice, whereas no changes were detected in mSAT. These data strongly support the idea that VAT but not SAT is crucial for impaired insulin sensitivity. Independent studies in lean mice demonstrate that Treg depletion induced by a genetic approach aggravated insulin resistance (17). Crossing adipose mouse strains with FoxP3-DTR mice is a difficult issue because of infertility of homozygous db/db mice. Therefore, we chose to transfer sorted FoxP3+ Tregs into db/db mice.
We provide the first direct evidence that FoxP3 Tregs have the capacity to improve insulin resistance. Transfer of CD4+FoxP3+ cells induces Treg infiltration and/or FoxP3 upregulation in mVAT. The increased Treg activity inhibited CD8+ T-cell infiltration into mVAT. CD8+ T cells can be divided in different subpopulations. Naïve T cells provide the potential to respond to new pathogens, whereas central-memory and effector-memory T cells can mount rapid and efficient secondary proliferate and effector responses to recall antigens (31). In our experimental setup, CD8+ T cells infiltrating mVAT mainly expressed the T-cell activation marker CD69. Since T-cell activation markers such as CD69 and CD25 are expressed only on a small fraction of the two memory subsets (32), it can be proposed that Treg transfer decreased the mVAT infiltration of effector CD8+ T cells, which originated from naïve CD8+ T cells. This is further supported by the recent observation that Tregs dramatically decrease the expansion and differentiation of naïve CD8+ T cell precursors in an experimental murine model of repopulation of lymphopenic environment (33). CD8+ effector T cells are involved in the pathogenesis of obesity and infiltrate adipose tissue (34). Nishimura et al. (34) provided evidence that CD8+ T cells lead to the recruitment of macrophages, thereby mediating insulin resistance. In contrast, Winer et al. (15) found CD8+ T cells did not influence diet-induced obesity in lymphocyte-free Rag1-null mice. In our murine model, transfer of CD4+FoxP3+ Tregs not only improved insulin resistance but also resulted in less end organ damage, such as diabetic nephropathy. In the kidney, we observed a shift toward a TH2 response, reflected by increased mRNA expression of Gata-3 and IL-10, as well as significantly increased Treg numbers. Thus, IL-10 produced by Tregs might be of crucial importance for the Treg-mediated protective effect, but further studies using Tregs from IL-10 knockout mice are necessary. The role of IL-10 needs further evaluation since renal IL-10 mRNA expression also tended to be increased in kidneys of Treg-depleted mice. Again, we found significantly decreased numbers of CD8+ T cells to infiltrate the kidneys, whereas no change in the number of renal CD4+ T cells was noted. It is interesting that no significant difference in the macrophage infiltration of kidneys was detected. It has been proposed that CD8+ T cells improve insulin resistance by recruiting macrophages (34), which seems not to be the case in the pathogenesis of diabetic nephropathy. Further studies depleting CD8+ T cells in diabetic nephropathy are needed to shed more light on the role of CD8+ T cells in diabetic nephropathy. It might be further speculated that Tregs, independently from improving insulin resistance, improve diabetic nephropathy by exerting their anti-inflammatory capacities, but our study design does not allow discrimination between the two mechanisms.
By transferring FoxP3+-GFP+ Tregs, we transferred a selective fluorescent Treg population. Contrary to the murine model of nephrotoxic nephritis (2), we failed to track these fluorescent Tregs by using molecular genetic methods and flow cytometry. The absence of FoxP3+-GFP+ Tregs in mVAT, liver, kidney, and spleen on day 56 is probably due to the long interval of 14 days after the final transfer. It seems that at the tested time point, all transferred Tregs were already apoptotic and that they had previously, in their short lifetime, primed the immune system toward an anti-inflammatory milieu.
Taken together, we prove that transfer of CD4+FoxP3+ Tregs improves insulin resistance and diabetic nephropathy by tipping the balance toward anti-inflammation and inhibiting CD8+ effector cell infiltration in VAT and kidneys. Our results confirm the importance of Tregs in the pathogenesis of insulin resistance and might lead to new therapeutic concepts of type 2 diabetes.
ACKNOWLEDGMENTS
W.P. was supported by the Wiener Wissenschafts-, Forschungs- und Technologiefonds (Project LS07-058). A.R.R. was supported by the Austrian Research Fund (P-21402).
No potential conflicts of interest relevant to this article were reported.
K.E. researched data and wrote the manuscript. A.K. researched data. A.M.W. and S.S. researched data and contributed to discussion. A.T. and U.S. researched data. D.W., W.P., and A.R.R. contributed to discussion and wrote the manuscript. P.E. researched data and wrote the manuscript.
REFERENCES
- 1.Siegmund K, Feuerer M, Siewert C, et al. Migration matters: regulatory T-cell compartmentalization determines suppressive activity in vivo. Blood 2005;106:3097–3104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolf D, Hochegger K, Wolf AM, et al. CD4+CD25+ regulatory T cells inhibit experimental anti-glomerular basement membrane glomerulonephritis in mice. J Am Soc Nephrol 2005;16:1360–1370 [DOI] [PubMed] [Google Scholar]
- 3.Read S, Malmström V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J Exp Med 2000;192:295–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mottet C, Uhlig HH, Powrie F. Cutting edge: cure of colitis by CD4+CD25+ regulatory T cells. J Immunol 2003;170:3939–3943 [DOI] [PubMed] [Google Scholar]
- 5.Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med 2004;199:971–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salama AD, Chaudhry AN, Holthaus KA, et al. Regulation by CD25+ lymphocytes of autoantigen-specific T-cell responses in Goodpasture’s (anti-GBM) disease. Kidney Int 2003;64:1685–1694 [DOI] [PubMed] [Google Scholar]
- 7.Cao D, van Vollenhoven R, Klareskog L, Trollmo C, Malmström V. CD25brightCD4+ regulatory T cells are enriched in inflamed joints of patients with chronic rheumatic disease. Arthritis Res Ther 2004;6:R335–R346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ehrenstein MR, Evans JG, Singh A, et al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med 2004;200:277–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seddon B, Mason D. Regulatory T cells in the control of autoimmunity: the essential role of transforming growth factor beta and interleukin 4 in the prevention of autoimmune thyroiditis in rats by peripheral CD4(+)CD45RC- cells and CD4(+)CD8(-) thymocytes. J Exp Med 1999;189:279–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foussat A, Cottrez F, Brun V, Fournier N, Breittmayer JP, Groux H. A comparative study between T regulatory type 1 and CD4+CD25+ T cells in the control of inflammation. J Immunol 2003;171:5018–5026 [DOI] [PubMed] [Google Scholar]
- 11.Groux H, O’Garra A, Bigler M, et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature 1997;389:737–742 [DOI] [PubMed] [Google Scholar]
- 12.Bluestone JA, Tang Q. Therapeutic vaccination using CD4+CD25+ antigen-specific regulatory T cells. Proc Natl Acad Sci USA 2004;101(Suppl. 2):14622–14626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thornton AM, Korty PE, Tran DQ, et al. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol 2010;184:3433–3441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McClymont SA, Putnam AL, Lee MR, et al. Plasticity of human regulatory T cells in healthy subjects and patients with type 1 diabetes. J Immunol 2011;186:3918–3926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Winer S, Chan Y, Paltser G, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med 2009;15:921–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ilan Y, Maron R, Tukpah AM, et al. Induction of regulatory T cells decreases adipose inflammation and alleviates insulin resistance in ob/ob mice. Proc Natl Acad Sci USA 2010;107:9765–9770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feuerer M, Herrero L, Cipolletta D, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med 2009;15:930–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deiuliis J, Shah Z, Shah N, et al. Visceral adipose inflammation in obesity is associated with critical alterations in tregulatory cell numbers. PLoS ONE 2011;6:e16376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeyda M, Huber J, Prager G, Stulnig TM. Inflammation correlates with markers of T-cell subsets including regulatory T cells in adipose tissue from obese patients. Obesity (Silver Spring) 2011;19:743–748 [DOI] [PubMed] [Google Scholar]
- 20.Chen H, Charlat O, Tartaglia LA, et al. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell 1996;84:491–495 [DOI] [PubMed] [Google Scholar]
- 21.Ninichuk V, Khandoga AG, Segerer S, et al. The role of interstitial macrophages in nephropathy of type 2 diabetic db/db mice. Am J Pathol 2007;170:1267–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oberkofler H, Pfeifenberger A, Soyal S, et al. Aberrant hepatic TRIB3 gene expression in insulin-resistant obese humans. Diabetologia 2010;53:1971–1975 [DOI] [PubMed] [Google Scholar]
- 23.Rosenkranz AR, Mendrick DL, Cotran RS, Mayadas TN. P-selectin deficiency exacerbates experimental glomerulonephritis: a protective role for endothelial P-selectin in inflammation. J Clin Invest 1999;103:649–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lumeng CN, Maillard I, Saltiel AR. T-ing up inflammation in fat. Nat Med 2009;15:846–847 [DOI] [PubMed] [Google Scholar]
- 25.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 1993;259:87–91 [DOI] [PubMed] [Google Scholar]
- 26.Stroopinsky D, Avivi I, Rowe JM, Avigan D, Katz T. Allogeneic induced human FOXP3(+)IFN-gamma(+) T cells exhibit selective suppressive capacity. Eur J Immunol 2009;39:2703–2715 [DOI] [PubMed] [Google Scholar]
- 27.Voo KS, Wang YH, Santori FR, et al. Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc Natl Acad Sci USA 2009;106:4793–4798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haeryfar SM, DiPaolo RJ, Tscharke DC, Bennink JR, Yewdell JW. Regulatory T cells suppress CD8+ T cell responses induced by direct priming and cross-priming and moderate immunodominance disparities. J Immunol 2005;174:3344–3351 [DOI] [PubMed] [Google Scholar]
- 29.Couper KN, Blount DG, de Souza JB, Suffia I, Belkaid Y, Riley EM. Incomplete depletion and rapid regeneration of Foxp3+ regulatory T cells following anti-CD25 treatment in malaria-infected mice. J Immunol 2007;178:4136–4146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kinsey GR, Huang L, Vergis AL, Li L, Okusa MD. Regulatory T cells contribute to the protective effect of ischemic preconditioning in the kidney. Kidney Int 2010;77:771–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol 2004;22:745–763 [DOI] [PubMed] [Google Scholar]
- 32.Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999;401:708–712 [DOI] [PubMed] [Google Scholar]
- 33.Almeida AR, Ciernik IF, Sallusto F, Lanzavecchia A. CD4+ CD25+ Treg regulate the contribution of CD8+ T-cell subsets in repopulation of the lymphopenic environment. Eur J Immunol 2010;40:3478–3488 [DOI] [PubMed] [Google Scholar]
- 34.Nishimura S, Manabe I, Nagasaki M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 2009;15:914–920 [DOI] [PubMed] [Google Scholar]