

Abstract
OBJECTIVE
Skeletal muscle myopathy is a common diabetes complication. One possible cause of myopathy is myocyte failure to repair contraction-generated plasma membrane injuries. Here, we test the hypothesis that diabetes induces a repair defect in skeletal muscle myocytes.
RESEARCH DESIGN AND METHODS
Myocytes in intact muscle from type 1 (INS2Akita+/−) and type 2 (db/db) diabetic mice were injured with a laser and dye uptake imaged confocally to test repair efficiency. Membrane repair defects were also assessed in diabetic mice after downhill running, which induces myocyte plasma membrane disruption injuries in vivo. A cell culture model was used to investigate the role of advanced glycation end products (AGEs) and the receptor for AGE (RAGE) in development of this repair defect.
RESULTS
Diabetic myocytes displayed significantly more dye influx after laser injury than controls, indicating a repair deficiency. Downhill running also resulted in a higher level of repair failure in diabetic mice. This repair defect was mimicked in cultured cells by prolonged exposure to high glucose. Inhibition of the formation of AGE eliminated this glucose-induced repair defect. However, a repair defect could be induced, in the absence of high glucose, by enhancing AGE binding to RAGE, or simply by increasing cell exposure to AGE.
CONCLUSIONS
Because one consequence of repair failure is rapid cell death (via necrosis), our demonstration that repair fails in diabetes suggests a new mechanism by which myopathy develops in diabetes.
The long-term complications of diabetes are diverse and include cardiac dysfunction, renal disease, stroke, nerve damage, blindness, and vascular damage (1). Many of these complications are the direct result of elevated blood glucose (2). However, the exact nature of the diverse cell defects that are induced by elevated blood glucose, and hence the molecular mechanisms underlying these defects, are often poorly understood.
Musculoskeletal abnormalities, a well-known medical complication of type 1 and 2 diabetes, are increasing in prevalence and are a common source of disability in the diabetic population (3). Indeed, muscle atrophy and necrosis in diabetes may now represent the most common clinical condition affecting human muscle growth and performance (3,4). Atrophy can be severe and debilitating (4), affecting most prominently the proximal limb-girdle muscle, a condition referred to as “diabetic amyotrophy” (5). Moreover, diabetic patients can experience acute muscle necrosis (6), also known as “diabetic myonecrosis” (7). Diabetic myopathy is demonstrable in animal models of diabetes, including mouse (8) and rat (9,10), where reductions in muscle mass and contractile force have been measured and increased susceptibility to contraction-induced injury have been demonstrated. However, the pathogenesis of diabetic myopathy remains unknown.
Plasma membrane disruptions are a naturally occurring phenomenon in mechanically active tissues. Skeletal muscle activity, especially that involving high-force eccentric contractions, causes disruptions to form in the plasma membrane of the contracting skeletal muscle myocytes (11). These disruptions are rarely fatal injuries because they trigger in the injured cell a rapid membrane repair response that is initiated by the influx of extracellular calcium (12). This influx initiates fusion of internal membrane compartments, thus producing a “patch vesicle” (13), which is then exocytotically fused to the disruption site to restore membrane continuity (14).
Numerous proteins have been shown to participate in this repair response, one of which is dysferlin (15). Dysferlin is a calcium-activated membrane-binding protein located in skeletal myocytes. Lack of functional dysferlin, in mice and humans, resulted in the development of a limb-girdle muscular dystrophy (15). We have shown that membrane repair is defective in myocytes isolated from dysferlin-deficient mouse muscle (16). Strikingly, the localization in diabetic amyotrophy is similar to that displayed in dysferlin-deficient atrophy, e.g., the limb-girdle musculature.
We hypothesize that diabetic myopathy, analogously to limb-girdle muscular dystrophy, is caused by defective membrane repair. As a test of this hypothesis, we compare the repair responses of normal and diabetic mouse skeletal muscle. We further use an in vitro model of diabetes to test whether the repair defect can be induced by elevated glucose and whether interactions of advanced glycation end products (AGEs) and the receptor for AGE (RAGE) are involved.
RESEARCH DESIGN AND METHODS
Mouse models.
The following mice strains were used: C57BL/J INS2Akita+/−, which display elevated glucose at 4–6 weeks of age; B6 (C57BL/6) (a gift from Dr. S. Smith, Georgia Health Sciences University) and db/db (BKS Cg-Dock7m+/+), which display elevated glucose at 4–8 weeks of age; and BLKS (C57BLKS/J) (Diabetes and Obesity Discovery Institute, Georgia Health Sciences University). Blood glucose was measured after a tail clip using Accu-Chek Aviva (Roche, Worldwide). Fibroblasts were obtained through trypsin digestion of mouse ears for 1 h and cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, Carlsbad, CA) from RAGE-deficient (RAGE−/−, n = 3) (17), C57BL/J INS2Akita+/− (n = 4), and B6 (n = 4) mice. All protocols for animal use and euthanasia were reviewed and approved by the Animal Use Committee at Georgia Health Sciences University and were in accordance with National Institutes of Health guidelines.
Laser repair assay.
Solei were dissected from mice, secured to 35-mm culture dishes by a drop of New-Skin (Medtech, Irvington, NY) liquid bandage, and kept (<1 h) in ice-cold Tyrode solution: 128 mmol/L NaCl, 4.7 mmol/L KCl, 1.36 mmol/L CaCl2, 20 mmol/L NaHCO3, 0.36 mmol/L NaH2PO4, 1 mmol/L MgCl2, and 10 mmol/L glucose, pH 7.4 (18). Laser analysis was carried out in 37°C Tyrode solution containing 1.36 mmol/L CaCl2, or Tyrode without added CaCl2. FM 1–43 dye (Invitrogen), a membrane impermeant dye, was added (2.5 μmol/L) before laser injury (19). Intact plasma membranes of healthy myocytes, highlighted by staining of FM dye, were targeted for laser injury using a two-photon laser scanning confocal microscope (LSM 510; Zeiss, Thornwood, NY) coupled with a 10-W Argon/Ti-sapphire laser (Spectra-Physics Lasers) operated at 100% power, 100 iterations, with a 5 × 35 pixels “bleach” area. Fluorescence, associated with the injured domain of the myocyte, was measured in a 120-μm diameter circular window centered on the myocyte injury site using Zeiss LSM 510 software. The “background” fluorescence measured before the injury was subtracted. After laser injury, myocyte contractions were often observed, moving fibers out of focus, and manual refocusing was used. Data were recorded only when the fiber was in focus. Cultured cells were wounded in phosphate-buffered saline (PBS) containing 137.9 mmol/L NaCl, 2.7 mmol/L KCl, 15.2 mmol/L Na2HPO4, 1.5 mmol/L KH2PO4, 1 mmol/L MgCl2 (with or without 1.2 mmol/L Ca2+) (Sigma, St. Louis, MO), and 2.5 μmol/L FM 1–43 or FM 4–64 (Invitrogen) for injury. The disruptions were made using 100% power and one laser iteration with a 15 × 15 pixels bleach area.
Downhill running procedure.
Mice in the age range of 6–7.5 months were used. INS2Akita+/− mice (n = 9) with blood glucose levels >600 mg/dL (Accu-Chek HI) and B6 mice (n = 9) with blood glucose levels of 151.11 ± 7.95 mg/dL were run on a treadmill set at a 15-degree decline as previously described (11) (Omnipacer Treadmill LC4/M-MGA/AT; AccuScan Instruments, Columbus, OH). These mice were subjected to a moderate downhill run of 10 m/min for 1 h. Control mice remained in their cages (n = 4/group). After completion of the downhill run, all mice were injected with Evan’s blue dye (EBD) (10 mg EBD/1 mL PBS, 100 μL/10 g body wt; Sigma) (20), and 24 h later, the quadriceps and gastrocnemius were excised, weighed, and frozen in O.C.T. (Tissue-Tek, Alphen aan den Rijn, the Netherlands). EBD stains albumin present within tissue, clearly highlighting the exterior of muscle fibers, while also staining albumin present within fibers that are no longer intact (16,20). Cross sections were cut at a 10-μm thickness and analyzed microscopically (10× magnification) for EBD-positive fibers. Fluorescently labeled cells were scored per cross-section taken at four random muscle locations and averaged per muscle.
In vitro diabetic cell model.
C2C12, BS-C-1, and HeLa cells were cultured in DMEM, passaged twice weekly, and supplemented with glucose (glucose at 5.5 mmol/L simulates a fasting blood glucose of 99 mg/dL, 7.5 mmol/L glucose represents fed blood glucose level of 135 mg/dL, and 30 mmol/L glucose elevated blood glucose level of 540 mg/dL) or mannitol (21) and, where noted, treated with 1 mmol/L aminoguanidine (AMG) (Sigma).
Mechanical injury: multi-well repair assay.
The 96-well plates of cells were subjected to scraping using a spring-loaded 96-well transfer device (22). This device was used in a circular motion to scrape cells off their substratum, therefore creating plasma membrane disruptions (23). These wounded cells (in suspension) were transferred to a fresh plate. Survival in this population was assessed using a live/dead viability/cytotoxicity kit according to the manufacturer’s instructions (Molecular Probes, Carlsbad, CA). The live/dead fluorescence ratio, calcein acetoxymethyl ester (excitation/emission 495 nm/515 nm) to ethidium homodimer-1 (excitation/emission 495 nm/635 nm) ratio, was determined after 1 h of incubation using a fluorescent plate reader (FLx800; BioTek, Winooski, VT). Alternatively, after replating cells from the multiwell scrape assay, fresh DMEM was added to each well. A DMEM rinse was used to remove dead nonadherent cells from the plate after 4 h at 37°C. The MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was then performed using the methods previously described (24). The cells surviving the scrape injury were compared with uninjured control populations to calculate percent survival.
RAGE construct and transfection.
A RAGE construct was produced by subcloning the full-length human RAGE gene into the enhanced green fluorescent protein (pEGFP)-N1 vector to generate pEGFR-N1-RAGE (EGFP is fused to the COOH terminus of RAGE). BS-C-1 cells were transfected with 1 μg pEFGP-N1-RAGE using Lipofectamine (Invitrogen). Cells were treated for 20 h with 5 mmol/L of AGE-modified BSA (AGE-BSA) (Sigma) 4 h after transfection.
Image processing.
Microscope images of laser injury were taken using a 40× IR-Acroplan lens, 0.8 numerical aperture; a Zeiss Axioplan 2 microscope; and LSM 510 software for image analysis. Microscope images of EBD were taken using a 10× Plan-Neofluar lens, 0.3 numerical aperture; a Zeiss Axioplan 2 microscope; and a Zeiss AxioCam high-resolution camera. Microscope images were rotated, cropped, and reduced in size. No false coloring or γ adjustments were performed for quantifications of fluorescence intensity.
Statistical analysis.
Comparisons were made with Prism 5.0 software using repeated-measures ANOVA and Tukey post-analysis tests for significance. A Student t test was used to determine the difference between muscle diameters (two-tailed), mouse weights (two-tailed), and decreased muscle weights (one-tailed). For all analysis, P < 0.05 was considered to be significant. In text and graphs, data are represented as the mean ± SEM.
RESULTS
In situ/in vivo analysis of membrane repair.
To directly assess repair in the diabetic skeletal muscle environment, we used a novel in situ assay. The intact solei of mice were placed in physiological saline containing a fluorescent dye, FM 1–43, which strongly stains the plasma membrane boundary only of intact cells. After laser disruption of the plasma membrane, FM 1–43 diffuses into the cytosol through the disruption site and begins to fluorescently stain the internal membrane. In the presence of calcium (Ca2+), membrane repair is activated, slowing dye entry into the cytosol and consequent staining of internal membranes (Supplementary Movie 1). A “hot spot” of fluorescence staining, the result of initial dye entry, marks the injury throughout the experiment. In the absence of Ca2+, membrane repair fails and the internal cellular compartments begin to saturate with dye, a process that continues throughout the 445-s time course (Supplementary Movie 2). Because of the contractions of the injured myocyte, refocusing is frequently necessary, so that measurement is centered on the initial disruption site. However, with this maneuver, fluorescence within the cell can be monitored and quantitated over time surrounding the disruption site.
Two diabetic mouse models, C57BL/J INS2Akita+/− (INS2Akita+/−) with type 1 diabetes and BKS Cg-Dock7m+/+ (db/db) with type 2 diabetes, were assessed using this in situ laser assay. In muscle from C57BL/6 (B6) and C57BLKS/J (BLKS) control mice, laser injured in the presence of Ca2+, a hot spot of FM 1–43 appeared at the membrane disruption site, but widespread staining of internal membrane was not observed, indicating repair had occurred (Fig. 1A, Control B6 + Ca, 445 s; and Supplementary Movie 3). However, when Ca2+ was absent, control fibers displayed continuous cytosolic filling with FM 1–43, characteristic of failed membrane repair (Fig. 1A, Control B6 – Ca). Strikingly, in both diabetic models, dye entered continuously into diabetic fibers, even when Ca2+ was present, indicating repair failed to occur (Fig. 1A, Type 1 INS2 + Ca; and Supplementary Movies 4 and 5). These results are illustrated quantitatively in Fig. 1B: all fibers initially displayed similar dye uptake kinetics in the presence and absence of Ca2+, indicating the size of the disruption made was equivalent. However, at time points thereafter (375 s for the INS2 and 175 s for the db/db), significantly more dye uptake, compared with wild-type controls, in the presence of Ca2+, was recorded in solei from both diabetic models. In fact, measured dye uptake in diabetic myocytes was indistinguishable, whether Ca2+ was present or absent. Thus, using a highly sensitive in situ assay, we show that Ca2+-dependent muscle membrane repair is deficient in both type 1 and type 2 diabetes mouse models.
FIG. 1.

Calcium-dependent repair fails in diabetic myocytes. A: A myocyte within the soleus muscle of a wild-type (Control B6, top two rows) and type 1 diabetic (Type 1 INS2, bottom two rows) mouse was imaged before (0 s) and after membrane irradiation with an infrared laser (arrow indicates injury site) in the presence of Ca2+ or its absence. FM 1–43 dye enters the myocyte through the injury site. Repair (Control B6 +Ca) impedes further dye uptake and confines the resultant fluorescence to a hot spot at the site of injury. Failed repair (Control B6 –Ca, Type 1 INS2 –Ca, and Type 1 INS2 +Ca) results in a sustained filling of the entire fiber with dye. B: Fluorescence signal was monitored over time to determine uptake of dye by myocytes from diabetic and control mice. The data presented represent five fiber measurements per soleus (Supplementary Table 1). The red circle indicates the time point where the first significant difference of P < 0.05 is observed (compared with all conditions). Data are presented as mean ± SEM. *P < 0.01; myofibers injured, n = 25 for B6 +Ca, n = 25 for B6 -Ca, n = 35 for INS2 +Ca, n = 30 for INS –Ca, n = 47 for BLKS +C, n = 45 for BLKS –Ca, n = 50 for db/db +Ca, and n = 49 for db/db –Ca. Scale bar, 50 μm and 40× magnification. (A high-quality digital representation of this figure is available in the online issue.)
Images obtained while performing the in situ laser assay suggested that there might be a size difference between normal and diabetic fibers. We therefore measured fiber diameters in diabetic and control mice. Decreased fiber diameter was measured in both the soleus and quadriceps of diabetic mice, and INS2Akita+/− mice also displayed significantly decreased body weight and muscle mass aged 6–7.5 months (Supplementary Fig. 1). These morphological changes confirm previous findings (25,26) that diabetes, as expected, negatively affected the skeletal muscle in these mouse models.
Plasma membrane disruptions are induced in vivo in muscle undergoing eccentric contractions, such as those produced by downhill running (11). To determine if membrane repair fails at a greater rate in diabetic mice during eccentric contraction-induced injury, we monitored myocyte permeability to EBD. EBD is a fluorescent dye that enters only into cells lacking an intact plasma membrane barrier and is effective at identifying injured muscle fibers without the presence of a counter-stain (16,20). We injected this dye into mice after running, so that myofibers that suffered a disruption and failed to repair would be labeled. The morphology of the EBD-positive fibers is illustrated in Fig. 2A (white arrows). As expected, in both normal and diabetic mice, running induced an increase in the number of EBD-positive fibers. However, running induced a significantly higher number of EBD-positive fibers in the diabetic mice (Fig. 2B, INS2 vs. control).
FIG. 2.
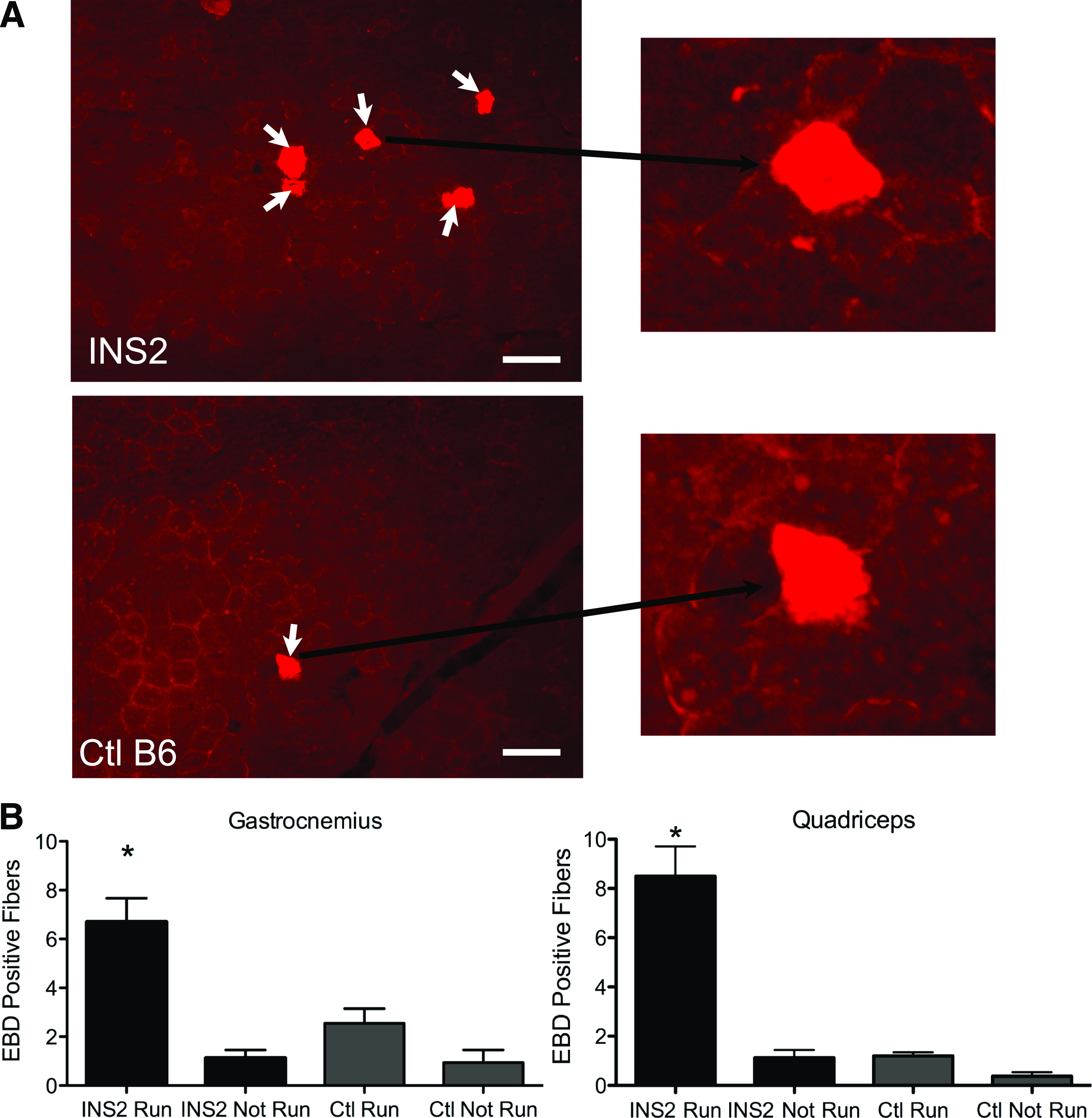
Exercised-induced myocyte disruptions are not repaired in diabetic mice. A: Paired transmission and fluorescence micrographs showing EBD-labeled myofibers (arrows) in the gastrocnemius muscle from diabetic (INS2) and control (Ctl) (B6) mice that run downhill for 60 min and then are injected with the EBD tracer. Myocytes that were injured by running and failed to repair are labeled. B: The number of EBD myofibers counted from such micrographs in the gastrocnemius and quadriceps muscles of diabetic INS2 and control B6 mice. Data are presented as the mean ± SEM. *P < 0.05; n = 5 mice for the “Run” groups and 4 mice for the “Not Run” groups. Scale bar, 100 μm and 20× magnification. (A high-quality digital representation of this figure is available in the online issue.)
Thus, both our in situ and in vivo measurements strongly support the hypothesis that membrane repair fails in diabetes.
In vitro analysis of membrane repair.
The diabetic environment in vivo is a complicated one, in which elevated blood glucose is just one pathophysiological element. To test whether glucose elevation alone is sufficient to induce a repair defect, we used an in vitro model mimicking the blood glucose levels found in our diabetic mouse models (Supplementary Table 1).
C2C12 cells, a skeletal muscle myocyte culture model, were assessed for membrane repair after growth for 1–8 weeks in medium containing normal glucose (7.5 mmol/L), elevated glucose (30 mmol/L), or mannitol (30 mmol/L). After 1 week of elevated glucose exposure, there was no qualitative change in repair (for example, dye entry pattern) (Fig. 3A, 1 week “+Ca” all conditions; and Supplementary Movie 6). However, after 8 weeks of high glucose exposure, dye entry after laser injury was clearly elevated, relative to the normal glucose and mannitol controls (Fig. 3A, 8 weeks N Gluc +Ca and Man +Ca), indicating an inability to repair (Fig. 3A, 8 weeks H Gluc +Ca; and Supplementary Movie 7). These observations are confirmed by quantitative analysis (Fig. 3B). Thus, prolonged exposure to high glucose is required for inducing a repair defect, a finding that is not compatible with other short-term responses, such as induction of insulin resistance.
FIG. 3.
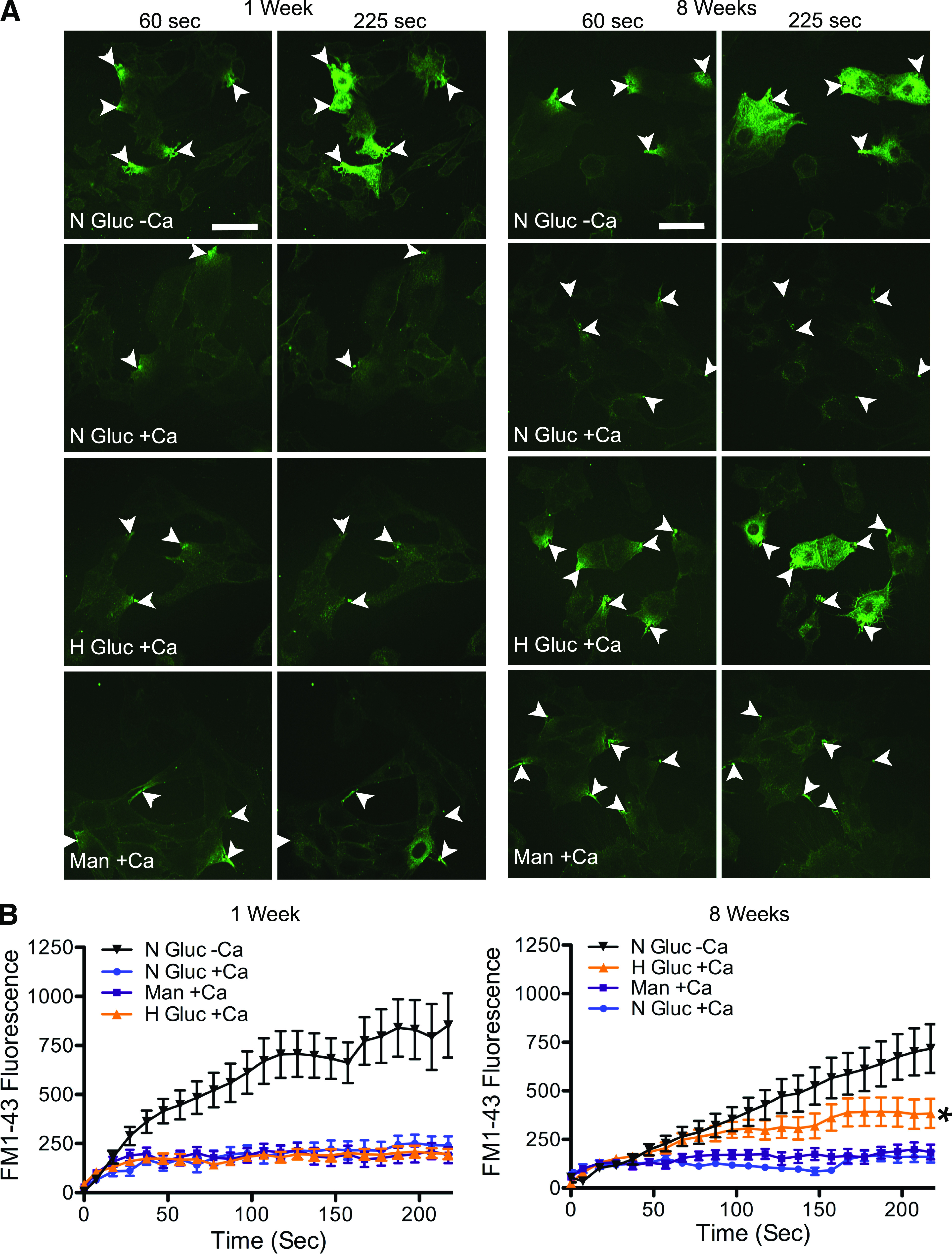
Myoblasts cultured in high glucose medium for prolonged periods display impaired membrane repair. A: C2C12 myoblasts were cultured in medium with 7.5 mmol/L glucose (N Gluc), 30 mmol/L glucose (H Gluc), or iso-osmolar control 30 mmol/L mannitol (Man) and then subject to laser analysis of repair (arrows indicate injury site) at 1 or 8 weeks of exposure. The interval (seconds) postinjury is indicated. Only cells grown in 30 mmol/L glucose for 8 weeks (H Gluc +Ca) were observed to fill with dye after laser injury in the presence of calcium. B: Dye uptake, fluorescence, measured within cells over time after injury. After 1 week of exposure to high glucose (left panel), all cells displayed calcium-dependent repair with no significant difference in dye uptake between groups. By contrast, cells cultured for 8 weeks in high glucose (right panel) showed a significant increase in dye uptake compared with normal glucose and mannitol controls. Data are presented as the mean ± SEM. *P < 0.05; 1 week, n = 15 for N Gluc +Ca, n = 14 for H Gluc +Ca, n = 13 for Man +Ca, and n = 17 for N Gluc –Ca; 8 weeks, n = 26 for N Gluc +Ca, n = 30 for H Gluc +Ca, n = 28 for Man +Ca, and n = 15 for N Gluc –Ca. Scale bar, 50 μm and 40× magnification. (A high-quality digital representation of this figure is available in the online issue.)
To confirm that this glucose-induced repair defect was not limited to laser-generated disruptions, adherent C2C12 cells were scraped to mechanically induce membrane disruptions (23). Survival was then assessed using two independent assays: live/dead and MTT. Consistent with the laser assay, there was no detectable difference in survival between treatment groups after 2 weeks of high glucose exposure (Fig. 4, 2 weeks). However, after 8 weeks of elevated glucose, cells displayed a significant decrease in survival when compared with those grown in normal glucose or mannitol (Fig. 4, 8 weeks H Gluc). Thus, using an independent method for inducing membrane disruptions and for monitoring repair failure, we confirmed that high glucose leads to the development of a membrane repair defect.
FIG. 4.

High glucose treatment also results in failure to repair mechanically induced disruptions. C2C12 myoblasts were cultured for 2 or 8 weeks in 7.5 mmol/L glucose (N Gluc), 30 mmol/L glucose (H Gluc), or iso-osmolar control 30 mmol/L mannitol (Man) and then scraped from 96-well plates. A live/dead or MTT assay was then performed to assess cell survival. No significant differences were demonstrated after 2 weeks of treatment. However, 8 weeks of high glucose treatment decreased survival as determined by both assays. Data are presented as the mean ± SEM. *P < 0.01; n = 24 wells for each condition.
To determine if other cell types develop a membrane repair defect after high glucose exposure, we applied the above analysis to BS-C-1 (monkey kidney epithelial) and HeLa (human cervical) cells. As was the case for C2C12 myoblasts, BS-C-1 cells developed a repair deficiency only after 8 weeks of high glucose treatment (Fig. 5A, H Gluc +Ca). BS-C-1 and HeLa survival of the scraping injury was also significantly decreased after 8 weeks of high glucose exposure, compared with cells grown in normal glucose or mannitol (Supplementary Fig. 2A, H Gluc HeLa and BS-C-1). This glucose-induced membrane repair defect remained even after cells were returned to a low glucose medium for over a week (Fig. 5B, H Gluc/L Gluc). Additionally, fibroblasts obtained from diabetic (INS2Akita+/−) mice also displayed a membrane repair defect after culturing in normal glucose medium for 1 week (Supplementary Fig. 2B, INS2).
FIG. 5.

An epithelium-derived cell line, BS-C-1, also develops repair failure in high glucose, and this defect is not readily reversible. A: BS-C-1 cells were cultured for 8 weeks in 7.5 mmol/L glucose (N Gluc), 30 mmol/L glucose (H Gluc), or iso-osmolar control 30 mmol/L mannitol (Man). Laser analysis revealed that cells cultured in high glucose displayed a significant increase in dye uptake when compared with normal glucose and mannitol. B: BS-C-1 cells were cultured for 10 weeks in H Gluc and then switched to L Gluc (5.5 mmol/L glucose) for 10 days (H Gluc/L Gluc), or cells were cultured for 11 weeks in L Gluc or H Gluc only. Laser analysis determined that the high glucose membrane repair defect remained 1 week after switching to low glucose (H Gluc/L Gluc). Data are presented as the mean ± SEM. *P < 0.01; 2 weeks, n = 18 for N Gluc +Ca, n = 14 for H Gluc +Ca, n = 12 for Man +Ca, and n = 13 for H Gluc –Ca; 10 weeks, n = 14 for L Gluc, n = 16 for H Gluc, and n = 15 for H Gluc/L Gluc.
Thus, our in vitro analysis revealed that the membrane repair defect is glucose induced, not exclusive to skeletal muscle cells, and does not appear to be readily reversible.
Is there an AGE/RAGE connection?
One well-established effect of elevated blood glucose is the production of AGEs, thought to contribute to the development of chronic diabetes complications (27,28). Numerous studies (29,30) have detected AGE generation in vitro as soon as 48 h after initiating high glucose incubation. To test if this AGE production is required for the development of the membrane repair defect, we simultaneously exposed BS-C-1 cells to elevated glucose and AMG, a potent inhibitor of AGE formation and accumulation (31). As expected, after 8 weeks of high glucose treatment in the absence of AMG, membrane repair was defective (Fig. 6A, H Gluc). However, this high glucose repair defect was completely eliminated by cotreatment with AMG (Fig. 6A, H Gluc AMG). Thus, glycation is implicated in the development of the membrane repair defect.
FIG. 6.

AGE/RAGE interaction is responsible for the repair defect. A: BS-C-1 cells were cultured in 30 mmol/L glucose supplemented with or without 1 mmol/L AMG (H Gluc AMG or H Gluc), or 30 mmol/L mannitol (Man) for 8 weeks. Cells were laser wounded in the presence of physiological saline containing calcium. Cells exposed to high glucose alone showed significantly increased (*) dye uptake when compared with mannitol controls. However, cells treated with high glucose and AMG did not exhibit this increase. B: RAGE-deficient fibroblasts (RAGE−/−) were cultured for 12 weeks in 30 mmol/L glucose (H Gluc), 5.5 mmol/L glucose (L Gluc), or 30 mmol/L mannitol (Man). Cells were laser wounded in the presence or absence of calcium. RAGE−/− cells exposed to high glucose and injured in the presence of calcium (H Gluc +Ca) did not exhibit a significant increase in dye uptake compared with controls (L Gluc +Ca and Man +Ca). C: BS-C-1 cells were transfected with a RAGE-EGFP expression vector (pEGFP-N1-RAGE). Cells displaying EGFP fluorescence (FL RAGE), and as controls, nonfluorescent cells in the same culture (NF), were selected for laser analysis of repair. One culture was exposed to 5 mmol/L AGE-BSA, and the other was not. Laser analysis showed membrane repair of fluorescent cells (FL RAGE) to be indistinguishable from nonfluorescent controls (NF) in cultures not exposed to AGE-BSA. However, fluorescent cells treated with AGE (FL RAGE AGE) showed a significant (**) elevation in dye uptake when compared with cells not treated with AGE, comparable to the dye uptake seen in cells wounded without calcium (–Ca). D: BS-C-1 cells were treated or not with 1 or 5 mmol/L AGE-BSA for 3 days before laser injury. Dye uptake was found to be significantly (*) greater for cells treated with AGE-BSA than cells left untreated and resembled the lack of repair for cells injured in the absence of Ca2+ (–Ca). Data are presented as the mean ± SEM. *P < 0.001 and **P < 0.05. BS-C-1: n = 14 cells for Man, n = 12 for H Gluc, and n = 12 for H Gluc AMG. Fibroblasts: n = 23 cells for L Gluc +Ca, n = 23 for H Gluc +Ca, n = 24 for Man +Ca, and n = 19 for L Gluc –Ca. Transfected: n = 12 cells for NF, n = 9 for NF AGE, n = 10 for FL RAGE, n = 10 for FL RAGE AGE, and n = 15 for –Ca. AGE treated: n = 21 cells for +Ca, n = 18 for –Ca, n = 22 for 1 mmol/L AGE, and n = 17 for 5 mmol/L AGE.
AGE potently binds to a cell surface RAGE (27,28), which in turn stimulates increased expression of this receptor and is hypothesized to contribute to the pathophysiology of diabetes (28,32). To test if RAGE is implicated in the development of the membrane repair defect, we cultured RAGE-deficient cells (RAGE−/−) in high glucose. After 12 weeks of elevated glucose exposure, RAGE−/− cells did not display the drastic increase in dye uptake, as previously determined in other cells treated to high glucose (Fig. 6B, H Gluc +Ca). This result indicates that RAGE is required for the glucose-induced repair defect.
To further test the involvement of RAGE in promoting repair failure, we transfected BS-C-1 cells with an expression vector (pEGFP-N1-RAGE, where EGFP is fused with the COOH terminus of RAGE) to force RAGE expression. Cells that were successfully transfected (as indicated by EGFP-derived fluorescence) were selected for laser injury. Dye entry kinetics into these RAGE-expressing cells were indistinguishable from nonfluorescent control cells in the same culture (Fig. 6C, FL RAGE and NF RAGE). However, when the mixed culture of EGFP-positive and -negative cells were incubated with AGE-BSA, fluorescent (EGFP-positive) cells displayed significantly enhanced dye entry relative to nonfluorescent cells and also relative to fluorescent cells not exposed to AGE-BSA (Fig. 6C, FL RAGE AGE). Thus, RAGE expression alone is not sufficient for inducing membrane repair deficiency in cultured cells, but RAGE binding of AGE is clearly implicated in defective membrane repair.
We noticed in these experiments a slight but not statistically significant increase in dye entry into the nonfluorescent cells exposed to AGE-BSA for 24 h (Fig. 6C, NF AGE). Therefore, we investigated further whether AGE exposure by itself could negatively influence membrane repair. When we increased the exposure level of cell to AGE-BSA, we found that dye entry was significantly elevated (Fig. 6D, 1 mmol/L AGE and 5 mmol/L AGE).
Thus, AGE formation, and not some other metabolic change induced by high glucose exposure, is responsible for repair failure, and RAGE binding of AGE is identified as one mechanism by which AGE exerts its deleterious effect.
DISCUSSION
The current study identifies a novel adverse effect of diabetes on skeletal muscle: we provide direct evidence that plasma membrane repair by myocytes is compromised in in vivo, in situ, and in vitro models. Diabetic myopathy, generally speaking, has been viewed as a consequence of underlying neuropathy (5,33). However, to our knowledge, there is no definitive evidence to suggest a causal relationship between diabetic neuropathy and myopathy. One study found that muscle contractions induced in diabetic rats by sciatic nerve stimulation resulted in “focal” (limited domains of abnormal myofiber structure at the light microscope level) muscle myocyte damage and elevated creatinine kinase levels (10). This result suggested that the diabetic condition could directly affect the contracting skeletal muscle myocyte. What element of the diabetic environment might be detrimental and what exactly fails when the diabetic myocyte contracts remain open questions. Using in vitro, in situ, and in vivo models, we demonstrate that elevated glucose results in defective plasma membrane repair and that, in diabetic mice, skeletal muscle myocytes are repair defective.
In this study, using a novel in situ laser assay, we show that myofibers of both type 1 and type 2 diabetic mouse models display an inability to repair membrane disruptions. Neuropathy cannot be implicated in this repair defect, since our analysis of isolated muscle is inherently independent of the nervous system functioning. Moreover, we use a vital dye (EBD) to show that repair of disruptions, induced by downhill running, is impaired in diabetic mice, indicating that the defect is not one unique to a laser injury, but one that arises under physiological conditions as well.
How could diabetes result in deficient membrane repair? Plasma membrane repair is a complex and dynamic cell survival response activated by influx of extracellular Ca2+ through the disruption. This Ca2+ entry rapidly elicits homotypic vesicle-vesicle fusion in the cytoplasm surrounding the site of disruption (12,13,34). A “patch” vesicle thus forms from cytoplasmic membranes, such as endosomes (35), lysosomes (36), enlargesomes (37), and yolk granules (38). Exocytotic annealing of this enlarging patch vesicle, also triggered by Ca2+, to plasma membrane surrounding the injury site, restores membrane continuity (14,39).
Membrane proteins are of course required for the diverse fusion events of repair. Not unexpectedly, these include known universal components of membrane fusion, such as the soluble n-ethylmaleimide–sensitive factor attachment protein receptor family members (12,40) and synaptotagmin (41). Also demonstrated to be required for successful repair are dysferlin (16), annexins (42), actin and myosin (43), calpain (44), and mitsugumin 53 (45).
For the present work, it is important to also consider the heterogeneous array of membrane glycoproteins that decorate cell surfaces. These too have been shown to play an important role in membrane repair. Multivalent plant lectins that bind these surface carbohydrates can cross-link these membrane glycoproteins. Such lectin cross-linking potently inhibits membrane repair, and it is the exocytotic step of repair that is blocked by lectins (46). This result suggested that AGE/RAGE interactions, which are elevated in diabetes, might be acting in an analogous fashion as an inhibitor of repair. In other words, RAGE binding and cross-linking of AGE (46) might be responsible for the repair defect in diabetic mice.
We report here the numerous tests of the possibility that AGE/RAGE interactions are responsible for the repair defect in diabetes. Our in vitro model system shows that development of the membrane repair defect can be driven by a prolonged (at least 8 weeks) exposure to high glucose. High glucose levels promote nonenzymatic cell surface hyperglycation of glycoproteins (47,48) (e.g., the production of AGE). We show that the accumulation of AGEs is responsible for the membrane repair defect seen in cell culture: the glucose-induced defect was not detectable when AGE generation was prevented by the addition of AMG to cell culture medium. Moreover, we show that AGE-BSA can, by virtue of its addition alone to culture medium, produces a repair defect in the absence of high glucose exposure. Finally, we show that cells expressing EGFP-RAGE, in the absence of high glucose exposure, develop a repair defect, but only if AGE-BSA is added to the culture medium, and that RAGE-deficient cells do not develop a repair defect when exposed to elevated glucose. Taken together, these results strongly suggest that AGE and AGE/RAGE binding is involved in the development of the repair defect in diabetes. Whether inhibition by AGE/RAGE occurs by a mechanism analogous to that mediated by lectin cross-linking (see above) remains a question for further research.
Normally produced at a relatively low rate under physiological conditions, reactive oxygen species are drastically elevated in diabetes (28). One explanation for this is that AGE/RAGE binding, elevated in diabetes, stimulates production of reactive oxygen species (49). Another is that intracellular glucose can by itself increase production of superoxide radicals (1). We have recently found that reactive oxygen species strikingly inhibit repair in skeletal muscle and that prior antioxidant treatment can prevent this (A.C.H., A.K.M., P.L.M., unpublished data). Thus, generation of reactive oxygen species in diabetes, whether by an AGE/RAGE-dependent or -independent mechanism, is a possible downstream mediator of membrane repair failure. Additionally, the high glucose environment induces a variety of alterations in the cell, some of which (such as hyperlipidemia, with consequent changes in membrane lipid composition) might influence the membrane fusion events of repair.
In summary, in vitro and in vivo assays show that the diabetic environment induces in the skeletal muscle myocyte a plasma membrane repair defect. Although it is conceivable that this repair defect could negatively affect muscle health in diabetes, via muscle cell death and resulting atrophy, this conclusion must await further work directly demonstrating the extent, if any, of the connection: the pathogenesis of diabetes is, after all, thought to be complex and multifactorial. However, we have identified for the first time the skeletal muscle myocyte as a cell directly affected in this disease and the molecular players involved in this cellular pathology (AGE/RAGE). Because, as we show here, elevated glucose also induces a repair defect in cell types other than the myocyte, it is possible that defective membrane repair may contribute to other diabetes complications as well.
ACKNOWLEDGMENTS
This work was supported by the Jain Foundation, the Medical College of Georgia, and the National Institutes of Health (R21-DK-090703-01A1). These financial contributors had no role in study design, data collection, and analysis; decision to publish; or preparation of the manuscript.
No potential conflicts of interest relevant to this article were reported.
A.C.H. designed and performed experiments and wrote the manuscript. A.K.M. reviewed and edited the manuscript. F.X. designed the RAGE construct. W.-C.X. contributed the RAGE−/− mice and RAGE construct. P.L.M. contributed to the design of experiments and reviewed and edited the manuscript.
The authors are grateful to Dr. Sylvia Smith and the Diabetes and Obesity Discovery Institute at Georgia Health Sciences University for supplying mice.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db11-0851/-/DC1.
REFERENCES
- 1.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001;414:813–820 [DOI] [PubMed] [Google Scholar]
- 2.Ceriello A. Postprandial hyperglycemia and diabetes complications: is it time to treat? Diabetes 2005;54:1–7 [DOI] [PubMed] [Google Scholar]
- 3.Ramchurn N, Mashamba C, Leitch E, et al. Upper limb musculoskeletal abnormalities and poor metabolic control in diabetes. Eur J Intern Med 2009;20:718–721 [DOI] [PubMed] [Google Scholar]
- 4.Park SW, Goodpaster BH, Lee JS, et al. Excessive loss of skeletal muscle mass in older adults with type 2 diabetes. Diabetes Care 2009;32:1993–1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor BV, Dunne JW. Diabetic amyotrophy progressing to severe quadriparesis. Muscle Nerve 2004;30:505–509 [DOI] [PubMed] [Google Scholar]
- 6.Naderi AS, Farsian FN, Palmer BF. Diabetic muscle necrosis. J Diabetes Complications 2008;22:150–152 [DOI] [PubMed] [Google Scholar]
- 7.Hoyt JR, Wittich CM. Diabetic myonecrosis. J Clin Endocrinol Metab 2008;93:3690. [DOI] [PubMed] [Google Scholar]
- 8.Vignaud A, Ramond F, Hourdé C, Keller A, Butler-Browne G, Ferry A. Diabetes provides an unfavorable environment for muscle mass and function after muscle injury in mice. Pathobiology 2007;74:291–300 [DOI] [PubMed] [Google Scholar]
- 9.Cotter MA, Cameron NE, Robertson S, Ewing I. Polyol pathway-related skeletal muscle contractile and morphological abnormalities in diabetic rats. Exp Physiol 1993;78:139–155 [DOI] [PubMed] [Google Scholar]
- 10.Copray S, Liem R, Brouwer N, Greenhaff P, Habens F, Fernyhough P. Contraction-induced muscle fiber damage is increased in soleus muscle of streptozotocin-diabetic rats and is associated with elevated expression of brain-derived neurotrophic factor mRNA in muscle fibers and activated satellite cells. Exp Neurol 2000;161:597–608 [DOI] [PubMed] [Google Scholar]
- 11.McNeil PL, Khakee R. Disruptions of muscle fiber plasma membranes: role in exercise-induced damage. Am J Pathol 1992;140:1097–1109 [PMC free article] [PubMed] [Google Scholar]
- 12.Steinhardt RA, Bi G, Alderton JM. Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science 1994;263:390–393 [DOI] [PubMed] [Google Scholar]
- 13.Terasaki M, Miyake K, McNeil PL. Large plasma membrane disruptions are rapidly resealed by Ca2+-dependent vesicle-vesicle fusion events. J Cell Biol 1997;139:63–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bi GQ, Alderton JM, Steinhardt RA. Calcium-regulated exocytosis is required for cell membrane resealing. J Cell Biol 1995;131:1747–1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bansal D, Campbell KP. Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol 2004;14:206–213 [DOI] [PubMed] [Google Scholar]
- 16.Bansal D, Miyake K, Vogel SS, et al. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 2003;423:168–172 [DOI] [PubMed] [Google Scholar]
- 17.Constien R, Forde A, Liliensiek B, et al. Characterization of a novel EGFP reporter mouse to monitor Cre recombination as demonstrated by a Tie2 Cre mouse line. Genesis 2001;30:36–44 [DOI] [PubMed] [Google Scholar]
- 18.Süzer O, Köseoğlu S, Ozüner Z. Human albumin enriched St. Thomas Hospital cardioplegic solution increases reperfusion injury in isolated perfused rat hearts. Pharmacol Res 1998;37:97–101 [DOI] [PubMed] [Google Scholar]
- 19.McNeil PL, Miyake K, Vogel SS. The endomembrane requirement for cell surface repair. Proc Natl Acad Sci U S A 2003;100:4592–4597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsuda R, Nishikawa A, Tanaka H. Visualization of dystrophic muscle fibers in mdx mouse by vital staining with Evans blue: evidence of apoptosis in dystrophin-deficient muscle. J Biochem 1995;118:959–964 [DOI] [PubMed] [Google Scholar]
- 21.Lei H, Romeo G, Kazlauskas A. Heat shock protein 90alpha-dependent translocation of annexin II to the surface of endothelial cells modulates plasmin activity in the diabetic rat aorta. Circ Res 2004;94:902–909 [DOI] [PubMed] [Google Scholar]
- 22.Yarrow JC, Perlman ZE, Westwood NJ, Mitchison TJ. A high-throughput cell migration assay using scratch wound healing, a comparison of image-based readout methods. BMC Biotechnol 2004;4:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McNeil PL, Murphy RF, Lanni F, Taylor DL. A method for incorporating macromolecules into adherent cells. J Cell Biol 1984;98:1556–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morgan DM. Tetrazolium (MTT) assay for cellular viability and activity. Methods Mol Biol 1998;79:179–183 [DOI] [PubMed] [Google Scholar]
- 25.Krause MP, Riddell MC, Gordon CS, Imam SA, Cafarelli E, Hawke TJ. Diabetic myopathy differs between Ins2Akita+/- and streptozotocin-induced type 1 diabetic models. J Appl Physiol 2009;106:1650–1659 [DOI] [PubMed] [Google Scholar]
- 26.Krause MP, Moradi J, Nissar AA, Riddell MC, Hawke TJ. Inhibition of plasminogen activator inhibitor-1 restores skeletal muscle regeneration in untreated type 1 diabetic mice. Diabetes 2011;60:1964–1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peyroux J, Sternberg M. Advanced glycation endproducts (AGEs): pharmacological inhibition in diabetes. Pathol Biol (Paris) 2006;54:405–419 [DOI] [PubMed] [Google Scholar]
- 28.Yan SF, Ramasamy R, Schmidt AM. Mechanisms of disease: advanced glycation end-products and their receptor in inflammation and diabetes complications. Nat Clin Pract Endocrinol Metab 2008;4:285–293 [DOI] [PubMed] [Google Scholar]
- 29.Li SY, Sigmon VK, Babcock SA, Ren J. Advanced glycation endproduct induces ROS accumulation, apoptosis, MAP kinase activation and nuclear O-GlcNAcylation in human cardiac myocytes. Life Sci 2007;80:1051–1056 [DOI] [PubMed] [Google Scholar]
- 30.Giardino I, Edelstein D, Brownlee M. Nonenzymatic glycosylation in vitro and in bovine endothelial cells alters basic fibroblast growth factor activity: a model for intracellular glycosylation in diabetes. J Clin Invest 1994;94:110–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brownlee M, Vlassara H, Kooney A, Ulrich P, Cerami A. Aminoguanidine prevents diabetes-induced arterial wall protein cross-linking. Science 1986;232:1629–1632 [DOI] [PubMed] [Google Scholar]
- 32.Hudson BI, Hofmann MA, Bucciarelli L, et al. Glycation and diabetes: the RAGE connection. Curr Sci 2002;83:1515–1521 [Google Scholar]
- 33.Andersen H, Stålberg E, Gjerstad MD, Jakobsen J. Association of muscle strength and electrophysiological measures of reinnervation in diabetic neuropathy. Muscle Nerve 1998;21:1647–1654 [DOI] [PubMed] [Google Scholar]
- 34.McNeil PL, Vogel SS, Miyake K, Terasaki M. Patching plasma membrane disruptions with cytoplasmic membrane. J Cell Sci 2000;113:1891–1902 [DOI] [PubMed] [Google Scholar]
- 35.Togo T, Alderton JM, Bi GQ, Steinhardt RA. The mechanism of facilitated cell membrane resealing. J Cell Sci 1999;112:719–731 [DOI] [PubMed] [Google Scholar]
- 36.Rodríguez A, Webster P, Ortego J, Andrews NW. Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J Cell Biol 1997;137:93–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Borgonovo B, Cocucci E, Racchetti G, Podini P, Bachi A, Meldolesi J. Regulated exocytosis: a novel, widely expressed system. Nat Cell Biol 2002;4:955–962 [DOI] [PubMed] [Google Scholar]
- 38.McNeil A, McNeil PL. Yolk granule tethering: a role in cell resealing and identification of several protein components. J Cell Sci 2005;118:4701–4708 [DOI] [PubMed] [Google Scholar]
- 39.Miyake K, McNeil PL. Vesicle accumulation and exocytosis at sites of plasma membrane disruption. J Cell Biol 1995;131:1737–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jahn R, Scheller RH. SNAREs: engines for membrane fusion. Nat Rev Mol Cell Biol 2006;7:631–643 [DOI] [PubMed] [Google Scholar]
- 41.Chakrabarti S, Kobayashi KS, Flavell RA, et al. Impaired membrane resealing and autoimmune myositis in synaptotagmin VII-deficient mice. J Cell Biol 2003;162:543–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McNeil AK, Rescher U, Gerke V, McNeil PL. Requirement for annexin A1 in plasma membrane repair. J Biol Chem 2006;281:35202–35207 [DOI] [PubMed] [Google Scholar]
- 43.Bement WM, Yu HY, Burkel BM, Vaughan EM, Clark AG. Rehabilitation and the single cell. Curr Opin Cell Biol 2007;19:95–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mellgren RL, Zhang W, Miyake K, McNeil PL. Calpain is required for the rapid, calcium-dependent repair of wounded plasma membrane. J Biol Chem 2007;282:2567–2575 [DOI] [PubMed] [Google Scholar]
- 45.Cai C, Masumiya H, Weisleder N, et al. MG53 nucleates assembly of cell membrane repair machinery. Nat Cell Biol 2009;11:56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miyake K, Tanaka T, McNeil PL. Lectin-based food poisoning: a new mechanism of protein toxicity. PLoS ONE 2007;2:e687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rellier N, Ruggiero D, Lecomte M, Lagarde M, Wiernsperger N. Advanced glycation end products induce specific glycoprotein alterations in retinal microvascular cells. Biochem Biophys Res Commun 1997;235:281–285 [DOI] [PubMed] [Google Scholar]
- 48.Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000;404:787–790 [DOI] [PubMed] [Google Scholar]
- 49.Yan SF, Ramasamy R, Schmidt AM. The RAGE axis: a fundamental mechanism signaling danger to the vulnerable vasculature. Circ Res 2010;106:842–853 [DOI] [PMC free article] [PubMed] [Google Scholar]