

Abstract
OBJECTIVE
To determine the subunit expression and functional activation of phagocyte-like NADPH oxidase (Nox), reactive oxygen species (ROS) generation and caspase-3 activation in the Zucker diabetic fatty (ZDF) rat and diabetic human islets.
RESEARCH DESIGN AND METHODS
Expression of core components of Nox was quantitated by Western blotting and densitometry. ROS levels were quantitated by the 2′,7′-dichlorofluorescein diacetate method. Rac1 activation was quantitated using the gold-labeled immunosorbent assay kit.
RESULTS
Levels of phosphorylated p47phox, active Rac1, Nox activity, ROS generation, Jun NH2-terminal kinase (JNK) 1/2 phosphorylation, and caspase-3 activity were significantly higher in the ZDF islets than the lean control rat islets. Chronic exposure of INS 832/13 cells to glucolipotoxic conditions resulted in increased JNK1/2 phosphorylation and caspase-3 activity; such effects were largely reversed by SP600125, a selective inhibitor of JNK. Incubation of normal human islets with high glucose also increased the activation of Rac1 and Nox. Lastly, in a manner akin to the ZDF diabetic rat islets, Rac1 expression, JNK1/2, and caspase-3 activation were also significantly increased in diabetic human islets.
CONCLUSIONS
We provide the first in vitro and in vivo evidence in support of an accelerated Rac1–Nox–ROS–JNK1/2 signaling pathway in the islet β-cell leading to the onset of mitochondrial dysregulation in diabetes.
Glucose-stimulated insulin secretion (GSIS) involves a series of metabolic and cationic events leading to translocation of insulin granules toward the plasma membrane for fusion and release of insulin into circulation (1–3). Insulin granule transport and fusion involve interplay between vesicle-associated membrane proteins on the insulin granules and docking proteins on the plasma membrane. In addition, a significant cross talk among multiple small G-proteins, including Arf6, Cdc42, and Rac1, was shown to be critical for GSIS (4–6). Several effector proteins for these G-proteins have been identified in the islet β-cell (4,7,8). We recently reported regulatory roles for Rac1 in the activation of phagocyte-like NADPH oxidase (Nox) and generation of reactive oxygen species (ROS) leading to GSIS (9).
Excessive ROS generation is considered central to the development of diabetes complications. The generation of free radicals is relatively low under physiologic conditions; however, increased levels of circulating glucose promote intracellular accumulation of superoxides, leading to cellular dysfunction. Although mitochondria remain the primary source for free radicals, emerging evidence implicates Nox as a major source of extra-mitochondrial ROS. Nox is a highly regulated membrane-associated protein complex that promotes a one-electron reduction of oxygen to superoxide anion involving oxidation of cytosolic NADPH. The Nox holoenzyme consists of membrane and cytosolic components (Fig. 1). The membrane-associated catalytic core consists of gp91phox and p22phox, and the cytosolic regulatory core includes p47phox, p67phox, p40phox, and Rac1. After stimulation, the cytosolic core translocates to the membrane for association with the catalytic core for functional activation of Nox. Immunologic localization and functional regulation of Nox have been described in clonal β-cells and in rat and human islets (10–13).
FIG. 1.

Schematic representation of Nox activation. Nox holoenzyme consists of cytosolic and membrane-associated components. Upon activation, Rac1, guanosine-5′-diphosphate (GDP) is converted to Rac1 guanosine-5′-triphosphate (GTP), which binds to p67phox, and the complex translocates to the membrane. Existing evidence in other cell types suggests that phosphorylation of p47phox also triggers its translocation to the membrane to form the Nox holoenzyme complex that culminates in the enzyme activation and associated increase in ROS.
Recent findings from studies of pharmacologic and molecular biologic approaches suggest that ROS derived from Nox play regulatory “second-messenger” roles in GSIS (9–11,13,14). In addition to the positive modulatory roles for ROS in islet function, recent evidence also implicates negative modulatory roles for ROS in the induction of oxidative stress and metabolic dysregulation of the islet β-cell under the duress of glucolipotoxicity, cytokines, and ceramide (15). The generation of ROS in these experimental conditions is largely due to the activation of Nox, because inhibition of Rac1 or Nox activation markedly attenuated deleterious effects of these stimuli (15–17). Despite this compelling evidence, potential roles of Nox in islet dysfunction in animal models of type 2 diabetes remain unexplored. We therefore undertook the current study to examine the functional status of Nox in islets from the ZDF rat, which develops obesity, hyperinsulinemia, hyperglycemia, and a decline in β-cell function. We present evidence to suggest significant activation of Nox, ROS generation, and caspase-3 activation in the ZDF islets. Our findings also suggest similar metabolic defects in islets from type 2 diabetic human islets.
RESEARCH DESIGN AND METHODS
Materials.
SP600125 and 2′,7′-dichlorofluorescein diacetate (DCHFDA) were from Sigma (St. Louis, MO). Antisera for p47phox and phospho-p47phox were from Santa Cruz Biotechnology (Santa Cruz, CA) and Abcam (Cambridge, MA), respectively. Rac1 antisera and gp91phox were from BD Bioscience (Rockville, MD). Antisera for caspase-3, JNK1/2, and extracellular signal–related kinase (ERK) 1/2 were from Cell Signaling Technology (Boston, MA). The gold-labeled immunosorbent assay (GLISA) Rac1 activation kit was from Cytoskeleton (Denver, CO).
Rodent and human pancreatic islets and INS 832/13 β-cells.
Male (9–11 weeks) ZDF and ZLC rats (Charles River Laboratories, Wilmington, MA) were maintained in a 12-h light/dark cycle with free access to water and food (Purina Diet 5008, Charles River Laboratories). All animal protocols were reviewed and approved by our Institutional Animal Care and Use Committee. Hyperglycemia in diabetic rats was confirmed by tail vein puncture using Glucometer Elite (Bayer, Germany). Body weights of the ZLC and the ZDF rats were 300 ± 6 and 396 ± 12 g, respectively (n = 11; P < 0.05).
Islets were isolated by collagenase digestion method (18). Human islets from normal and diabetic donors were obtained from Prodo Laboratories, Inc. (Irvine, CA). Control islets from a 54-year-old male donor (85–90% purity) and diabetic islets from a 45-year-old male donor (∼60% purity) and a 56-year-old male donor ( 85–90% purity) were homogenized in Tris-HCl (50 mmol/L, pH 7.4) containing sucrose (250 mmol/L), EDTA (1 mmol/L), dithiothreitol (1 mmol/L), and protease inhibitor cocktail and used in this study. INS 832/13 cells (provided by Dr. Chris Newgard) were cultured and processed using previously described protocols (17).
Quantitation of ROS.
Control and diabetic (rodent and human) islets and INS 832/13 cells were incubated with DCHFDA (10 μmol/L) for 30 min in RPMI-1640 media without serum and glucose (9). After incubation, islets were washed with ice-cold phosphate-buffered saline and sonicated. Equal amounts of protein were used for fluorescence measurements (λem 485 nm and λex 535 nm) using a luminescence spectrophotometer (PerkinElmer, Waltham, MA).
Rac1 activation assay.
Activated Rac1 was quantitated using the GLISA activation assay kit according to the manufacturer's instructions. Briefly, lysates were clarified by centrifugation at 14,000 rpm for 2 min. Equal amounts of islet lysate protein were incubated in the Rac1-GTP affinity plate for 30 min at 4°C. The wells were washed twice with washing buffer and incubated with anti-Rac1 primary antibody and secondary antibody, followed by additional incubation with horseradish peroxidase-detection reagent. Horseradish peroxidase-stop buffer was added to stop the reaction, and the absorbance was measured at 490 nm using a microplate reader.
Other assays and statistical analysis of data.
Western blot protein bands were visualized by Kodak Imaging System (Rochester, NY) and analyzed densitometrically using UN-SCAN-IT software (Orem, UT). Statistical significance of differences between control and experimental groups was determined by the Student t test and ANOVA analysis. P < 0.05 was considered significant.
RESULTS
ROS levels and expression and phosphorylation of p47phox are significantly increased in ZDF islets.
The ZDF rats presented a fourfold increase in blood glucose levels compared with age-matched ZLC rats (323 ± 15 vs. 85 ± 1 mg/dL). A significant increase (>60%) in ROS generation was observed in the ZDF rat islets compared with the ZLC islets (Fig. 2A). Because recent evidence indicated a significant increase in Nox-derived ROS generation in isolated β-cells after exposure to high glucose, palmitate, or cytokines (15–17), we next examined Nox as a potential source of increased ROS in the ZDF rat islets.
FIG. 2.

Increased expression and phosphorylation of p47phox and ROS generation in the ZDF rat islets compared with the ZLC islets ROS. Levels were measured in isolated islets from ZLC and ZDF rats after incubation with DCHFDA (10 μmol/L) for 30 min. Islets were washed with ice-cold phosphate-buffered saline and sonicated. An equal amount of protein was used to quantitate 2′,7′-dichlorofluorescin fluorescence. A: Data are expressed as percent control and are mean ± SEM (error bars) from islets from four rats in each group. In a separate experiment, islets from the ZLC or the ZDF rats were lysed using radioimmunoprecipitation assay buffer. Equal amounts of lysate proteins were resolved by SDS-PAGE. Expression of phosphorylated and total p47phox was determined by Western blotting. A representative blot is provided for total p47phox (B) and phospho-p47phox (D). Densitometric quantitation of total p47phox (C) and phosphorylated p47phox (E). Data are mean ± SEM (error bars) from islets from four rats in each group. *P < 0.05 vs. ZLC rat islets.
The Nox holoenzyme consists of cytosolic and membranous components (Fig. 1). Recent evidence also suggests that the cytosolic components require post-translational modifications, including phosphorylation of p47phox and prenylation of Rac1 for optimal holoenzyme assembly (9,19). The expression of p47phox is also significantly increased in isolated β-cells after exposure to high glucose, palmitate, or cytokines (15–17). Therefore, we determined the expression levels and the degree of phosphorylation of p47phox in islets from the control and diabetic rats. Pooled data accrued from multiple islet preparations, as determined by Western blotting and densitometry (Fig. 2B and C), indicated a significant increase (∼40%) in the expression of p47phox in the ZDF islets compared with the ZLC islets. Levels of the phosphorylated p47phox were also significantly higher (∼50%) in the ZDF islets (Fig. 2D and E).
Rac1, a cytosolic component of Nox, is activated in the ZDF islets.
We next quantitated Rac1 expression and activation in the ZLC and the ZDF islets. The underlying premise here is that an increase in the Nox-derived ROS generation in the diabetic islets (Fig. 2A) requires activation of Rac1. Data showed a marked increase (>60%) in the expression of Rac1 in the diabetic islets compared with the control islets (Fig. 3A). Further, the abundance of the activated Rac1 is significantly higher (∼2.25-fold) in the diabetic islets than in the control islets (Fig. 3B). The observed increase in Rac1 activation (Fig. 3C) may not be a reflection of increased Rac1 expression in the ZDF islets (Fig. 3A) because the ratio of activated to total Rac1 also indicated a significant increase (>40%) in the diabetic rat islets compared with the control islets (Fig. 3D). Together the data (Figs. 2 and 3) indicate an increase in the phosphorylation status of p47phox and activation of Rac1 in the ZDF islets, which are required for holoenzyme assembly and activation of Nox and subsequent increase in ROS generation (Fig. 2A).
FIG. 3.

Expression and activation of Rac1 are significantly increased in ZDF rat islets. Total Rac1 expression in islets from the ZLC and the ZDF rats was determined by Western blotting (A) and quantitated densitometrically (B). C: The degree of Rac1 activation was quantitated by the GLISA method. D: Data are expressed as percent change in Rac1 activation over total Rac1 and are mean ± SEM (error bars) from islets from six rats in each group. *P < 0.05 vs. ZLC rat islets.
Increased expression of gp91phox in the ZDF islets.
Numerous studies have focused on potential alterations in the expression of the cytosolic components of Nox in β-cells under the duress of glucolipotoxicity and cytokines (16,17,19,20); however, relatively little is known about alterations in the expression of the membrane components of Nox under such conditions. We therefore quantitated expression levels of gp91phox in islets from the ZLC or the ZDF rats and noticed an increase in the expression of the gp91phox subunit in the ZDF islets (Fig. 4A). Densitometric quantitation of protein bands indicated an increase of >40% in gp91phox expression in the ZDF islets (Fig. 4B), thus supporting the overall hypothesis that an increase in the intracellular ROS in diabetic islet may be partly due to increased activation of Nox via an increase in the expression and phosphorylation of individual subunits.
FIG. 4.

Increased expression of gp91phox and caspase-3 activation in the ZDF rat islets. A: Lysates derived from control and diabetic rats were used for the determination of expression of gp91phox by Western blotting. β-Actin was used as loading control. B: The protein bands were analyzed densitometrically, expressed as percent increase over lean control. Data are mean ± SEM (error bars) from islet preparations from five rats in each group. *P < 0.05 vs. ZLC islets. In a separate set of studies, islet lysates from the ZLC and the ZDF rats were resolved by SDS-PAGE and immunoprobed for caspase-3 activation. β-Actin was used as loading control. C: A representative blot from three independent experiments yielding similar results is shown. D: The density of the procaspase and its hydrolytic product-bands was quantitated and expressed as percent control. Data are mean ± SEM (error bars) from islet lysates from three rats in each group. *P < 0.05 vs. procaspase values of lean control. **P < 0.05 vs. caspase cleavage product of ZLC islets.
Assessment of mitochondrial dysregulation in the ZDF islets.
Using in vitro models systems of glucolipotoxicity or cytokine exposure, we have recently proposed that Nox activation leads to loss of mitochondrial membrane potential and caspase-3 activation (16,17). We also reported that inhibition of Rac1 activation by using NSC23776 to attenuate the function of T-cell lymphoma invasion and metastasis–inducing protein 1 (Tiam1), a guanine nucleotide exchange factor for Rac1, or using GGTI-2147 to inhibit prenylation of Rac1, leads to partial restoration of mitochondrial dysfunction induced by a mixture of cytokines (16). We therefore quantitated caspase-3 activation in the control and diabetic rat islets. Our findings (Fig. 4C and D) indicated a significant activation of caspase-3 in islets from the ZDF but not from the ZLC islets. These data are suggestive of mitochondrial defects in the ZDF islets at an age where significant changes in Nox activation are observed (see above).
Differential regulation of JNK1/2 and ERK1/2 in the ZDF islets.
Stress-activated JNK activation lies upstream to caspase-3 activation (21). Further, constitutive activation of Rac1 promotes JNK phosphorylation and activation (22,23). Existing evidence also implicates significant cross talk between ROS and JNK1/2 (24). Therefore, we quantitated the phosphorylation status of JNK1/2 in islets from the ZLC and ZDF rats. Western blot analysis of lysates from the control and diabetic rats indicated consistently higher levels of phosphorylated JNK1 and JNK2 in ZDF rat islets (Fig. 5A). The ratios of phosphorylated to total JNK1 and JNK2, determined by densitometric quantitation of the protein bands (Fig. 5B) indicated a significant increase (>60%) in the phosphorylation of JNK1/2 in the diabetic islets.
FIG. 5.
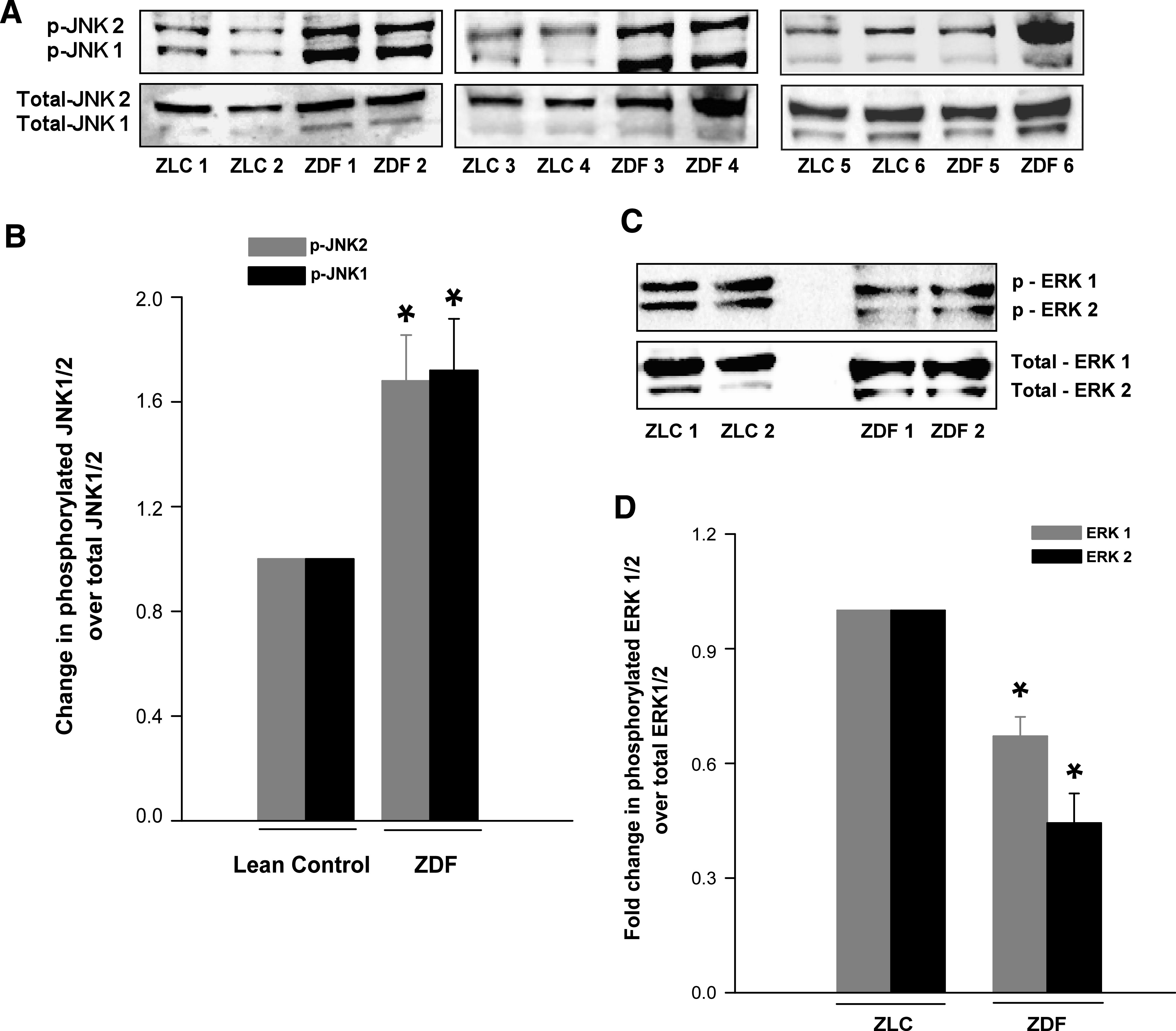
Phosphorylation of JNK1/2 and ERK1/2 in the ZLC or the ZDF rat islets. Islet lysates from control and diabetic rats were prepared in RIPA buffer. A: Total and phospho-JNK1/2 were determined by Western blotting and analyzed densitometrically. B: Data are expressed as fold change in phosphorylation over total JNK1/2. Data are mean ± SEM (error bars) from islet lysates derived from six rats in each group. *P < 0.05 vs. the ZLC islets. Lysates of islets from control and diabetic rats were prepared in radioimmunoprecipitation assay buffer. An equal amount of lysate protein was resolved by SDS-PAGE. Relative abundance of total and phospho-ERK1/2 were determined by Western blotting (C), followed by densitometry (D). Data are expressed as fold change in phosphorylation over total ERK1/2 and are mean ± SEM (error bars) from islets from six rats in each group. *P < 0.05 vs. ZLC islets.
We next quantitated ERK1/2 phosphorylation in the ZLC and the ZDF islets to further determine if diabetic conditions elicit regulatory effects on activation of this enzyme because it has been implicated in islet β-cell function at multiple levels, including insulin gene expression, GSIS, and β-cell proliferation (25,26). ERK1/2 phosphorylation in the ZDF islets was significantly attenuated compared with the control islets (Fig. 5C and D). Together, these findings (Fig. 5) suggest differential regulation of JNK1/2 and ERK1/2 in diabetic islets, conditions that might favor proapoptotic and nonproliferative events in the diabetic islets. Our recently published observations on increased Nox activity in β-cells under the duress of glucolipotoxic conditions (17) and our current observations in the ZDF islets led us to hypothesize that glucolipotoxic distress may elicit such dual regulatory effects on JNK1/2 and ERK1/2 phosphorylation and activation. This hypothesis was further tested in clonal β-cells via studies described in the next section.
In vitro exposure to high glucose or palmitate exerts differential effects on JNK1/2 and ERK1/2.
To assess if glucotoxicity or lipotoxicity are responsible for the differential regulatory effects on JNK1/2 and ERK1/2 seen in the ZDF islets, INS 832/13 cells were incubated for 48 h with high glucose (20 mmol/L) or palmitate (400 μmol/L), and the relative abundance of total and phospho JNK1/2 and ERK1/2 was determined by Western blotting, followed by densitometry. Pooled data (Fig. 6A) indicated a marked increase (∼40–87%) in JNK1 and JNK2 phosphorylation in β-cells treated with high glucose (lanes 3 and 4) or palmitate (∼30–34%; lanes 5 and 6) compared with their levels under basal conditions (lanes 1 and 2). However, total levels of JNK1/2 remained unchanged under these conditions. We also observed a significant reduction in ERK1/2 phosphorylation in INS 832/13 cells treated with high glucose (∼22–48%) or palmitate (∼60%); but these conditions did not affect the abundance of total ERK1/2 (Fig. 6B). Together, these in vitro findings in INS 832/13 cells are compatible to those observed in the ZDF islets (Fig. 5) and suggest differential regulation of JNK1/2 and ERK1/2 under the duress of glucotoxic or lipotoxic conditions.
FIG. 6.
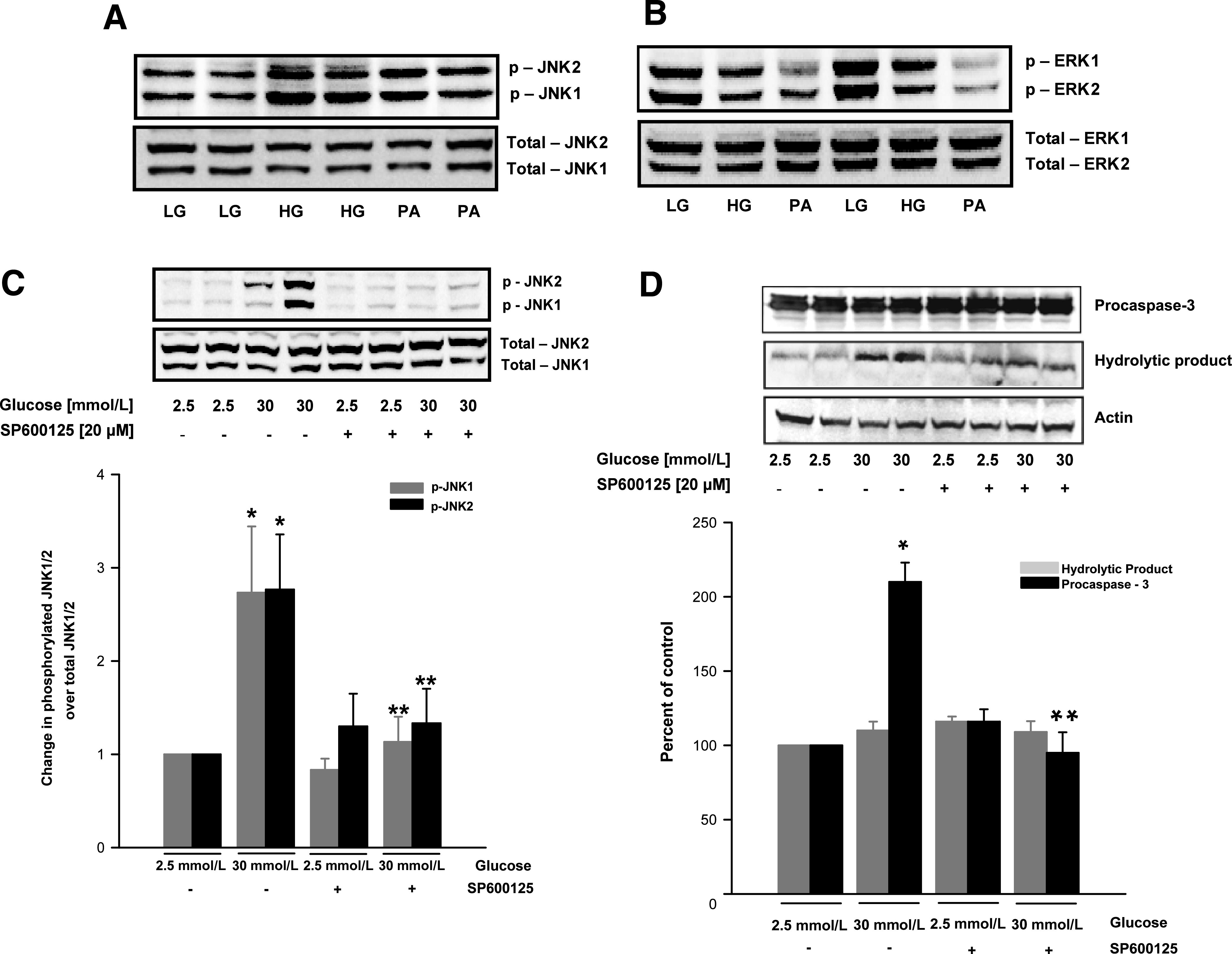
Glucotoxic or lipotoxic conditions differentially regulate JNK1/2 and ERK1/2 and mitochondrial dysfunction in INS 832/13 pancreatic β-cells. INS 832/13 cells were cultured in the presence of low glucose (LG; 2.5 mmol/L), high glucose (HG; 20 mmol/L), or palmitate (PA; 400 μmol/L) for 48 h. At the end of incubation, cells were lysed and the expression of total and phosphorylated JNK1/2 (A) and ERK1/2 (B) was determined by Western blotting. In a separate set of studies, INS 832/13 cells were incubated with glucose (30 mmol/L) with or without SP600125 (20 μmol/L) for 24 h. Cell lysates were prepared in radioimmunoprecipitation assay buffer for Western blot analysis to determine the degree of JNK1/2 (C) and caspase-3 activation (D). Data were quantitated densitometrically and are expressed as mean ± SEM (error bars) from three independent experiments. *P < 0.05 vs. 2.5 mmol/L glucose; **P < 0.05 vs. 30 mmol/L glucose alone.
To verify if inhibition of JNK1/2 phosphorylation would restore high glucose-induced caspase-3 activation, INS 832/13 cells were cultured under basal or high glucose (30 mmol/L) conditions in the absence or presence of SP600125 (20 μmol/L; 24 h), a selective inhibitor of JNK1/2. Degrees of JNK1/2 and caspase-3 activation were determined by Western blotting, followed by densitometry. There was a marked attenuation, by SP600125, of high glucose-induced JNK1/2 phosphorylation (Fig. 6C), and caspase-3 activation (Fig. 6D). These data implicate JNK1/2 activation is upstream to caspase-3 activation seen under the duress of glucotoxicity.
Regulation of Nox in human islets.
We next studied regulation of Nox under glucotoxic conditions in human islets. At the outset, ROS generation and Rac1 activation were quantitated in normal human islets incubated with glucose (5.8 or 30 mmol/L) for 48 h. The data indicated a ∼2.2-fold increase in ROS generation in human islets after incubation with high glucose (Fig. 7A). These data were compatible with our observations in INS 832/13 cells and normal rat islets (9) and ZDF rat islets (current studies). Incubation of human islets with high glucose resulted in a significant (∼1.5-fold) activation of Rac1 (Fig. 7B). A consistent increase in Rac1 expression, JNK1/2 activation and caspase-3 degradation were also demonstrated in type 2 diabetic human islets (Fig. 7C, D, and F, respectively), findings compatible with data in the ZDF islets. However, the levels of phosphorylated or total p47phox and gp91phox (Fig. 7C and E, respectively) were comparable between normal and diabetic human islets. Limited availability of diabetic human islets precluded us from quantitation of Nox and Rac1 activities. Nonetheless, our preliminary findings in human islets support our current findings in the diabetic ZDF islets or in INS 832/13 cells after exposure to glucolipotoxic conditions.
FIG. 7.

Regulation of Nox in human islets. Normal human islets were cultured in PMI medium in the presence of 5.8 or 30 mmol/L glucose for 48 h. A: Generation of ROS (mean ± SEM from triplicate measurements) was quantitated by 2′,7′-dichlorofluorescin fluorescence. B: Rac1 activation (mean ± variance from two determinations) was quantitated by GLISA. *P < 0.05 vs. 5.8 mmol/L glucose. In a separate set of studies, islets derived from control or diabetic human donors were lysed in RIPA buffer, and lysate proteins were resolved by SDS-PAGE. The expression of total Rac1 and gp91phox (C), phosphorylated and total JNK1/2 (D), phosphorylated and total p47phox (E), and caspase-3 (F) were determined by Western blotting. Corresponding housekeeping genes were also measured in parallel to confirm equal loading.
DISCUSSION
Existing evidence in multiple cell types, including the pancreatic β-cell, implicates post-translational phosphorylation and prenylation of individual components as a requisite for the optimal activation of Nox (9,19). The main objective of the current study was to determine the functional status of Nox in islets derived from the ZDF rat, a well-studied model for obesity and type 2 diabetes, and to determine potential regulation of Nox components in human islets under the duress of glucolipotoxicity and diabetes. Our data suggested a significant activation of Nox and associated ROS generation in the ZDF islets compared with those derived from the ZLC islets. Lastly, our data in diabetic human islets corroborated our findings in the ZDF islets.
Several recent studies have demonstrated activation of Nox after exposure to physiologic concentrations of glucose in a variety of insulin-secreting cells (10–13). Data from studies of pharmacologic and molecular biologic inhibition of Nox revealed that a tonic increase in Nox-derived ROS is necessary for GSIS (10,20) and that prenylation of Rac1 appears to be necessary for glucose-induced Nox activation and ROS generation in isolated β-cells (9). Emerging evidence also implicates Nox in metabolic dysfunction of the islet β-cell under conditions of glucolipotoxicity and exposure to cytokines (16,17). These studies demonstrated an increase in the expression and phosphorylation of Nox subunits (i.e., p47phox), together with significant activation of Rac1. In addition, the activation status of Rac1 was under the precise control of Tiam1, a guanine nucleotide exchange factor for Rac1 in β-cells (27). In further support of this, we reported a marked reduction in high glucose-, high palmitate-, and cytokine-induced Rac1 and Nox activation and ROS generation in isolated β-cells after exposure to NSC23766, a selective inhibitor of the Tiam1/Rac1 signaling axis (16,17). Taken together, previous in vitro findings implicated participatory roles of Nox in exerting effects at the mitochondrial level, including loss in membrane potential, cytochrome C release, and activation of caspase-3 culminating in islet β-cell dysfunction (16).
In addition to an increase in p47phox and gp91phox expression, Rac1 activation, and ROS generation, we observed a significant increase in the phosphorylation of JNK1/2 in the ZDF islets compared with the control islets. Similar changes in the activation of JNK1/2 were demonstrable in INS 832/13 cells after incubation with high glucose or palmitate. Selective inhibition of JNK1/2 using SP600125 markedly attenuated caspase-3 activation under glucotoxic conditions, suggesting that JNK1/2 activation lies upstream to mitochondrial dysfunction and caspase-3 activation. These data are in accord with findings of Cunha et al. (28) demonstrating significant inhibition of palmitate-induced JNK activation and cell apoptosis in INS-1E cells by SP600125 and L-TAT-JNKi, a small peptide inhibitor of JNK. Along these lines, several recent studies have also demonstrated inhibition of caspase-3 activation after inhibition of JNK1/2 activation in models of cellular apoptosis (21,29–31).
The observed reduction of ERK1/2 activation under glucolipotoxic conditions in the ZDF rat islets in vivo and in the INS 832/13 cells in vitro are indicative of impaired metabolic function and β-cell proliferation. Our current findings on reduction in ERK1/2 phosphorylation in INS 832/13 cells are in accord with studies of Costes et al. (32), who demonstrated a significant reduction in ERK1/2 phosphorylation in MIN6 cells after exposure to 25 mmol/L glucose for 24 h. From the results of further studies, these investigators concluded that glucotoxic conditions downregulate the ERK1/2-cAMP-responsive element–binding protein-signaling pathway, leading to the apoptotic demise of the β-cell.
Recent studies by Zhang et al. (33) demonstrated a significant increase in JNK1/2 phosphorylation and reduction in ERK1/2 phosphorylation during mevastatin-induced apoptosis of salivary adenoid carcinoma cells, suggesting a potential inverse relationship between JNK1/2 and ERK1/2 phosphorylation in the induction of cellular apoptosis.
Together, our observations in INS 832/13 cells, ZDF islets, and diabetic human islets support involvement of the Nox–ROS stress-activated signaling axis in the metabolic dysfunction; however, additional studies are needed to substantiate this formulation. Recent studies by Nakayama et al. (34) demonstrated the functional activation of Nox in islets of db/db mice and in Otsuka Long-Evans Tokushima Fatty rats. Treatment of these animals with angiotensin II type-1 receptor antagonists reduced Nox activation and oxidative stress. It may be germane to point out that Valle et al. (35) recently examined potential changes in Nox in islets derived from obese animals fed a high-fat diet. In contrast to islets from db/db mice, Otsuka Long-Evans Tokushima Fatty rats (34), and ZDF rat (current study), islets from animals fed a high-fat diet exhibited markedly lower expression levels of p47phox and gp91phox subunits and ROS production compared with control rat islets. These investigators attributed this toward increased glucose oxidation and GSIS seen in islets from animals fed a high-fat diet in response to glucose (35).
On the basis of the existing information and our current findings, we propose the following model for Nox-mediated induction of β-cell dysfunction in diabetes (Fig. 8): Exposure of isolated β-cells to glucolipotoxic conditions or islets derived from the diabetic condition in ZDF rats or humans results in increased activation of Rac1 and Nox. Consequential generation of ROS and the associated oxidative stress, in turn, promote activation of JNK1/2 and mitochondrial dysregulation. Alternatively, activation of the cytosolic Nox–ROS–JNK1/2 signaling pathway increases superoxide generation that impairs the functional efficiency of mitochondria. This proposal is supported by findings of Bindokas et al. (36) that demonstrated excessive superoxide levels in islet mitochondria from the ZDF rat.
FIG. 8.

Proposed model for Nox-induced ROS-mediated mitochondrial dysregulation in diabetes. Based on the data accrued from the current studies, we propose a model for the Nox–ROS–JNK signaling in the metabolic dysfunction of the pancreatic β-cell under the duress of hyperglycemia and hyperlipidemia. Glucotoxicity or lipotoxicity induces Nox activation by promoting the phosphorylation of p47phox and Rac1 activation. We have recently demonstrated that inhibition of Rac1 activation by NSC23766, or prenylation inhibitors, attenuates high glucose- or palmitate-induced Nox activation and ROS generation (15,17). Likewise, inhibition of Nox action by apocynin, diphenylene iodonium, or siRNA-p47phox alleviates ROS generation and oxidative stress under the duress of high glucose, high palmitate, or cytokines (15–17). Nox activation and excessive ROS generation leads to the activation of stress-activated kinases (JNK1/2), culminating in mitochondrial dysfunction and caspase-3 activation. In support of this formulation, our current studies using SP600125 demonstrated significant inhibition in glucose-induced JNK1/2 phosphorylation and caspase-3 activation. On the basis of these data, we propose that the collective effects of Tiam1-mediated Rac1 activation, p47phox phosphorylation, Nox holoenzyme assembly, and associated ROS generation, followed by inhibition of ERK1/2 and activation of JNK1/2, result in mitochondrial dysregulation and caspase-3 activation leading to the islet β-cell dysfunction and demise in diabetes. DPI, diphenylene iodonium; siRNA, short interfering RNA; T2DM, type 2 diabetes mellitus.
In summary, our current findings implicate Nox as one of the sources of oxidative stress in the diabetic islet. It will be interesting to determine if pharmacologic intervention of Nox activation seen in islets under diabetic conditions can be restored to its normal function. Such intervention modalities include NSC23766, a selective inhibitor of Tiam1/Rac1, which we have used in in vitro experiments to restore mitochondrial function in β-cells exposed to elevated glucose, lipids, and cytokines (16,17). In this context, recent investigations have successfully used NSC23766, a selective inhibitor of the Tiam1–Rac1 signaling axis, to correct Nox-mediated effects on cellular function in vitro and in vivo (15). Using the streptozotocin diabetic mouse model, Shen et al. (37) demonstrated a regulatory role for Rac1 in hyperglycemia-induced apoptosis in cardiomyocytes. They demonstrated that upregulation of Rac1, Nox activity, and increased ROS generation led to apoptosis of cardiomyocytes under the duress of hyperglycemia. Treatment of diabetic db/db mice with NSC23766 significantly inhibited Nox activity and cell apoptosis (37). Additional studies are needed to pinpoint the regulatory roles of Tiam1–Rac1–Nox–ROS signaling in the metabolic dysfunction in the diabetic islet.
ACKNOWLEDGMENTS
This research was supported by a Merit Review Award from the Department of Veterans Affairs and by grants from the National Institutes of Health to C.J.R. (DK-55267) and to A.K. (DK-74921). A.K. is the recipient of the Senior Research Career Scientist Award from the Department of Veterans Affairs.
No potential conflicts of interest relevant to this article were reported.
I.S., C.N.K., B.J., and S.G. conducted the experimental work. C.J.R. and R.A.K. planned the studies and reviewed the manuscript. A.K. reviewed the literature, planned the studies, supervised experimental work, and wrote and revised the manuscript.
REFERENCES
- 1.MacDonald MJ. Elusive proximal signals of β-cells for insulin secretion. Diabetes 1990;39:1461–1466 [DOI] [PubMed] [Google Scholar]
- 2.Newgard CB, Lu D, Jensen MV, et al. Stimulus/secretion coupling factors in glucose-stimulated insulin secretion: insights gained from a multidisciplinary approach. Diabetes 2002;51(Suppl. 3):S389–S393 [DOI] [PubMed] [Google Scholar]
- 3.Prentki M, Matschinsky FM. Ca2+, cAMP, and phospholipid-derived messengers in coupling mechanisms of insulin secretion. Physiol Rev 1987;67:1185–1248 [DOI] [PubMed] [Google Scholar]
- 4.Kowluru A. Small G proteins in islet beta-cell function. Endocr Rev 2010;31:52–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lawrence JT, Birnbaum MJ. ADP-ribosylation factor 6 regulates insulin secretion through plasma membrane phosphatidylinositol 4,5-bisphosphate. Proc Natl Acad Sci USA 2003;100:13320–13325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jayaram B, Syed I, Kyathanahalli CN, Rhodes CJ, Kowluru A. Arf nucleotide binding site opener (ARNO) promotes sequential activation of Arf6, Cdc42 and Rac1 and insulin secretion in INS 832/13 β-cells and rat islets. Biochem Pharmacol 2011;81:1016–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takai Y, Sasaki T, Matozaki T. Small GTP-binding proteins. Physiol Rev 2001;81:153–208 [DOI] [PubMed] [Google Scholar]
- 8.Wang Z, Thurmond DC. Mechanisms of biphasic insulin-granule exocytosis - roles of the cytoskeleton, small GTPases and SNARE proteins. J Cell Sci 2009;122:893–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Syed I, Kyathanahalli CN, Kowluru A. Phagocyte-like NADPH oxidase generates ROS in INS 832/13 cells and rat islets: role of protein prenylation. Am J Physiol Regul Integr Comp Physiol 2011;300:R756–R762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morgan D, Rebelato E, Abdulkader F, et al. Association of NAD(P)H oxidase with glucose-induced insulin secretion by pancreatic beta-cells. Endocrinology 2009;150:2197–2201 [DOI] [PubMed] [Google Scholar]
- 11.Oliveira HR, Verlengia R, Carvalho CR, Britto LR, Curi R, Carpinelli AR. Pancreatic beta-cells express phagocyte-like NAD(P)H oxidase. Diabetes 2003;52:1457–1463 [DOI] [PubMed] [Google Scholar]
- 12.Uchizono Y, Takeya R, Iwase M, et al. Expression of isoforms of NADPH oxidase components in rat pancreatic islets. Life Sci 2006;80:133–139 [DOI] [PubMed] [Google Scholar]
- 13.Graciano MF, Santos LR, Curi R, Carpinelli AR. NAD(P)H oxidase participates in the palmitate-induced superoxide production and insulin secretion by rat pancreatic islets. J Cell Physiol 2011;226:1110–1117 [DOI] [PubMed] [Google Scholar]
- 14.Pi J, Collins S. Reactive oxygen species and uncoupling protein 2 in pancreatic β-cell function. Diabetes Obes Metab 2010;12(Suppl. 2):141–148 [DOI] [PubMed] [Google Scholar]
- 15.Kowluru A. Friendly, and not so friendly, roles of Rac1 in islet β-cell function: lessons learnt from pharmacological and molecular biological approaches. Biochem Pharmacol 2011;81:965–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Subasinghe W, Syed I, Kowluru A. Phagocyte-like NADPH oxidase promotes cytokine-induced mitochondrial dysfunction in pancreatic β-cells: evidence for regulation by Rac1. Am J Physiol Regul Integr Comp Physiol 2011;300:R12–R20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Syed I, Jayaram B, Subasinghe W, Kowluru A. Tiam1/Rac1 signaling pathway mediates palmitate-induced, ceramide-sensitive generation of superoxides and lipid peroxides and the loss of mitochondrial membrane potential in pancreatic beta-cells. Biochem Pharmacol 2010;80:874–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kowluru A, Veluthakal R. Rho guanosine diphosphate-dissociation inhibitor plays a negative modulatory role in glucose-stimulated insulin secretion. Diabetes 2005;54:3523–3529 [DOI] [PubMed] [Google Scholar]
- 19.Hirata AE, Morgan D, Oliveira-Emilio HR, et al. Angiotensin II induces superoxide generation via NAD(P)H oxidase activation in isolated rat pancreatic islets. Regul Pept 2009;153:1–6 [DOI] [PubMed] [Google Scholar]
- 20.Morgan D, Oliveira-Emilio HR, Keane D, et al. Glucose, palmitate and pro-inflammatory cytokines modulate production and activity of a phagocyte-like NADPH oxidase in rat pancreatic islets and a clonal beta cell line. Diabetologia 2007;50:359–369 [DOI] [PubMed] [Google Scholar]
- 21.Kim BC, Kim HG, Lee SA, et al. Genipin-induced apoptosis in hepatoma cells is mediated by reactive oxygen species/c-Jun NH2-terminal kinase-dependent activation of mitochondrial pathway. Biochem Pharmacol 2005;70:1398–1407 [DOI] [PubMed] [Google Scholar]
- 22.Minden A, Lin A, Claret FX, Abo A, Karin M. Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell 1995;81:1147–1157 [DOI] [PubMed] [Google Scholar]
- 23.Shen YH, Godlewski J, Zhu J, et al. Cross-talk between JNK/SAPK and ERK/MAPK pathways: sustained activation of JNK blocks ERK activation by mitogenic factors. J Biol Chem 2003;278:26715–26721 [DOI] [PubMed] [Google Scholar]
- 24.Ohashi N, Urushihara M, Satou R, Kobori H. Glomerular angiotensinogen is induced in mesangial cells in diabetic rats via reactive oxygen species—ERK/JNK pathways. Hypertens Res 2010;33:1174–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kowluru A, Veluthakal R, Rhodes CJ, Kamath V, Syed I, Koch BJ. Protein farnesylation-dependent Raf/extracellular signal-related kinase signaling links to cytoskeletal remodeling to facilitate glucose-induced insulin secretion in pancreatic beta-cells. Diabetes 2010;59:967–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fontés G, Semache M, Hagman DK, et al. Involvement of Per-Arnt-Sim Kinase and extracellular-regulated kinases-1/2 in palmitate inhibition of insulin gene expression in pancreatic beta-cells. Diabetes 2009;58:2048–2058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Veluthakal R, Madathilparambil SV, McDonald P, Olson LK, Kowluru A. Regulatory roles for Tiam1, a guanine nucleotide exchange factor for Rac1, in glucose-stimulated insulin secretion in pancreatic beta-cells. Biochem Pharmacol 2009;77:101–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cunha DA, Hekerman P, Ladrière L, et al. Initiation and execution of lipotoxic ER stress in pancreatic β-cells. J Cell Sci 2008;121:2308–2318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramiro-Cortés Y, Morán J. Role of oxidative stress and JNK pathway in apoptotic death induced by potassium deprivation and staurosporine in cerebellar granule neurons. Neurochem Int 2009;55:581–592 [DOI] [PubMed] [Google Scholar]
- 30.Ramin M, Azizi P, Motamedi F, Haghparast A, Khodagholi F. Inhibition of JNK phosphorylation reverses memory deficit induced by β-amyloid (1-42) associated with decrease of apoptotic factors. Behav Brain Res 2011;217:424–431 [DOI] [PubMed] [Google Scholar]
- 31.Kuo WW, Wang WJ, Lin CW, Pai P, Lai TY, Tsai CY. NADPH oxidase-derived superoxide anion-induced apoptosis is mediated via the JNK-dependent activation of NF-κB in cardiomyocytes exposed to high glucose. J Cell Physiol. 20 May 2011 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 32.Costes S, Longuet C, Broca C, et al. Cooperative effects between protein kinase A and p44/p42 mitogen-activated protein kinase to promote cAMP-responsive element binding protein activation after beta cell stimulation by glucose and its alteration due to glucotoxicity. Ann N Y Acad Sci 2004;1030:230–242 [DOI] [PubMed] [Google Scholar]
- 33.Zhang S, Wang XL, Gan YH, Li SL. Activation of c-Jun N-terminal kinase is required for mevastatin-induced apoptosis of salivary adenoid cystic carcinoma cells. Anticancer Drugs 2010;21:678–686 [PubMed] [Google Scholar]
- 34.Nakayama M, Inoguchi T, Sonta T, et al. Increased expression of NAD(P)H oxidase in islets of animal models of Type 2 diabetes and its improvement by an AT1 receptor antagonist. Biochem Biophys Res Commun 2005;332:927–933 [DOI] [PubMed] [Google Scholar]
- 35.Valle MM, Graciano MF, Lopes de Oliveira ER, et al. Alterations of NADPH oxidase activity in rat pancreatic islets induced by a high-fat diet. Pancreas 2011;40:390–395 [DOI] [PubMed] [Google Scholar]
- 36.Bindokas VP, Kuznetsov A, Sreenan S, Polonsky KS, Roe MW, Philipson LH. Visualizing superoxide production in normal and diabetic rat islets of Langerhans. J Biol Chem 2003;278:9796–9801 [DOI] [PubMed] [Google Scholar]
- 37.Shen E, Li Y, Li Y, et al. Rac1 is required for cardiomyocyte apoptosis during hyperglycemia. Diabetes 2009;58:2386–2395 [DOI] [PMC free article] [PubMed] [Google Scholar]