

Abstract
OBJECTIVE
Previous studies show that polyunsaturated fatty acids (PUFAs) increase the insulin secretory capacity of pancreatic β-cells. We aimed at identifying PUFA-derived mediators and their cellular targets that are involved in the amplification of insulin release from β-cells preexposed to high glucose levels.
RESEARCH DESIGN AND METHODS
The content of fatty acids in phospholipids of INS-1E β-cells was determined by lipidomics analysis. High-performance liquid chromatography was used to identify peroxidation products in β-cell cultures. Static and dynamic glucose-stimulated insulin secretion (GSIS) assays were performed on isolated rat islets and/or INS-1E cells. The function of peroxisome proliferator–activated receptor-δ (PPAR-δ) in regulating insulin secretion was investigated using pharmacological agents and gene expression manipulations.
RESULTS
High glucose activated cPLA2 and, subsequently, the hydrolysis of arachidonic and linoleic acid (AA and LA, respectively) from phospholipids in INS-1E cells. Glucose also increased the level of reactive oxygen species, which promoted the peroxidation of these PUFAs to generate 4-hydroxy-2E-nonenal (4-HNE). The latter mimicked the GSIS-amplifying effect of high glucose preexposure and of the PPAR-δ agonist GW501516 in INS-1E cells and isolated rat islets. These effects were blocked with GSK0660, a selective PPAR-δ antagonist, and the antioxidant N-acetylcysteine or by silencing PPAR-δ expression. High glucose, 4-HNE, and GW501516 also induced luciferase expression in a PPAR-δ–mediated transactivation assay. Cytotoxic effects of 4-HNE were observed only above the physiologically effective concentration range.
CONCLUSIONS
Elevated glucose levels augment the release of AA and LA from phospholipids and their peroxidation to 4-HNE in β-cells. This molecule is an endogenous ligand for PPAR-δ, which amplifies insulin secretion in β-cells.
The effect of glucose on β-cell function depends on its concentration and duration of exposure (1). An acute exposure to high glucose triggers the classical biphasic insulin secretion as a result of membrane depolarization and enhanced exocytosis of insulin granules. Chronic hyperglycemia, however, induces β-cell dysfunction as a result of excessive generation of glucose-derived reactive oxygen species (ROS) and accumulation of glycated proteins, which induce endoplasmic reticulum stress and apoptosis. In addition, intermittent or intermediate periods of hyperglycemia influence β-cells differently; a prior exposure of human subjects and animals to high glucose activates “priming” or “memory” pathways that amplify insulin release in comparison with naïve cells (2–4). The amplification of insulin release was described in the compensatory and adaptive phases of type 2 diabetes (1,5).
The hypothesis that lipid mediators are involved in this adaptive stage as a result of their amplifying effects on insulin secretion has gained experimental support (6,7). Specifically, the nonesterified polyunsaturated fatty acids (PUFAs), arachidonic acid (AA) and linoleic acid (LA), per se and/or their cyclooxygenase- and lipoxygenase (LO)-derived metabolites were suggested to be such mediators (6,8–10). The idea that phospholipases regulate insulin secretion was introduced by Metz (11) and Dunlop and Larkins (12). Others have reported that high glucose increased the expression of and activated some PLA2 isotypes, which release fatty acids from the sn-2 position in phospholipids (13).
Of interest is the finding that ROS, generated in β-cells under high glucose conditions, also function as insulinotropic signals (14). Among their multiple cellular functions, ROS catalyze the peroxidation of PUFAs and their LO-derived hydroperoxy metabolites by multiple nonenzymatic pathways and initiate cascade reactions yielding reactive aldehyde products, known as 4-hydroxyalkenals (15,16). The peroxidation of AA and LA and of their respective 12- and 15-LO–derived hydroperoxy metabolites results in the generation of 4-hydroxy-2E-nonenal (4-HNE) and/or 4-hydroxy-2E, 6Z-dodecadienal (4-HDDE) (15). Moreover, their formation is enhanced in diabetes as a result of a decrease of the glutathione peroxidase activity (15). Of interest, hyperglycemia promotes the production of these two compounds in diabetic patients and animal models of diabetes (17–19). These 4-hydroxyalkenals exert dual dose-dependent effects in cells; at high levels, they are cytotoxic because of their inherent tendency to form covalent adducts with proteins, DNA, and phospholipids (20). Yet at physiological, nontoxic levels, they act as signaling molecules. Coleman et al. (21) and Riahi et al. (18) have recently found that 4-HNE and 4-HDDE are selective endogenous ligands for peroxisome proliferator–activated receptor-δ (PPAR-δ). Recent reports suggest a role of PPAR-δ in regulating β-cell function; Winzell et al. (22) reported that prolonged treatment of diabetic db/db mice with a PPAR-δ agonist reduced blood glucose levels in association with improved insulin sensitivity and pancreatic islet function. Furthermore, Ravnskjaer et al. (23) attributed to PPAR-δ a fatty acid–sensor role, improving insulin secretion in β-cells.
The current study shows increased 4-HNE levels in β-cells exposed to high glucose, coupled to a marked release of AA and LA from membrane phospholipids. This lipid peroxidation product of AA and LA functions as an endogenous ligand for PPAR-δ, augmenting insulin secretion from β-cells. Detrimental effects of high levels of 4-HNE in mediating β-cell damage are also addressed.
RESEARCH DESIGN AND METHODS
Tissue culture reagents were from Biological Industries (Beit-Haemek, Israel). 4-HDDE and 4-hydroxynonenoic acid (4-HNA) were synthesized as described (24,25). Compounds and reagents included GW501516 and 4-HNE (Calbiochem, Darmstadt, Germany); GSK0660, troglitazone, WY14643, PPAR-δ primer sequences, scrambled RNA sequences, and anti-tubulin antibody (Sigma-Aldrich, Rehovot, Israel); carboxy-DCFDA [5-(and-6)-carboxy-2′,7′-dichlorofluorescein diacetate], OptiMEM, and lipofectamine 2000 (Invitrogen, Carlsbad, CA); collagenase-P (Roche Diagnostics, Mannheim, Germany); polyclonal antibodies against the various PPAR isotypes (Cayman Chemicals, Ann Arbor, MI); horseradish peroxidase–conjugated anti-rabbit- and anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA); anti-cPLA2 and anti-pSer505-cPLA2 antibodies (Cell Signaling, Boston, MA); monoclonal anti-4-HNE histidine adduct antibody (Abcam, Cambridge, MA); TransIT-LT1 reagent (Mirus Bio-Corporation, Madison, WI); dual luciferase reporter assay (Promega, Madison, WI); real-time PCR reagents (Applied Biosystems, Carlsbad, CA); All-blue ROX PCR-mix (Thermo Scientific, Epsom, Surrey, U.K.); and PPAR-δ small interfering RNA (siRNA) sequences (Dharmacon, Chicago, IL). The pcDNA–hPPAR-δ expression vector was constructed as described (18).
Animals, islet isolation, and INS-1E β-cell culture.
Male Wistar rats (150–250 g) and diabetes-prone male Psammomys obesus (P. obesus) gerbils (150–200 g) were from Harlan laboratories (Jerusalem, Israel). The gerbils were fed a low-energy (LE) diet (9.96 kJ/g; Koffolk, Petach-Tikva, Israel) to maintain normoglycemia (random nonfasted blood glucose <5.6 mmol/L). Some animals were fed a high-energy (HE) diet (14.23 kJ/g; Teklad Global Diets, Boston, MA) for at least 7 days to induce postprandial hyperglycemia (nonfasted blood glucose >8.6 mmol/L). The joint ethics committee for animal welfare of the Hebrew University and Hadassah Medical Center approved the study protocol. The Hebrew University is an Association for Assessment and Accreditation of Laboratory Animal Care International–accredited institute. Pancreatic islets were isolated from rats after collagenase digestion, as previously described (26). Pooled islets from three to five animals were preincubated in RPMI-1640 medium (11 mmol/L glucose) for a 16-h recovery period and then divided into the experimental groups, each consisting of 15 islets of a similar size. INS-1E cells (passages 70–90) were grown and maintained as described (27).
Lipidomic analysis.
INS-1E cells were collected in PBS from 100-mm tissue culture plates; 6 × 106 cell pellets were suspended in 1-mL double-distilled water, vortexed, and centrifuged at 14,000g for 30 min at 4°C to separate membrane pellets from the mixture. Phospholipid extracts of these pellets were obtained after extraction with 2:1 chloroform-to-methanol according to Ferreri and Chatgilialoglu (28) and Bligh and Dyer (29). The purity of the obtained fraction was analyzed by thin layer chromatography, using the bidimensional method according to Mangold and Malins (30). Fatty acid methyl esters of membrane phospholipids were prepared as described (31) and were then extracted with n-hexane and analyzed by gas chromatography (GC). Saturated fatty acids (SFAs) and unsaturated fatty acids, including the cis and trans geometrical fatty acids, were identified by comparison with standard references either commercially available or obtained by synthesis, as already described (32). The quantitative determination of the fatty acids was also obtained using the calibration curves of reference compounds in the GC apparatus.
Glucose-stimulated insulin secretion and insulin radioimmunoassay
Static assays.
Isolated rat islets and INS-1E cells were preincubated for 30 min in Krebs-Ringer bicarbonate HEPES–BSA buffer containing 3.3 mmol/L glucose, followed by a 1-h incubation at 3.3 and an additional 1 h at 16.7 mmol/L glucose, as described (26). Aliquots from the incubation buffers were collected, cleared by centrifugation, and frozen until used for insulin radioimmunoassay. Total insulin content in β-cells was measured in aliquots of cell extracts (26).
Dynamic assay.
Rat islets were treated for 48 h, followed by a dynamic assay, performed as previously described (33). Briefly, 40–50 islets per group were placed in a 25-mm Swinnex chamber (Millipore Corp., Billerica, MA) and perifused (0.5 mL/min) with Krebs-Ringer bicarbonate HEPES–BSA buffer containing 3.3 mmol/L glucose and saturated with 95% O2/5% CO2 at 37°C for a 1-h equilibration period. The islets were then perifused with 16.7 mmol/L glucose for 40 min, followed by 10 min at 3.3 mmol/L glucose. Samples were collected throughout the perifusion period for insulin determination. Suitable insulin radioimmunoassay kits for P. obesus insulin (26) and rat insulin (Linco Research, St. Charles, MO) were used according to manufacturers’ protocols.
Western blot analyses.
Cell lysates were prepared and used for Western blot analyses of PPARs, tubulin, cPLA2, pSer505/515-cPLA2, and 4-HNE–protein adducts according to the suppliers’ protocols.
Real-time PCR analysis.
The RNeasy kit (Qiagen, Valencia, CA) was used for RNA isolation. The RevertAid kit (Fermentas, Glen Burnie, MD) was used for cDNA synthesis, using oligo(dT) primers, according to the kit’s instructions. Real-time PCR was performed in Stratagene MX3000P system (Stratagene, Santa Clara, CA) according to the manufacturer’s guidelines. Oligonucleotide primers were designed using Primer Express program (Applied Biosystems) and were synthesized by Sigma-Aldrich. Primer sequences were PPAR-δ: FW-CCCTTCATCATCCACGACATT; RV-TGGACTGGCAGCGGTAGAAC; and glyceraldehyde-3-phosphate dehydrogenase (GAPDH): FW-GGCACAGTCAAGGCTGAGAAT; RV-GCCTTCTCCATGGTGGTGAA. PCR was performed in 20-μL volumes containing SyBr Green ROX Mix, 175 nmol/L of sense and anti-sense primers, and 62.5 pg cDNA. The thermal cycling program consisted of 15 min at 95°C activation, followed by 40 cycles of 15 s at 95°C, 30 s at 60°C, and 30 s at 72°C. The last cycle of 30 s at 95°C and 30 s at 60°C was used to detect nonspecific amplification products. A cycle threshold was calculated for each sample using MXpro software (Stratagene). Results were normalized against those obtained for GAPDH and the internal ROX control.
Transient transfections.
INS-1E cells were cotransfected with expression vectors for human (h)PPAR-α, hPPAR-γ1, hPPAR-γ2, or hPPAR-δ, along with the retinoid X receptor (hRXR), green fluorescent protein and Renilla luciferase expression vectors, and the 3XPPRE-TK-luciferase reporter, as previously described (18). The yield of the transfection, assessed by green fluorescent protein fluorescence, was >80%. Luciferase-induced luminescence was determined in cell lysates with the dual luciferase reporter assay using the Mithras LB-940 luminometer (Berthold Technologies, Bad Wildbad, Germany); results were normalized to the Renilla luciferase activity, used as an internal control, according to the kit’s instructions.
Extraction of polar lipids and high-performance liquid chromatography analysis.
Polar lipids from INS-1E culture media or animal sera were extracted, and high-performance liquid chromatography (HPLC) analysis of 4-HDDE in the extracts was performed as described (18). The HPLC analysis of 4-HNE was as follows: elution was at a flow rate of 1 mL/min with a two-solvent gradient; the initial solvent mixture was 30:70 (A: acetonitrile, B: water) and a linear gradient progressed over 25 min to 100% of A. The elution peak (223 nm) was at 4.2 min. The recovery of the standards, added to fresh samples prior to extraction, was 85–90%. Peaks in the HPLC profiles were monitored and quantified by using Clarity-Lite software (DataApex Co., Prague, Czech Republic).
PPAR-δ silencing with siRNA.
INS-1E cell cultures at 40% confluency were maintained in antibiotic-free RPMI-1640 medium for 24 h before transfection. The cells were then transfected with 15 pmol/L siRNA or scrambled RNA sequences using lipofectamine 2000 in OptiMEM according to the manufacturer’s instructions. Cells were harvested 72 h after transfection and processed for further analysis. Target sequences for PPAR-δ siRNA were ACGAGAAGUGCGAUCGGAU, CCUCAAGUACGGCGUGCAU, CCACAACGCUACCGCUUU, and CAUGAGUUCUUGCGCAGUA.
Cell viability assays.
The trypan blue exclusion test was performed on dispersed rat islet cells and INS-1E cells, as described (34). Apoptosis was determined in INS-1E cells with the Poly Caspases FLICA Kit (ImmunoChemistry Technologies, Bloomington, MN) according to the manufacturer’s instructions.
Determination of intracellular ROS.
The green fluorescence dye carboxy-H2DCFDA was used in INS-1E cell cultures, according to the manufacturer’s instructions.
Statistical analysis.
Results are given as mean ± SEM. Statistical analyses were performed using single factor ANOVA. P < 0.05 is considered significant.
RESULTS
High glucose induces AA and LA release from membrane phospholipids of INS-1E cells.
The fatty acid composition in membrane phospholipids of INS-1E cells exposed to 5, 11, or 25 mmol/L glucose for 16 h was determined by lipidomic analysis. The overall analysis (Supplementary Table 1) shows the relative abundance of major PUFAs, monounsaturated fatty acids (MUFAs), and SFAs in the phospholipid compartment of the cells under increasing glucose levels; while the relative content of SFAs was not significantly altered, MUFA content was elevated and PUFA content reduced.
These data were used to quantify the content of AA and LA. Figure 1A shows a marked decrease in their content in the phospholipid compartment of cells maintained at 11 and 25 mmol/L glucose in comparison with the 5 mmol/L glucose incubation; AA was reduced by 45 and 59% and LA by 64 and 88%, respectively. The high number of cells (>106 cells per determination) required for the lipidomic analysis precluded its application to purified islet-derived β-cells.
FIG. 1.

Effect of high glucose on AA and LA content in INS-1E cells. A: INS-1E cells were incubated with the indicated glucose (Glc) levels for 16 h and processed for analysis of fatty acid residues in membrane phospholipids. The absolute content of AA and LA in phospholipids of INS-1E cells exposed to 5, 11, and 25 mmol/L glucose was calculated as follows: 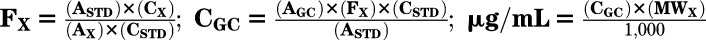 , where ASTD is the area of the standard reference of the fatty acid from the calibration run, CSTD is the concentration of the standard reference from the calibration run, Ax is the area of the compound (x) from the calibration run, Cx is the concentration of the compound (x) from the calibration run, Fx is the conversion factor of the compound (x), CGC is the concentration of the compound in the sample from the GC trial run, AGC is the area of the compound (x) obtained from the GC trial run, and MWx is the molecular weight of the compound (x). Results are mean ± SEM, n = 4. *P < 0.05 for the difference from the respective 5 mmol/L glucose values. B: Lysates were prepared from similarly treated INS-1E cells and taken for Western blot analysis of total cPLA2, pSer505-cPLA2 (white bars), and pSer515-cPLA2 (black bars). Representative Western blots are shown (inset). Tubulin was used for equal protein loading control. Results are mean ± SEM, n = 3. *P < 0.05 for the difference from the respective 5 mmol/L glucose control.
, where ASTD is the area of the standard reference of the fatty acid from the calibration run, CSTD is the concentration of the standard reference from the calibration run, Ax is the area of the compound (x) from the calibration run, Cx is the concentration of the compound (x) from the calibration run, Fx is the conversion factor of the compound (x), CGC is the concentration of the compound in the sample from the GC trial run, AGC is the area of the compound (x) obtained from the GC trial run, and MWx is the molecular weight of the compound (x). Results are mean ± SEM, n = 4. *P < 0.05 for the difference from the respective 5 mmol/L glucose values. B: Lysates were prepared from similarly treated INS-1E cells and taken for Western blot analysis of total cPLA2, pSer505-cPLA2 (white bars), and pSer515-cPLA2 (black bars). Representative Western blots are shown (inset). Tubulin was used for equal protein loading control. Results are mean ± SEM, n = 3. *P < 0.05 for the difference from the respective 5 mmol/L glucose control.
The release of these fatty acids from membrane phospholipids is mediated by enzymes of the PLA2 family. We found that incubation of INS-1E cells with 11 and 25 mmol/L glucose activated cPLA2 (Ser505 phosphorylation) by 2.5 ± 0.4 and 3.9 ± 0.7 fold, respectively, compared with cells at 5 mmol/L glucose; total cPLA2 content was not significantly altered. In a similar manner, Ser515 phosphorylation, which is required for full cPLA2 activation (35), was concomitantly increased 2.3 ± 0.5 and 3.3 ± 0.7 fold, respectively (Fig. 1B).
High glucose augments the peroxidation of PUFAs in INS-1E cells.
The major peroxidation products of AA and LA are 4-HNE and 4-HDDE (15). We measured their levels in culture media of INS-1E cells exposed to increasing glucose concentrations in a serum-free medium during the last 16 h of incubation. This procedure eliminates the formation of 4-HNE adducts with serum proteins. Figure 2A shows a clear glucose-dependent increase in 4-HNE generation, up to ninefold at 25 mmol/L glucose. In contrast, 4-HDDE was not detectable in INS-1E culture medium extracts at all glucose concentrations used (data not shown). The gerbil P. obesus is an established animal model of diet-induced diabetes (5). Figure 2B shows that hyperglycemia induced by HE diet was accompanied by a marked increase in serum 4-HNE. The plasma insulin levels in the normo- and hyperglycemic animal were 1.41 ± 0.16 and 6.38 ± 0.59 nmol/L, respectively. ROS mediate the initiation step in the peroxidation cascade of AA and LA (15). Figure 2C shows increased ROS production in INS-1E cells incubated at 11 and 25 mmol/L glucose.
FIG. 2.

Effect of high glucose on PUFA peroxidation in β-cells. A: INS-1E cells were exposed to the indicated d-glucose (D-Glc) and l-glucose (L-Glc) concentrations for 48 h; during the last 16 h, the cells were incubated with serum-free culture medium with the same additions. The media (10 mL) were then collected, extracted, and analyzed by HPLC. Data are given as nanogram 4-HNE per milligram cellular protein. Representative HPLC tracings are depicted (inset) and the arrows point to 4-HNE peaks. Results are mean ± SEM, n = 3–4. *P < 0.05 for the difference from the 5 mmol/L glucose controls. B: P. obesus gerbils fed LE or HE diet were killed, and sera were collected for glucose and 4-HNE determinations, as described in research design and methods. Serum glucose levels were 3.9 ± 0.1 (LE group) and 14.3 ± 0.7* mmol/L (HE group). Results are mean ± SEM, n = 5–12 animals. *P < 0.05 for the differences from the LE-diet control group. C: ROS production in INS-1E cells incubated for 16 h with the indicated glucose levels was determined by the carboxy-DCF-fluorescence method. Results are mean ± SEM, n = 3–4. *P < 0.05 for the difference from the 5 mmol/L glucose controls.
High glucose and 4-HNE amplify insulin secretion.
The insulin secretory capacity of isolated rat islets and INS-1E cells after preexposure to increasing glucose concentrations was determined. The preexposure periods for rat islets (48 h) and INS-1E cells (24 h) were selected empirically to allow maximal glucose-stimulated insulin secretion (GSIS) with no apparent depletion of cellular insulin content. As expected, GSIS was significantly amplified in a glucose-dependent manner in both β-cell preparations (Fig. 3A). The marked reduction of the response in the presence of the selective PPAR-δ antagonist GSK0660 indicated that this nuclear receptor plays a role in the insulin secretion amplifying action of glucose preexposure.
FIG. 3.

4-HNE mimics high glucose amplification of insulin secretion. A: Rat islets and INS-1E cells were incubated with RPMI-1640 medium containing the indicated glucose levels in the absence or presence of 1 μmol/L GSK0660 for 48 and 24 h, respectively. GSIS was evaluated by 1-h static incubations at 3.3 mmol/L glucose (white bars), followed by a 1-h incubation at 16.7 mmol/L glucose (black bars). Insulin secretion is presented as percent of insulin content. Glc, glucose. Results are mean ± SEM, n = 3. *P < 0.05 for the difference of stimulated secretion (16.7 mmol/L glucose) compared with untreated controls maintained at 5 mmol/L glucose. #P < 0.05 for differences from the respective GSK0660-free incubation. B: Isolated rat islets and INS-1E cells were incubated for 48 or 24 h, respectively, in standard RPMI-1640 medium (11 mmol/L glucose) in the absence or presence of increasing concentrations of 4-HNE. GSIS was measured as in A. The vehicle ethanol, at a 1:1,000 dilution, did not affect GSIS in either β-cell preparation. Insulin secretion is presented as percent of insulin content. Results are mean ± SEM, n = 3. *P < 0.05 relative to the stimulated secretion of vehicle-treated controls. #P < 0.05 relative to the maximal stimulatory level. C: Rat islets and INS-1E cells were incubated for 48 and 24 h, respectively, at 5, 11, or 25 mmol/L glucose in the absence or presence of 4-HNE (15 or 1 µmol/L, respectively) followed by GSIS analysis. Results are mean ± SEM, n = 3. *P < 0.05 for difference from the respective untreated controls, exposed to the same glucose concentration.
The effect of exogenously added 4-HNE on GSIS was then investigated; isolated rat islets and INS-1E cells were maintained for 48 or 24 h, respectively, in standard RPMI-1640 medium (11 mmol/L glucose) without or with increasing 4-HNE concentrations, followed by a static GSIS assay (Fig. 3B). The addition of 4-HNE amplified GSIS in a concentration-dependent manner; maximal effects were observed with 25 and 1 μmol/L 4-HNE in rat islets and in INS-1E cells, respectively. Above these levels, GSIS was reduced, possibly as a result of cytotoxic effects of 4-HNE (see Fig. 7).
FIG. 7.

High 4-HNE concentrations compromise β-cell survival. A: Rat islets and INS-1E cells were incubated for 48 or 24 h, respectively, with increasing concentrations of 4-HNE. Islets were pooled from three animals and divided into 10–12 islets per group. After incubation, islet cells were dispersed by mild trypsin digestion. Cell viability was determined at the end of the incubation by the trypan blue exclusion test. Cell viability in the absence of 4-HNE was >95% in both preparations. Results are mean ± SEM, n = 3. *P < 0.05 for the difference from untreated cells. B: INS-1E cells were taken for the FLICA apoptosis assay, as described in research design and methods. Results are mean ± SEM of the relative apoptotic levels in three different slides. C: Western blot analysis of 4-HNE–protein adducts in lysates prepared from INS-1E cells incubated for 48 h at 11 mmol/L glucose and the indicated concentrations of 4-HNE.
Next, we compared the effects of 4-HNE in β-cells exposed to 5, 11, and 25 mmol/L glucose (Fig. 3C). Treatment with 4-HNE amplified GSIS in both β-cell preparations preexposed to 5 and 11 mmol/L glucose. However, it did not increase further GSIS in cells preexposed to 25 mmol/L glucose.
The enzyme fatty aldehyde dehydrogenase (FALDH) converts 4-HNE to its acidic metabolite 4-HNA (25). We asked whether this metabolite mediates the effect on insulin secretion attributed to 4-HNE. To answer this question, we synthesized 4-HNA; studies at different doses and incubation times showed that it lacked insulin secretory activity in INS-1E cells (Supplementary Fig. 1A).
4-HNE activates PPAR-δ in β-cells.
Recent reports show that 4-HNE is an endogenous ligand of PPAR-δ (21); therefore, we tested its ability to activate PPAR-δ in INS-1E cells. The cells were transfected with hPPAR-α, hPPAR-γ1, hPPAR-γ2, or hPPAR-δ expression vectors along with hRXR- and 3XPPRE (PPAR response element)-luciferase expression vectors, as described before (18). Each hPPAR isotype was successfully expressed in INS-1E cells (Supplementary Fig. 2). Furthermore, WY14643, troglitazone, and GW501516, the pharmacological agonists of PPAR-α, PPAR-γ, and PPAR-δ, respectively (18), transactivated luciferase expression in an hPPAR isotype–specific manner (Fig. 4A). Exogenously added 4-HNE mimicked GW501516 and induced luciferase activity only in cells ectopically expressing hPPAR-δ. The finding that 4-HNE activated PPAR-δ in β-cells was further confirmed by the use of the selective PPAR-δ antagonist GSK0660; it abolished 4-HNE–induced luciferase activity in hPPAR-δ–expressing cells (Fig. 4B). The corresponding analysis of GSK0660 inhibitory effects on GW501516-treated INS-1E cells is shown in Supplementary Fig. 3. Moreover, 4-HNA, the acidic metabolite of 4-HNE, lacked any PPAR-δ stimulating activity (Supplementary Fig. 1B). Figure 4C shows a similar transactivation assay with increasing glucose concentrations in the absence or presence of GSK0660. Glucose stimulated luciferase activity in a concentration-dependent manner, which was abolished by the PPAR-δ antagonist.
FIG. 4.

High glucose and 4-HNE activate PPAR-δ in INS-1E cells. A: INS-1E cells were transfected with the hPPAR-α, hPPAR-γ1, hPPAR-γ2, or hPPAR-δ expression vectors; the hRXR expression vector; and the 3XPPRE-TK-luciferase reporter plasmid. Renilla luciferase plasmid was transfected for normalizing the luciferase activity data. The transfected cells were treated with 60 μmol/L WY14643, 30 μmol/L troglitazone, 0.1 μmol/L GW501516 (GW), or 1.0 μmol/L 4-HNE for 24 h and luciferase activity was then measured. The 100% values were assigned to the respective untreated control groups. *P < 0.05 for differences from the respective controls. B: INS-1E cells were transfected as described in A using the hPPAR-δ expression vector. The cells were then incubated for 24 h with increasing concentrations of 4-HNE without (●) or with (○) 1 μmol/L GSK0660. Luciferase activity was then measured. The value of light units measured in lysates of vehicle-treated cells was taken as 100%. *P < 0.05 for differences from untreated controls. #P < 0.05 for differences from the respective 4-HNE–treated cells. C: INS-1E cells were transfected as described in B and incubated for 48 h in RPMI-1640 medium containing the indicated glucose levels. GSK0660 (1 μmol/L) was present during the last 24 h of incubation. *P < 0.05 for the difference in luciferase activity in comparison with the control incubation at 5 mmol/L glucose in the absence of GSK0660. #P < 0.05 for difference from the cells incubated in the same glucose concentration without GSK0660. The 100% value was taken as the light units of control cells at 5 mmol/L glucose. Results are mean ± SEM, n = 3. The vehicle DMSO, used at a 1:1,000 dilution in the incubation medium, had no significant effect on GSIS. Glc, glucose.
The role of PPAR-δ in glucose- and 4-HNE–induced amplification of insulin secretion.
To establish the role of PPAR-δ in mediating high glucose-induced amplification of insulin secretion, we silenced its expression. Supplementary Fig. 4A and B shows that siRNA targeted to PPAR-δ mRNA reduced the mRNA and protein levels by 65–75 and 45–60%, respectively. Figure 5A shows that this treatment resulted in loss of the cells’ ability to amplify GSIS after preexposure to high glucose levels. Noteworthy, the activation of PPAR-δ by increasing glucose concentrations was not associated with increased expression of this nuclear receptor (Supplementary Fig. 4A and B).
FIG. 5.
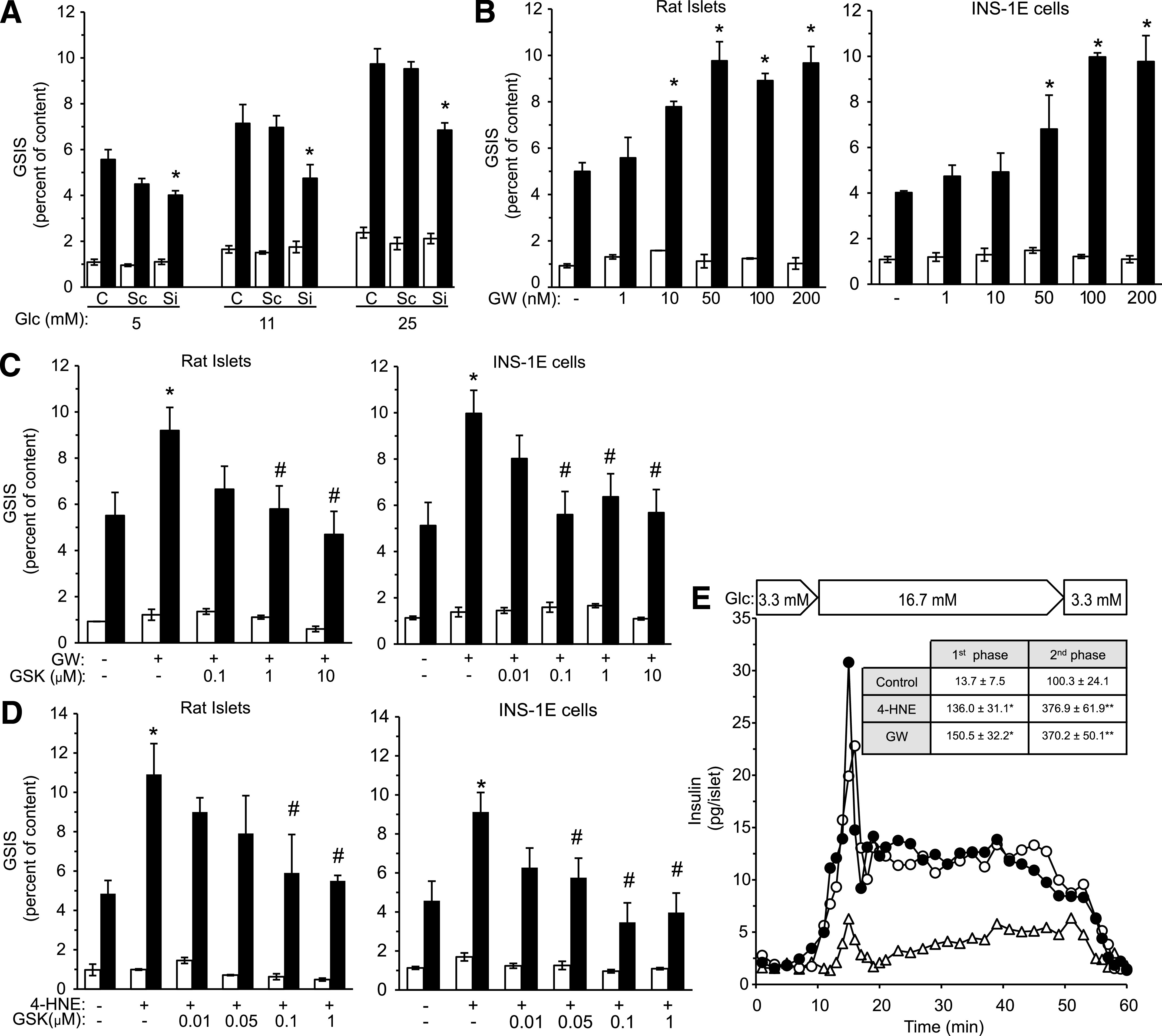
PPAR-δ mediates 4-HNE effects in β-cells. A: PPAR-δ expression was silenced in INS-1E cells with targeted siRNA sequences (Si), as described in research design and methods. The cells were then incubated for 24 h at the indicated glucose concentrations, washed, processed, and taken for the standard GSIS analysis, as described in the legend to Fig. 3A. Cells transfected with scrambled RNA (Sc) served as controls. C, nontransfected cells; Glc, glucose. *P < 0.05 for differences from the respective controls. B: Rat islets and INS-1E cells were incubated in RPMI-1640 medium containing 11 mmol/L glucose with increasing GW501516 concentrations for 48 and 24 h, respectively, and taken for GSIS analysis. *P < 0.05 for difference from untreated cells. C: Similar β-cell preparations were incubated as described in A for 48 and 24 h, respectively, without or with 0.1 μmol/L GW501516 and increasing concentrations of GSK0660, and taken for GSIS analysis. *P < 0.05 for the difference from untreated cells. #P < 0.05 for the difference from GW501516-treated cells in the absence of GSK0660. D: The β-cell preparations were incubated at 11 mmol/L glucose without or with 15 (rat islets) or 1 μmol/L 4-HNE (INS-1E cells) with increasing concentrations of GSK0660. At the end of incubations, the islets and INS-1E cells were taken for GSIS analysis. *P < 0.05 for difference from untreated cells. #P < 0.05 for the differences from 4-HNE–treated cells in the absence of GSK0660. Results (A–D) are mean ± SEM, n = 3. E: Isolated rat islets were preincubated for 48 h in RPMI-1640 medium (11 mmol/L glucose) with 15 μmol/L 4-HNE (○), 0.1 μmol/L GW501516 (●), or the vehicle (△) and taken for perfusion experiments. AUC of the first and second phases of insulin release of each treatment are shown (inset). P < 0.05 for differences of the AUC values of the first (*) and second phase (**) insulin secretion from 4-HNE– and GW501516-treated islets in comparison with control islets. Results are mean ± SEM, n = 3.
To ascertain the role of PPAR-δ in insulin secretion, we tested the effect of GW501516. This PPAR-δ agonist mimicked the effect of high glucose preexposure; it increased GSIS dose-dependently by up to twofold in isolated rat islets and INS-1E cells, with maximal stimulatory effect at 50–100 nmol/L (Fig. 5B). This effect of GW501516 was abolished by GSK0660 (Fig. 5C). GSK0660 also fully antagonized the amplifying effect of 4-HNE on insulin secretion in both β-cell preparations (Fig. 5D).
Further support for the role of 4-HNE and PPAR-δ in regulating insulin secretion comes from dynamic insulin secretion studies. Isolated rat islets were preincubated for 48 h in the absence or presence of effective concentrations of 4-HNE or GW501516; both agents considerably augmented both first and second phases of insulin secretion, in comparison with the secretion profile of the vehicle-treated islets, as shown in the representative experiment depicted in Fig. 5E. The inset in Fig. 5E gives the values of the areas under the curve (AUC) of four independent experiments that are summarized in Supplementary Fig. 5.
N-acetylcysteine blocks high glucose-induced generation of 4-HNE, PPAR-δ activation, and insulin secretion.
The role of endogenous 4-HNE in regulating insulin secretion was further substantiated by measuring insulin secretion after the blocking of 4-HNE production in β-cells by the antioxidant N-acetylcysteine (NAC), as described previously (18). Figure 6A and B shows that glucose-induced generation of ROS and 4-HNE in INS-1E cells was nearly abolished in the presence of 1 mmol/L NAC. This treatment also prevented high glucose-induced expression of luciferase in the PPRE-dependent transactivation assay in cells overexpressing PPAR-δ. Finally, glucose-amplified insulin secretion was markedly reduced in NAC-treated rat islets and in INS-1E cells (Fig. 6D).
FIG. 6.
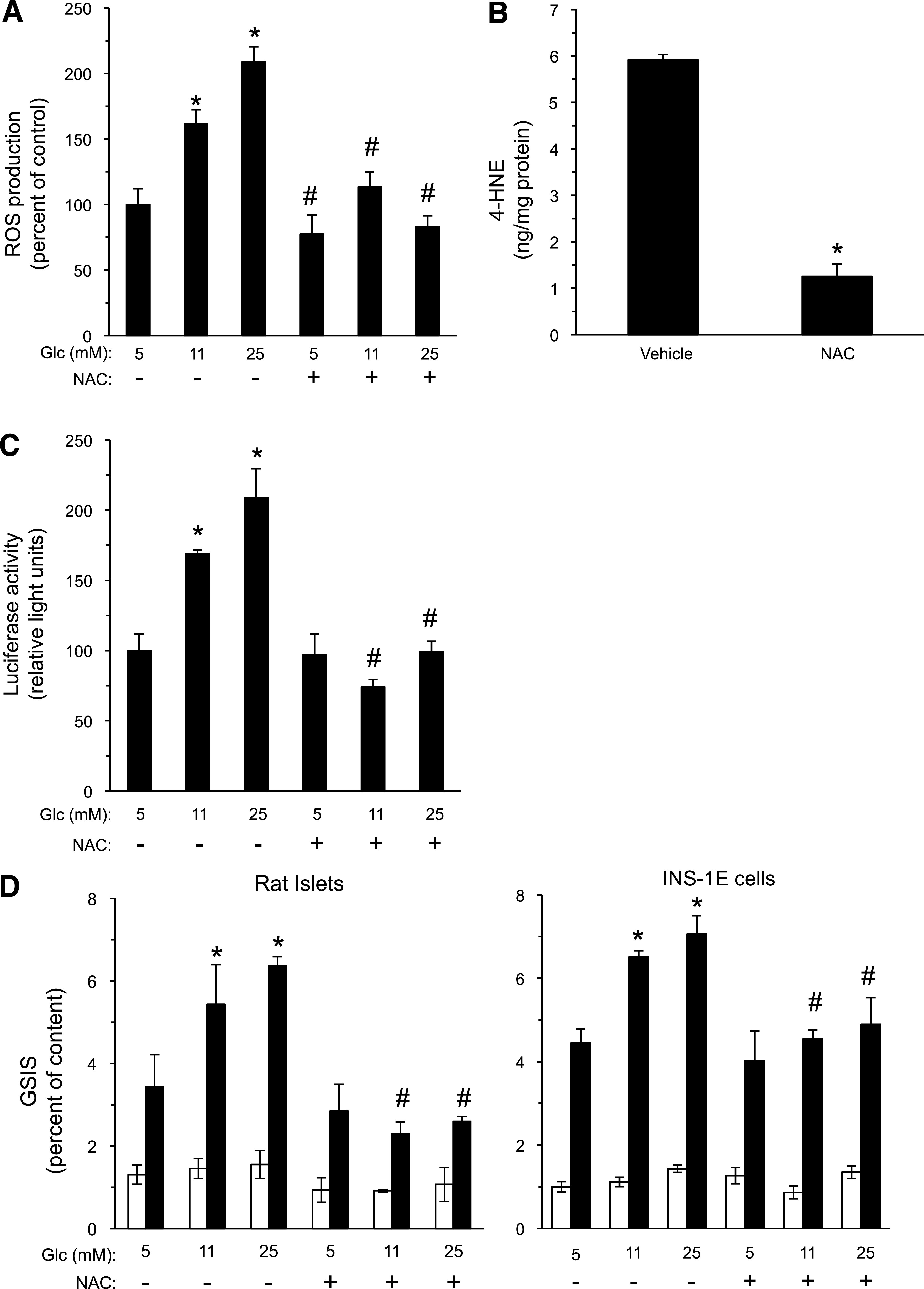
NAC reduces ROS production, 4-HNE generation, and insulin secretion in β-cells. A: INS-1E cells were incubated for 16 h with the indicated glucose levels without or with 1 mmol/L NAC. ROS production was determined with the carboxy-DCF-fluorescence assay. Glc, glucose. *P < 0.05 for the difference from the 5 mmol/L glucose controls. #P < 0.05 for the difference from the cells incubated in the same glucose concentration without NAC. B: HPLC determination of 4-HNE in INS-1E cultures maintained at 25 mmol/L glucose, without or with 1 mmol/L NAC for 16 h, was performed as described in the legend to Fig. 2A. *P < 0.05 for the difference from the vehicle-treated controls. C: INS-1E cells were prepared for the PPRE-luciferase transactivation assay, as described in the legend to Fig. 4B, and incubated with the indicated glucose levels without or with 1 mmol/L NAC; luciferase activity was determined after 24 h of incubation. *P < 0.05 for the difference in luciferase activity in comparison with the control incubation at 5 mmol/L glucose in the absence of NAC. #P < 0.05 for the difference from cells incubated in the same glucose concentration without NAC. D: Rat islets and INS-1E cells were incubated with RPMI-1640 medium containing the indicated glucose concentrations in the absence or presence of 1 mmol/L NAC for 48 or 24 h, respectively. The islets and INS-1E cells were then washed and GSIS evaluated, as described in the Fig. 3A legend (white bars, 3.3 mmol/L glucose; dark bars, 16.7 mmol/L glucose). Insulin secretion is presented as percent of insulin content. *P < 0.05 for the difference in stimulated secretion compared with cells maintained at 5 mmol/L glucose. #P < 0.05 for differences from the respective NAC-free incubation. Results are mean ± SEM, n = 3.
4-HNE cytotoxicity in β-cells.
The data shown in Fig. 2B suggest that 4-HNE levels above the physiological effective range are deleterious to the cells, impairing insulin secretion. Therefore, the trypan blue exclusion test was used to determine the viability of rat islets and INS-1E cells exposed to increasing concentrations of 4-HNE (Fig. 7A). We observed that INS-1E cells were more susceptible to 4-HNE–induced death than rat islet cells; the former tolerated 4-HNE up to 20 μmol/L, while the latter sustained it well at 50 μmol/L. Figure 7B shows marked apoptosis of INS-1E cells exposed to 50 μmol/L 4-HNE. Representative images of apoptotic INS-1E cells treated with increasing 4-HNE concentrations are shown in Supplementary Fig. 6.
A key feature of 4-HNE–induced cell damage is the accumulation of 4-HNE–protein adducts. A Western blot analysis of histidine–4-HNE adducts in lysates of INS-1E cells incubated without or with increasing 4-HNE concentrations is depicted in Fig. 7C. A modest accumulation of adducts was observed up to 20 μmol/L, whereas a marked increase was apparent at 50 μmol/L 4-HNE, the same concentration that induced substantial apoptosis and cell death.
DISCUSSION
A role for PUFAs and their LO-derived metabolites in regulating insulin secretion has long been suggested (6–9). Our results link the glucose-induced activation of cPLA2, the release of PUFAs from membrane phospholipids in β-cells, and their peroxidation to this phenomenon. In particular, PPAR-δ and its endogenous ligand 4-HNE amplify the adaptive insulin secretory response of β-cells upon exposure to increasing glucose concentrations.
The current lipidomic analysis shows that preexposure of β-cells to high glucose modifies the distribution of various fatty acids in membrane phospholipids. While the abundance of SFAs was not significantly altered, the relative content of MUFAs increased and that of PUFAs decreased. Within the latter group, the relative abundance of n-3 PUFAs increased, whereas that of n-6 PUFAs decreased. It is interesting that a reduced ratio of n-6–to–n-3 PUFAs in β-cells of the fat-1 mouse, genetically engineered to produce n-3 PUFAs, has recently been associated with an enhanced insulin secretion (36); however, the molecular mechanisms behind this phenomenon and its applicability to other metabolic conditions is not clear.
The abundance of palmitic acid in membrane phospholipids was moderately reduced under high glucose incubation, whereas that of oleic acid increased. The observed increase in the monounsaturated palmitoleic acid in β-cell phospholipids after exposure to high glucose could be mediated by stearoyl-CoA desaturase-1. Noteworthy, recent studies suggest that palmitoleic acid functions as a lipid mediator in organ cross talk and metabolic abnormalities (37). Whether these changes in the content of SFAs and MUFAs in phospholipids affect β-cell function under high glucose conditions remains to be investigated. It has been shown that insulin content and the abundance of secretory granule membranes are lower in INS-1E cells than in intact rat β-cells (27). In a similar manner, the lipidome of β-cells in rat islets may differ from that of INS-1E cells.
We suggest that AA-derived 4-HNE participates in the regulation of insulin secretion. Keane and Newsholme (6) have recently reported that AA has regulatory and protective effects that improve β-cell function and survival. Recent studies and our present work show that hyperglycemia in human and animal models of diabetes is associated with an increased generation of 4-hydroxyalkenals (17–19). The present findings on hyperglycemic P. obesus gerbils correlate the increased 4-HNE plasma levels to the increased compensatory plasma insulin level. The antioxidant NAC, which markedly reduced the generation of endogenous 4-HNE, also blocked PPAR-δ activation and abolished the amplifying effect of high glucose. These data suggest a key role for 4-HNE in glucose amplification of insulin secretion.
Various signaling and regulatory functions were attributed to 4-HNE, yet very few genuine pharmacological targets (e.g., receptors) have been identified (15). Both the study by Coleman et al. (21) and the current study demonstrate that 4-HNE is an endogenous activating ligand of PPAR-δ. In silico analysis suggests specific binding interactions between 4-HNE and certain amino acid moieties within the ligand-binding domain of PPAR-δ (21). The discovery of 4-HNE as a PPAR-δ ligand is important in view of the regulatory functions that this receptor mediates in β-cells; it potentiates GSIS by regulating the expression of enzymes involved in anaplerotic glucose metabolism and mitochondrial oxidative reactions, as well as increasing fatty acid transport and metabolism (23,38,39). In contrast to the stimulatory effects of PPAR-δ, both PPAR-α and PPAR-γ suppress insulin secretion from β-cells by either attenuating glucose-induced Ca+2 signals (40) or inducing endoplasmic reticulum stress (41,42). However, the suppressive effects of PPAR-α and PPAR-γ may not be physiologically relevant because their expression level in β-cells is normally low (23).
Upon activation, PPAR-δ dimerizes with its cognate partner, the retinoic X receptor (RXR). The resulting complex interacts with PPREs in promoters of target genes and activates the transcription program that ultimately enhances GSIS. Recent studies point to pyruvate dehydrogenase kinase 4 (PDK4) and carnitine palmitoyltransferase 1 (CPT1) as targets for PPAR-δ in β-cells; Ravnskjaer et al. (23) and Wan et al. (43) have associated PPAR-δ activation in different β-cell lines to an increased expression of PDK4, which can be linked to increased mitochondrial activity and insulin secretion. The latter group has also shown that the activation of PPAR-δ significantly increased CPT1 mRNA levels. Of interest, Sol et al. (44) have demonstrated that overexpression of CPT1 counteracts glucolipotoxicity in INS-1E cells. Additional studies are needed to identify the gene network that mediates high glucose-primed insulin secretion in β-cells in a PPAR-δ–dependent manner. Also of interest, Xu et al. (45) have shown that PPAR-δ activation induces phosphorylation of cPLA2 in cholangiocarcinoma cell lines. It remains to be investigated whether such a regulatory loop functions in β-cells.
We report that the amplifying effect of 4-HNE was more pronounced in INS-1E cells and isolated rat islets that were preexposed to 11 mmol/L compared with 5 mmol/L glucose. This disparity may result from high glucose-regulated expression of coactivators or corepressors of the PPAR-δ–RXR transcription complex, and/or factors involved in the intracellular trafficking of insulin granules. The lack of effect of exogenous 4-HNE in β-cells exposed to 25 mmol/L glucose most likely results from the endogenous 4-HNE production, which already amplifies insulin secretion.
The duality of 4-HNE function as an endogenous signaling molecule on one hand and a cytotoxic agent on the other hand has long been recognized (46). The cytotoxic effects result from an excessive accumulation of covalent 4-HNE adducts with proteins, phospholipids, and DNA (15). Our results indicate that the level of 4-HNE in INS-1E cultures exposed to increasing glucose levels remains below the cytotoxic threshold. The current study agrees with Miwa et al. (47) who reported that 100 μmol/L 4-HNE, which is above this threshold, impeded insulin secretion from isolated rat islets. The disparate susceptibility of primary islet cells and the immortalized β-cell line to 4-HNE, observed in our study, may result from differences in the expression and function of 4-hydroxyalkenal–neutralizing enzymes (FALDH and glutathione-S-transferase) (48,49) between these β-cell preparations. Nonetheless, 4-HNA, the FALDH metabolite of 4-HNE, does not activate PPAR-δ or augment insulin secretion.
Chronic high glucose-induced β-cell dysfunction may be associated with an excessive generation of 4-hydroxyalkenals, which are deleterious to cells. Of interest is the finding that increased LO activity induces β-cell dysfunction (50). Whether this results in an excessive production of hydroperoxy metabolites of AA and LA, which are subsequently peroxidized to cytotoxic levels of 4-HNE, remains to be investigated. Finally, our findings on glucose-mediated generation of 4-HNE and the subsequent activation of PPAR-δ that leads to an amplified insulin secretion from β-cells are summarized in the model shown in Fig. 8.
FIG. 8.

A model for the dual function of 4-HNE in β-cells. Exposure to high glucose markedly enhances ROS production as well as the activation of cPLA2 in β-cells by inducing Ser505 and Ser515 phosphorylations and the subsequent release of arachidonic acid (ARA) and linoleic acid (LNA) from phospholipids. ROS-mediated peroxidation of these PUFAs results in the generation of 4-HNE. This molecule affects β-cell function in two major ways; when present at nontoxic concentrations, it amplifies insulin secretion in a PPAR-δ–RXR–dependent manner. RXR is activated by cis-retinoic acid (cRA). However, chronic hyperglycemia may lead to an excessive generation of 4-HNE with the accumulation of 4-HNE adducts, causing β-cell dysfunction, characteristic of the advanced stages of type 2 diabetes. (A high-quality color representation of this figure is available in the online issue.)
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the Israel Science Foundation (44/10) and the Yedidut Foundation, Mexico, at the Hebrew University. The support and sponsorship of COST Actions BM0602 on “Adipose Tissue: A Key Target for Prevention of the Metabolic Syndrome,” CM0603 on “Free Radicals in Chemical Biology (CHEMBIORADICAL),” and B35 on “Lipid Peroxidation Associated Disorders: LPO” are gratefully acknowledged. G.C., Y.R., and O.S. have received fellowships from the Hebrew University Center for Diabetes Research. S.S. is affiliated with the David R. Bloom Center for Pharmacy and the Dr. Adolf and Klara Brettler Center for Research of Molecular Pharmacology and Therapeutics.
No potential conflicts of interest relevant to this article were reported.
G.C. researched data, contributed to discussion, and wrote and edited the manuscript. Y.R. and O.S. researched data. M.G. contributed to discussion and reviewed the manuscript. C.C. and C.F. researched data, contributed to discussion, and reviewed the manuscript. N.K. contributed to discussion and reviewed the manuscript. S.S. wrote and edited the manuscript.
The authors are grateful to Mrs. Y. Ariav (Endocrinology and Metabolism Service of the Hebrew University–Hadassah Medical Center) for sharing her technical expertise in islet studies; Dr. B.M. Spiegelman (Dana Farber Cancer Institute, Boston, MA), Dr. R. Evans (Howard Hughes Medical Institute, La Jolla, CA), and Dr. B. Staels (Université Lille Nord de France, Lille, France) for kindly providing the pSVPORT1-hRXR vector and the 3XPPRE-TK-luciferase plasmid, the pCMX-hPPAR-γ1 and pCMX-hPPAR-γ2 plasmids, and the pSG5-hPPAR-α plasmid, respectively; Dr. K.U. Malik (University of Tennessee, Memphis, TN) for the generous gift of purified anti-phospho-Ser515-cPLA2 antibody; and Prof. E. Cerasi (Endocrinology and Metabolism Service of the Hebrew University–Hadassah Medical Center) for critically reading the manuscript and providing valuable comments.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db11-0347/-/DC1.
REFERENCES
- 1.Weir GC, Bonner-Weir S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes 2004;53(Suppl. 3):S16–S21 [DOI] [PubMed] [Google Scholar]
- 2.Grill V, Adamson U, Cerasi E. Immediate and time-dependent effects of glucose on insulin release from rat pancreatic tissue. Evidence for different mechanisms of action. J Clin Invest 1978;61:1034–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ward WK, Halter JB, Beard JC, Porte D, Jr. Adaptation of B and A cell function during prolonged glucose infusion in human subjects. Am J Physiol 1984;246:E405–E411 [DOI] [PubMed] [Google Scholar]
- 4.Nesher R, Cerasi E. Modeling phasic insulin release: immediate and time-dependent effects of glucose. Diabetes 2002;51(Suppl. 1):S53–S59 [DOI] [PubMed] [Google Scholar]
- 5.Kaiser N, Leibowitz G. Failure of beta-cell adaptation in type 2 diabetes: lessons from animal models. Front Biosci 2009;14:1099–1115 [DOI] [PubMed] [Google Scholar]
- 6.Keane D, Newsholme P. Saturated and unsaturated (including arachidonic acid) non-esterified fatty acid modulation of insulin secretion from pancreatic beta-cells. Biochem Soc Trans 2008;36:955–958 [DOI] [PubMed] [Google Scholar]
- 7.Poitout V. Phospholipid hydrolysis and insulin secretion: a step toward solving the Rubik’s cube. Am J Physiol Endocrinol Metab 2008;294:E214–E216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dixon G, Nolan J, McClenaghan NH, Flatt PR, Newsholme P. Arachidonic acid, palmitic acid and glucose are important for the modulation of clonal pancreatic beta-cell insulin secretion, growth and functional integrity. Clin Sci (Lond) 2004;106:191–199 [DOI] [PubMed] [Google Scholar]
- 9.Turk J, Gross RW, Ramanadham S. Amplification of insulin secretion by lipid messengers. Diabetes 1993;42:367–374 [DOI] [PubMed] [Google Scholar]
- 10.Persaud SJ, Muller D, Belin VD, et al. The role of arachidonic acid and its metabolites in insulin secretion from human islets of langerhans. Diabetes 2007;56:197–203 [DOI] [PubMed] [Google Scholar]
- 11.Metz SA. Is phospholipase A2 a “glucose sensor” responsible for the phasic pattern of insulin release? Prostaglandins 1984;27:147–158 [DOI] [PubMed] [Google Scholar]
- 12.Dunlop ME, Larkins RG. Activity of endogenous phospholipase C and phospholipase A2 in glucose stimulated pancreatic islets. Biochem Biophys Res Commun 1984;120:820–827 [DOI] [PubMed] [Google Scholar]
- 13.Ramanadham S, Song H, Bao S, et al. Islet complex lipids: involvement in the actions of group VIA calcium-independent phospholipase A(2) in beta-cells. Diabetes 2004;53(Suppl. 1):S179–S185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leloup C, Tourrel-Cuzin C, Magnan C, et al. Mitochondrial reactive oxygen species are obligatory signals for glucose-induced insulin secretion. Diabetes 2009;58:673–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Riahi Y, Cohen G, Shamni O, Sasson S. Signaling and cytotoxic functions of 4-hydroxyalkenals. Am J Physiol Endocrinol Metab 2010;299:E879–E886 [DOI] [PubMed] [Google Scholar]
- 16.Schneider C, Tallman KA, Porter NA, Brash AR. Two distinct pathways of formation of 4-hydroxynonenal. Mechanisms of nonenzymatic transformation of the 9- and 13-hydroperoxides of linoleic acid to 4-hydroxyalkenals. J Biol Chem 2001;276:20831–20838 [DOI] [PubMed] [Google Scholar]
- 17.Toyokuni S, Yamada S, Kashima M, et al. Serum 4-hydroxy-2-nonenal-modified albumin is elevated in patients with type 2 diabetes mellitus. Antioxid Redox Signal 2000;2:681–685 [DOI] [PubMed] [Google Scholar]
- 18.Riahi Y, Sin-Malia Y, Cohen G, et al. The natural protective mechanism against hyperglycemia in vascular endothelial cells: roles of the lipid peroxidation product 4-hydroxydodecadienal and peroxisome proliferator-activated receptor delta. Diabetes 2010;59:808–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orioli M, Aldini G, Benfatto MC, Facino RM, Carini M. HNE Michael adducts to histidine and histidine-containing peptides as biomarkers of lipid-derived carbonyl stress in urines: LC-MS/MS profiling in Zucker obese rats. Anal Chem 2007;79:9174–9184 [DOI] [PubMed] [Google Scholar]
- 20.Negre-Salvayre A, Auge N, Ayala V, et al. Pathological aspects of lipid peroxidation. Free Radic Res 2010;44:1125–1171 [DOI] [PubMed] [Google Scholar]
- 21.Coleman JD, Prabhu KS, Thompson JT, et al. The oxidative stress mediator 4-hydroxynonenal is an intracellular agonist of the nuclear receptor peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta). Free Radic Biol Med 2007;42:1155–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winzell MS, Wulff EM, Olsen GS, Sauerberg P, Gotfredsen CF, Ahrén B. Improved insulin sensitivity and islet function after PPARdelta activation in diabetic db/db mice. Eur J Pharmacol 2010;626:297–305 [DOI] [PubMed] [Google Scholar]
- 23.Ravnskjaer K, Frigerio F, Boergesen M, Nielsen T, Maechler P, Mandrup S. PPARdelta is a fatty acid sensor that enhances mitochondrial oxidation in insulin-secreting cells and protects against fatty acid-induced dysfunction. J Lipid Res 2010;51:1370–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soulère L, Queneau Y, Doutheau A. An expeditious synthesis of 4-hydroxy-2E-nonenal (4-HNE), its dimethyl acetal and of related compounds. Chem Phys Lipids 2007;150:239–243 [DOI] [PubMed] [Google Scholar]
- 25.Bacot S, Bernoud-Hubac N, Baddas N, et al. Covalent binding of hydroxy-alkenals 4-HDDE, 4-HHE, and 4-HNE to ethanolamine phospholipid subclasses. J Lipid Res 2003;44:917–926 [DOI] [PubMed] [Google Scholar]
- 26.Attali V, Parnes M, Ariav Y, Cerasi E, Kaiser N, Leibowitz G. Regulation of insulin secretion and proinsulin biosynthesis by succinate. Endocrinology 2006;147:5110–5118 [DOI] [PubMed] [Google Scholar]
- 27.Merglen A, Theander S, Rubi B, Chaffard G, Wollheim CB, Maechler P. Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology 2004;145:667–678 [DOI] [PubMed] [Google Scholar]
- 28.Ferreri C, Chatgilialoglu C. Membrane lipidomics and the geometry of unsaturated fatty acids: from biomimetic models to biological consequences. In Lipidomics, Vol. 1: Methods and Protocols. Armstrong D, Ed. New York, Humana Press, 2009, p. 391–412 [DOI] [PubMed] [Google Scholar]
- 29.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 1959;37:911–917 [DOI] [PubMed] [Google Scholar]
- 30.Mangold H, Malins D. Fractionation of fats, oils and waxes on thin layers of silicic acid. J Am Oil Chem Soc 1960;37:383–385 [Google Scholar]
- 31.Kramer JK, Fellner V, Dugan ME, Sauer FD, Mossoba MM, Yurawecz MP. Evaluating acid and base catalysts in the methylation of milk and rumen fatty acids with special emphasis on conjugated dienes and total trans fatty acids. Lipids 1997;32:1219–1228 [DOI] [PubMed] [Google Scholar]
- 32.Ferreri C, Kratzsch S, Brede O, Marciniak B, Chatgilialoglu C. Trans lipid formation induced by thiols in human monocytic leukemia cells. Free Radic Biol Med 2005;38:1180–1187 [DOI] [PubMed] [Google Scholar]
- 33.Dov A, Abramovitch E, Warwar N, Nesher R. Diminished phosphodiesterase-8B potentiates biphasic insulin response to glucose. Endocrinology 2008;149:741–748 [DOI] [PubMed] [Google Scholar]
- 34.Okuya S, Tanabe K, Tanizawa Y, Oka Y. Leptin increases the viability of isolated rat pancreatic islets by suppressing apoptosis. Endocrinology 2001;142:4827–4830 [DOI] [PubMed] [Google Scholar]
- 35.Pavicevic Z, Leslie CC, Malik KU. cPLA2 phosphorylation at serine-515 and serine-505 is required for arachidonic acid release in vascular smooth muscle cells. J Lipid Res 2008;49:724–737 [DOI] [PubMed] [Google Scholar]
- 36.Wei D, Li J, Shen M, et al. Cellular production of n-3 PUFAs and reduction of n-6-to-n-3 ratios in the pancreatic beta-cells and islets enhance insulin secretion and confer protection against cytokine-induced cell death. Diabetes 2010;59:471–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cao H, Gerhold K, Mayers JR, Wiest MM, Watkins SM, Hotamisligil GS. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell 2008;134:933–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kharroubi I, Lee CH, Hekerman P, et al. BCL-6: a possible missing link for anti-inflammatory PPAR-delta signalling in pancreatic beta cells. Diabetologia 2006;49:2350–2358 [DOI] [PubMed] [Google Scholar]
- 39.Jiang L, Wan J, Ke LQ, Lü QG, Tong NW. Activation of PPARδ promotes mitochondrial energy metabolism and decreases basal insulin secretion in palmitate-treated β-cells. Mol Cell Biochem 2010;343:249–256 [DOI] [PubMed] [Google Scholar]
- 40.Tordjman K, Standley KN, Bernal-Mizrachi C, et al. PPARalpha suppresses insulin secretion and induces UCP2 in insulinoma cells. J Lipid Res 2002;43:936–943 [PubMed] [Google Scholar]
- 41.Weber SM, Chambers KT, Bensch KG, Scarim AL, Corbett JA. PPARgamma ligands induce ER stress in pancreatic beta-cells: ER stress activation results in attenuation of cytokine signaling. Am J Physiol Endocrinol Metab 2004;287:E1171–E1177 [DOI] [PubMed] [Google Scholar]
- 42.Terauchi Y, Kadowaki T. Peroxisome proliferator-activated receptors and insulin secretion. Endocrinology 2005;146:3263–3265 [DOI] [PubMed] [Google Scholar]
- 43.Wan J, Jiang L, Lü Q, Ke L, Li X, Tong N. Activation of PPARdelta up-regulates fatty acid oxidation and energy uncoupling genes of mitochondria and reduces palmitate-induced apoptosis in pancreatic beta-cells. Biochem Biophys Res Commun 2010;391:1567–1572 [DOI] [PubMed] [Google Scholar]
- 44.Sol EM, Sargsyan E, Akusjärvi G, Bergsten P. Glucolipotoxicity in INS-1E cells is counteracted by carnitine palmitoyltransferase 1 over-expression. Biochem Biophys Res Commun 2008;375:517–521 [DOI] [PubMed] [Google Scholar]
- 45.Xu L, Han C, Wu T. A novel positive feedback loop between peroxisome proliferator-activated receptor-delta and prostaglandin E2 signaling pathways for human cholangiocarcinoma cell growth. J Biol Chem 2006;281:33982–33996 [DOI] [PubMed] [Google Scholar]
- 46.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med 1991;11:81–128 [DOI] [PubMed] [Google Scholar]
- 47.Miwa I, Ichimura N, Sugiura M, Hamada Y, Taniguchi S. Inhibition of glucose-induced insulin secretion by 4-hydroxy-2-nonenal and other lipid peroxidation products. Endocrinology 2000;141:2767–2772 [DOI] [PubMed] [Google Scholar]
- 48.Srivastava S, Chandra A, Wang LF, et al. Metabolism of the lipid peroxidation product, 4-hydroxy-trans-2-nonenal, in isolated perfused rat heart. J Biol Chem 1998;273:10893–10900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Canuto RA, Ferro M, Muzio G, et al. Role of aldehyde metabolizing enzymes in mediating effects of aldehyde products of lipid peroxidation in liver cells. Carcinogenesis 1994;15:1359–1364 [DOI] [PubMed] [Google Scholar]
- 50.Prasad KM, Thimmalapura PR, Woode EA, Nadler JL. Evidence that increased 12-lipoxygenase expression impairs pancreatic beta cell function and viability. Biochem Biophys Res Commun 2003;308:427–432 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.