

Abstract
OBJECTIVE
Islet-specific glucose-6-phosphatase catalytic subunit–related protein (IGRP), now known as G6PC2, is a major target of autoreactive T cells implicated in the pathogenesis of type 1 diabetes in both mice and humans. This study aimed to determine whether suppression of G6p2 gene expression might therefore prevent or delay disease progression.
RESEARCH DESIGN AND METHODS
G6pc2−/− mice were generated on the NOD/ShiLtJ genetic background, and glycemia was monitored weekly up to 35 weeks of age to determine the onset and incidence of diabetes. The antigen specificity of CD8+ T cells infiltrating islets from NOD/ShiLtJ G6pc2+/+ and G6pc2−/− mice at 12 weeks was determined in parallel.
RESULTS
The absence of G6pc2 did not affect the time of onset, incidence, or sex bias of type 1 diabetes in NOD/ShiLtJ mice. Insulitis was prominent in both groups, but whereas NOD/ShiLtJ G6pc2+/+ islets contained CD8+ T cells reactive to the G6pc2 NRP peptide, G6pc2 NRP-reactive T cells were absent in NOD/ShiLtJ G6pc2−/− islets.
CONCLUSIONS
These results demonstrate that G6pc2 is an important driver for the selection and expansion of islet-reactive CD8+ T cells infiltrating NOD/ShiLtJ islets. However, autoreactivity to G6pc2 is not essential for the emergence of autoimmune diabetes. The results remain consistent with previous studies indicating that insulin may be the primary autoimmune target, at least in NOD/ShiLtJ mice.
The G6PC gene family is composed of three members, G6PC, G6PC2, and G6PC3 (1), each encoding multispanning membrane proteins located in the endoplasmic reticulum that catalyze the hydrolysis of glucose-6-phosphate (G6P) to glucose and inorganic phosphate. G6PC (G6Pase) is predominantly expressed in liver and kidney, where it catalyzes the terminal step in the gluconeogenic and glycogenolytic pathways (1). G6PC3 (UGRP; G6Pase-β) is ubiquitously expressed, although its function in vivo is unclear (1). G6PC2 (IGRP) is expressed specifically in pancreatic islet β-cells (1). G6PC2 is proposed to act as a negative component of the β-cell glucose sensor, opposing the action of glucokinase and thereby modulating glucose-stimulated insulin secretion (2). Accordingly, G6pc2−/− mice exhibit a 15% reduction of fasting blood glucose but are otherwise metabolically and developmentally indistinguishable from wild-type littermates. Likewise, genome-wide association studies have linked polymorphic variants in G6PC2 to variations in fasting blood glucose levels in humans (3,4).
G6pc2 was implicated as a major autoantigen in the NOD mouse model of type 1 diabetes, following its identification as the molecular target of a CD8+ T-cell clone (NY8.3) isolated from NOD/ShiLtJ mice (5,6). This clone, cognate mimotope peptides, and 8.3 T cell receptor (TCR) transgenic mice have provided insight into the role of CD8+ T cells in type 1 diabetes and the importance of phenomena such as TCR avidity and maturation as determinants of autoimmunity and escape from immune tolerance (6). In NOD/ShiLtJ mice, up to 40% of the CD8+ cells infiltrating the islet were found to recognize the cognate peptide of the 8.3 TCR (G6pc2 amino acids 206–214) (5). Subsequent reports have shown that G6pc2 also is recognized by CD4+ T cells in NOD/ShiLtJ mice (7). Most importantly, T-cell responses to G6PC2 peptides have been reported in human type 1 diabetes (8,9) and in humanized NOD/ShiLtJ mice (10). The observation that the administration of G6pc2-derived peptides abrogate or delay the disease process in NOD/ShiLtJ mice (7,11) offers the prospect that the protein or its encoding DNA, or the suppression of G6PC2 gene expression, might be used as tolerogenic therapeutic strategies to delay or prevent the onset of type 1 diabetes in humans. A key question in this context is where G6PC2 stands in the sequence of immunological events that characterize the initiation of autoimmunity, epitope spreading, and skewing of effector and regulatory T-cell subsets that characterize the progression of the disease from benign peri-insulitis to infiltrating β-cell destruction. We address this issue here by investigating the incidence and progression of type 1 diabetes in NOD/ShiLtJ mice lacking G6pc2.
RESEARCH DESIGN AND METHODS
Animal care.
The animal housing and surgical facilities used for the mice in these studies meet the Association for Assessment and Accreditation of Laboratory Animal Care International standards. All animal protocols were approved by the Vanderbilt University Medical Center Animal Care and Use Committee.
Generation and genotyping of NOD/ShiLtJ G6pc2−/+ mice.
The generation of G6pc2 knockout mice on a mixed 129/SvEvBrd × C57BL/6 background has been previously described (2). The NOD/ShiLtJ G6pc2−/− congenic strain was developed using a speed congenic breeding strategy (12) combined with the analysis of single nucleotide polymorphisms (SNPs) that distinguished between the NOD/ShiLtJ, 129/SvEv, and C57BL/6 genomes. Using this approach, backcrossing was complete after eight generations, although diabetes appeared after four generations and backcrossing continued for nine generations. Mice were genotyped as previously described (2). Wild-type animals for these studies were generated by interbreeding NOD/ShiLtJ G6pc2−/+ mice.
Determination of disease status.
Intercrossed NOD/ShiLtJ G6pc2−/+ mice were monitored for the development of diabetes from 10 weeks of age by weekly testing for morning glycosuria (Chemstrip 2 GP urinalysis strips; Roche). Mice with urine glucose concentrations >250 mg/dL, subsequently confirmed 24 h later, were designated as having diabetes, at which time the mice were killed.
Insulitis scoring.
Serial paraffin sections from wild-type and NOD/ShiLtJ G6pc2−/− mice were stained using immunoperoxidase methods for the presence of insulin and glucagon before counterstaining with methyl green. Slides were then scanned in Aperio Scanscope (Aperio Technologies, Vista, CA), and varying degrees of insulitis were visualized and scored on ImageScope software using an arbitrary scale (0 = no insulitis, 1 = peri-islet insulitis, 2 = intermediate insulitis, 3 = intraislet insulitis, and 4 = atropic islets [residual glucagon cells only]) in a double-blind study.
Isolation of pancreatic islets and analysis of antigen-specific CD8+ T cells using major histocompatibility complex–class I peptide tetramer staining.
Islet isolation following collagenase perfusion of the common bile duct was carried out by the Islet Procurement and Analysis Core of the Vanderbilt Diabetes Research and Training Center (http://www.mc.vanderbilt.edu/diabetes/drtc). Isolated islets were washed and then resuspended in Hanks’ buffered salt solution containing DNase I (Worthington Biochemical, Lakewood, NJ) and handpicked using a silinized micropipet. Isolated islets were resuspended in RPMI-1640 medium supplemented with 10% FBS (HyClone, Logan, UT) and 50 units/mL human recombinant interleukin-2 (BD Pharmingen, San Diego, CA) and cultured intact in 24-well tissue-culture plates (~50 islets per well) for 9 days.
The following phycoerythrin (PE)-conjugated, peptide-loaded major histocompatibility complex (MHC)-class I tetramers were obtained from the National Institutes of Health Tetramer Core Facility (Emory University, Atlanta, GA): MimA2, a mimotope peptide of dystrophia myotonica kinase that is recognized by AI4-like T cells (13,14) (YAIENYLEL)/H-2Db; NRP-V7, a peptide mimetic of G6pc2206–214 (5) (KYNKANVFL)/H-2Kd; INS-L9, the G9 L variant of murine insulin B15–23 (LYLVCGERL)/H-2Kd; Flu-NP366–374 (ASNENMETM)/H-2Db; and Flu-NP147–155 (TYQRTRALV)/H-2Kd. Fluorescein isothiocyanate (FITC)-conjugated anti-CD8α was purchased from BD Pharmingen. Cells were stained with tetramer and anti-CD8α antibody in a fluorescence-activated cell sorter (FACS) buffer (PBS containing 3% FBS, 1 mmol/L EDTA, and 10 mmol/L HEPES) in a 96-well U-bottom plate at 37°C for 15 min and an additional 15 min at room temperature. Flow cytometry was performed using a FACS Calibur instrument (BD Pharmingen), and the acquired data were analyzed using FlowJo software (Tree Star, Ashland, OR).
Analysis of interferon-γ secretion by islet-resident CD8+ T cells.
Islets were cultured in complete medium as described above, and the culture supernatant was collected at day 9 for measurement of interferon (IFN)-γ by ELISA (BD Biosciences). Alternatively, for intracellular detection of IFN-γ, cells were harvested and stimulated with ionomycin (1 μmol/L) and phorbol myristate acetate (100 ng/mL) for 5 h in the presence of Golgi Plug (BD Biosciences). The cells were then surface stained with anti–CD8-FITC, followed by intracellular staining with anti–IFNγ-PE, according to the manufacturer’s protocol (BD Biosciences).
Peptide immunization of mice.
Wild-type or G6pc2−/− mice were immunized with NRP-V7 peptide (100 μg per mouse) emulsified in complete Freund’s adjuvant. Two weeks later, splenic and draining lymph-node cells were prepared and stained with anti–CD8α-FITC together with PE-labeled NRP-V7 or control Flu-NP tetramer, as described above.
Statistical analyses.
Progression to diabetes and diabetes incidence were calculated using the product-limit method (Kaplan Meier analysis). Statistical significance of the difference between pairs of curves was determined by the log-rank test (Mantel-Cox test using Prism 5 software; GraphPad). P values ≤ 0.05 were considered statistically significant. Data derived from T-cell studies were analyzed with a two-tailed Student t test. P values ≤ 0.05 were considered statistically significant.
RESULTS
Generation and analysis of diabetes in congenic NOD/ShiLtJ G6pc2−/− mice.
A previously described modified G6pc2 allele, generated by replacement of exons 1–3 plus 10 bp of the third intron by a LacZ/Neo cassette (2), was backcrossed onto the NOD/ShiLtJ genetic background using a speed congenic strategy followed by the analysis of specific SNPs surrounding the G6pc2 locus. Intercrosses of heterozygous animals generated 259 pups (137 male) with an approximating Mendelian genotype distribution at 3 weeks (78 G6pc2+/+, 116 G6pc2−/+, and 65 G6pc2−/−).
Monitoring for diabetes in this mouse colony began at 10 weeks of age. Figures 1 and 2 show survival analysis data. A clear sex difference was evident between female and male mice in the median onset of disease in G6pc2+/+ (18 vs. 26 weeks, P = 0.0002), G6pc2−/+ (19 vs. 29 weeks, P < 0.0001), and G6pc2−/− (21 vs. 28 weeks, P = 0.0021) animals (Fig. 1), which was in accord with reported studies on wild-type NOD/ShiLtJ mice (15). However, the key observation was that neither the absence of G6pc2 nor G6pc2 gene dosage affected the median onset of diabetes or final incidence at 35 weeks. Post hoc analysis of data up to 24 weeks indicated a weak trend (P = 0.0924) toward retarded onset of diabetes in male G6pc2−/− animals versus wild-type animals; however, this was not apparent in females (P = 0.6470).
FIG. 1.
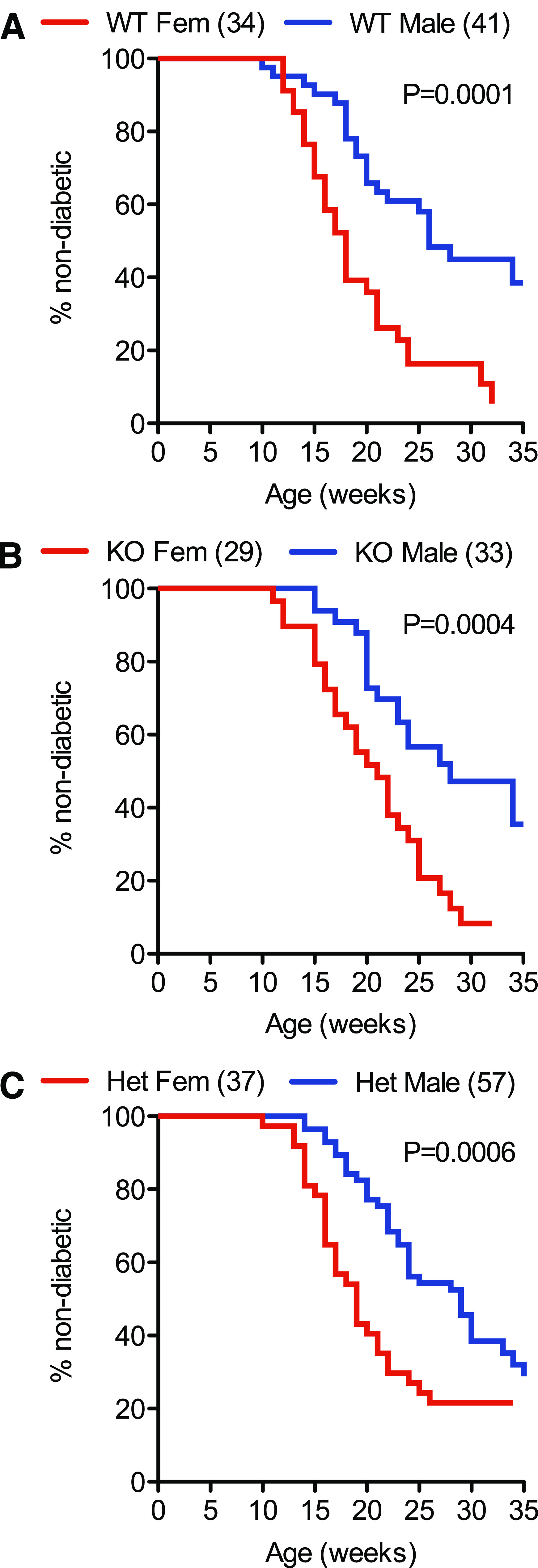
Comparison of the incidence and time of onset of type 1 diabetes in wild-type NOD/ShiLtJ and NOD/ShiLtJ G6pc2−/− mice: the effect of sex. The incidence and time of onset of type 1 diabetes in male and female wild-type NOD/ShiLtJ, NOD/ShiLtJ G6pc2−/+, and NOD/ShiLtJ G6pc2−/− mice was compared starting at the age of 10 weeks. Clear differences were observed between male and female wild-type NOD/ShiLtJ mice (A), between male and female NOD/ShiLtJ G6pc2−/− mice (B), and between male and female NOD/ShiLtJ G6pc2−/+ mice (C). The number of animals studied is indicated in parentheses. Statistical significance between curves was determined by the log-rank (Mantel-Cox) test. Het, heterozygous; KO, knockout; WT, wild-type.
FIG. 2.

Comparison of the incidence and time of onset of type 1 diabetes in wild-type NOD/ShiLtJ and NOD/ShiLtJ G6pc2−/− mice: the effect of G6pc2 gene deletion. The cumulative incidence of type 1 diabetes in male and female wild-type NOD/ShiLtJ, NOD/ShiLtJ G6pc2−/+, and NOD/ShiLtJ G6pc2−/− mice was compared starting at the age of 10 weeks. No differences were observed between female wild-type, NOD/ShiLtJ G6pc2−/+, and NOD/ShiLtJ G6pc2−/− mice (A) or between male wild-type, NOD/ShiLtJ G6pc2−/+, and NOD/ShiLtJ G6pc2−/− mice (B). The number of animals studied is indicated in parentheses. Statistical significance between curves was determined by the log-rank (Mantel-Cox) test. Het, heterozygous; KO, knockout; WT, wild-type.
Histological analyses performed on a series of male G6pc2+/+ and G6pc2−/− mice at 25 weeks of age that exhibited no glycosuria revealed insulitis in both groups, which ranged from apparent normal islets, islets with pronounced peri-insulitis yet strong insulin immunoreactivity, and islets with reduced insulin immunoreactivity and invasive mononuclear cell infiltrates (Fig. 3A and B). Such histopathological lesions typify the presence of autoimmune diabetes in NOD/ShiLtJ animals from as early as 8 weeks of age (16,17). No major differences were observed between G6pc2+/+ and G6pc2−/− mice (Fig. 3B).
FIG. 3.

Insulitis in normoglycemic wild-type and G6pc2-deficient NOD/ShiLtJ mice. Paraffin sections of pancreata removed from 25-week-old normoglycemic male animals were stained by immunoperoxidase methods for the presence of insulin (gray) and glucagon (brown). Varying degrees of peri-islet mononuclear cell infiltration were evident, as visualized by the methyl green counter stain. Representative images (scale bar = 50 μm) are shown in A, whereas insulitis scores from five wild-type and five NOD/ShiLtJ G6pc2−/− mice are shown in B, based on the examination of a total of 127 wild-type and 151 G6pc2−/− islets. KO, knockout; WT, wild-type. (A high-quality digital representation of this figure is available in the online issue.)
Analysis of antigen-specific T-cell responses in congenic NOD/ShiLtJ G6pc2−/− mice.
Diabetes in the NOD/ShiLtJ mouse results from the specific destruction of pancreatic β-cells by a cell-mediated process involving an autoreactive antigen-specific CD4+ and CD8+ T-cell attack in an environment where regulatory T-cell responses are perturbed (6). Molecularly defined targets of pathogenic CD8+ T cells have been well established (6) and include an H-2Kd–restricted peptide of G6pc2206–214 and its mimotope NRP-V7 (6) and a H-2Kd–restricted peptide of the insulin B chain (insulin B15–23 and its variant INS-L9), both of which have been implicated in disease pathogenesis and both of which are recognized by an appreciable proportion of CD8+ T cells residing in islet infiltrates in NOD/ShiLtJ mice (6). We compared the prevalence of islet autoantigen-specific CD8+ T cells in islets isolated from 12-week-old wild-type and NOD/ShiLtJ G6pc2−/− mice. Islets were cultured for 9 days ex vivo in the presence of interleukin-2 to expand CD8+ T cells residing in them. Cells were then stained with a panel of peptide-loaded MHC–class I tetramers bearing the abovementioned G6pc2 and insulin peptides, or influenza peptides as negative controls, and analyzed by flow cytometry. As previously reported, CD8+ T cells reactive to NRP-V7 were the dominant population of CD8+ T cells in wild-type mouse islets, accompanied by a variable, lesser percentage of insulin B15–23–and MimA2-specific T cells (Fig. 4A and B). In contrast, G6pc2−/− mouse islets completely lacked CD8+ T cells reactive to NRP-V7 (Fig. 4A and B) but retained a high proportion of CD8+ T cells specific for insulin B (Fig. 4A and B). The CD8+ T cells in the islets of G6pc2−/− mice were functionally competent, as demonstrated by their capacity to secrete IFN-γ in the culture supernatant (Supplementary Fig. 1A) and by their intracellular expression of IFN-γ (Supplementary Fig. 1B).
FIG. 4.

Islet autoantigen–specific CD8+ T-cell responses in wild-type and G6pc2-deficient NOD/ShiLtJ mice. A and B: Islets were isolated from individual female wild-type or G6pc2−/− NOD/ShiLtJ mice at 12 weeks of age and cultured in the presence of interleukin-2 for 9 days. Cells were then stained with FITC-labeled anti-CD8α and PE-labeled MHC–class I tetramers loaded with the indicated peptides and analyzed by flow cytometry. Tetramers loaded with Flu-NP366–374 or Flu-NP147–155 were used as a negative control for tetramer staining. Data were electronically gated for CD8α-expressing cells. A: Representative flow cytometry data for one wild-type mouse and one G6pc2-deficient mouse. Numbers indicate the percentage of cells within the gated area. B: Summary of the flow cytometry data for four wild-type and four G6pc2-deficient mice from two experiments. C: Splenic and lymph-node cells were prepared from female wild-type or G6pc2−/− NOD/ShiLtJ mice at 12 weeks of age, and the frequency of NRP-V7–specific CD8+ T cells was determined by tetramer staining. Data were electronically gated for CD8α-expressing cells. Representative flow cytometry data are shown. D: Wild-type or G6pc2−/− NOD/ShiLtJ female mice were immunized with NRP-V7 peptide (100 μg per mouse) emulsified in complete Freund’s adjuvant. Two weeks later, splenic and draining lymph-node cells were prepared and stained with anti–CD8α-FITC and NRP-V7 or control Flu-NP tetramer and analyzed by flow cytometry. Data were electronically gated for CD8α-expressing cells. Representative flow cytometry data are shown. NS, not significant; WT, wild-type. (A high-quality color representation of this figure is available in the online issue.)
Similar to what we observed in the islets, G6pc2−/− mice also lacked NRP-V7–reactive CD8+ T cells in the spleen and lymph nodes (Fig. 4C). Nevertheless, these animals were capable of generating NRP-V7–specific CD8+ T cells following immunization with NRP-V7 peptide emulsified in complete Freund’s adjuvant (Fig. 4D).
DISCUSSION
The NOD/ShiLtJ mouse has proven to be a robust and valuable experimental model for the investigation of the pathogenesis of type 1 diabetes and exhibits many features of the human clinical disease, including overlapping autoantigen specificity at the humoral and cellular level and shared genetic susceptibility, most notably the MHC–class II loci (16,17). In the current study, we used gene ablation to examine the role of G6pc2 in the development of type 1 diabetes in NOD/ShiLtJ mice.
CD8+ T cells infiltrating the islets of NOD/ShiLtJ G6pc2−/− mice failed to recognize the mimotope NRP-V7 peptide, indicating that the cognate peptide encoded by exon 5 is not expressed (Fig. 4). This also indicates that CD8+ T-cell reactivity to NRP-V7 is not the result of reactivity or molecular mimicry afforded by another gene family member or unrelated protein. However, the diabetes that emerged in G6pc2−/− and G6pc2−/+ offspring on the NOD/ShiLtJ background was indistinguishable from that in G6pc2+/+ animals in terms of its acute clinical onset (Fig. 2). Histopathological analysis showing monocytic infiltrates and attrition of β-cell numbers and insulin immunoreactivity were indicative of type 1 autoimmune disease (Fig. 3). It is thus apparent that G6pc2−/− animals progressed to invasive insulitis. It is concluded that although G6pc2-reactive CD8+ T cells are a prominent feature in wild-type NOD/ShiLtJ mice and the dominant population of CD8+ T cells that invade islets (Fig. 4) (6), they are not necessary for the emergence of disease in the absence of autoreactivity to the protein. A similar conclusion was reached by Wang et al. (18), who examined the incidence and kinetics of diabetes in NOD/ShiLtJ mice expressing a nonimmunoreactive form of G6pc2.
Similar conclusions also have been reached from gene-ablation studies in NOD/ShiLtJ mice with GAD65, IA2, IA2β (phogrin), and IA2/IA2β double-knockout animals (16,17). In contrast, NOD/ShiLtJ mice with deletion of both the endogenous insulin I and insulin II alleles carrying an insulin transgene with an inactivated CD4+ T-cell epitope do not develop diabetes (19). In addition, transgenic NOD/ShiLtJ mice tolerized to proinsulin do not develop proinsulin or G6pc2 autoreactive T cells or diabetes; whereas although transgenic NOD/ShiLtJ mice tolerant to G6pc2 do not develop G6pc2 autoreactive T cells, they still develop proinsulin autoreactive T cells and diabetes (20). Even in transgenic mice with excess G6pc2 autoreactive T cells, diabetes development is markedly reduced in the absence of proinsulin autoreactive T cells (21). These observations are consistent with the concept that proinsulin is the primary autoantigen in type 1 diabetes in the NOD/ShiLtJ mouse (15). Whether this also is the case in humans is another issue given the complexity of the MHC association with the disease and differences in the pathophysiology of type 1 diabetes in mice and humans (22).
ACKNOWLEDGMENTS
Research in the laboratory of R.M.O. was supported by National Institutes of Health (NIH) Grant DK-076027 and by NIH Grant P60-DK-20593, to the Vanderbilt Diabetes Research Training Center. Research in the laboratory of J.C.H. was supported by the American Diabetes Association (9901-116), NIH grants DK-076027 and DK-52068, Juvenile Diabetes Research Foundation Grant 4-2007-1056, and the Barbara Davis Center Diabetes and Endocrinology Research Center (P30-DK-57516). Research in the laboratory of L.V.K. was supported by a postdoctoral fellowship from the National Multiple Sclerosis Society (to V.V.P.) and NIH Grant HL-089667 (to L.V.K.). L.D.P. was supported by the Vanderbilt Molecular Endocrinology Program Grant 5T32-DK-07563.
No potential conflicts of interest relevant to this article were reported.
J.K.O. performed genotyping and diabetes incidence analyses. V.V.P. performed isolated islet T-cell analyses and wrote parts of the manuscript. Y.W. performed genotyping and diabetes incidence analyses. N.K.J. performed diabetes incidence analyses. S.A.S. performed immunohistochemical analyses and wrote parts of the manuscript. R.W. performed speed congenic analyses. C.E.L. performed immunohistochemical analyses. L.D.P. performed diabetes incidence analyses. J.C.H. was the principal investigator for the immunohistochemical analyses and wrote parts of the manuscript. L.V.K. was the principal investigator for the isolated islet T-cell analyses and wrote parts of the manuscript. R.M.O. was the principal investigator for the diabetes incidence analyses and wrote parts of the manuscript.
The authors thank P. Jacobson (Abbott Pharmaceuticals) for assistance with the initiation of this project and Marcia McDuffie and Jerry L. Nadler (Strelitz Diabetes Center, Eastern Virginia Medical School, Norfolk, VA) for advice on the performance of diabetes incidence studies. The authors also thank Erin Stewart (Barbara Davis Center for Childhood Diabetes, Anschutz Medical Center, University of Colorado Denver, Aurora, CO) for help with the speed congenic backcrossing procedure and Dobromir Slavov (Department of Cardiology, Anschutz Medical Center, University of Colorado Denver, Aurora, CO) for performing the SNP analyses. The authors thank the NIH tetramer facility at Emory University (Atlanta, GA) for providing MHC–class I peptide tetramers.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db11-0220/-/DC1.
REFERENCES
- 1.Hutton JC, O’Brien RM. Glucose-6-phosphatase catalytic subunit gene family. J Biol Chem 2009;284:29241–29245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Y, Martin CC, Oeser JK, et al. Deletion of the gene encoding the islet-specific glucose-6-phosphatase catalytic subunit-related protein autoantigen results in a mild metabolic phenotype. Diabetologia 2007;50:774–778 [DOI] [PubMed] [Google Scholar]
- 3.Bouatia-Naji N, Rocheleau G, Van Lommel L, et al. A polymorphism within the G6PC2 gene is associated with fasting plasma glucose levels. Science 2008;320:1085–1088 [DOI] [PubMed] [Google Scholar]
- 4.Chen WM, Erdos MR, Jackson AU, et al. Variations in the G6PC2/ABCB11 genomic region are associated with fasting glucose levels. J Clin Invest 2008;118:2620–2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lieberman SM, Evans AM, Han B, et al. Identification of the beta cell antigen targeted by a prevalent population of pathogenic CD8+ T cells in autoimmune diabetes. Proc Natl Acad Sci USA 2003;100:8384–8388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsai S, Shameli A, Santamaria P. CD8+ T cells in type 1 diabetes. Adv Immunol 2008;100:79–124 [DOI] [PubMed] [Google Scholar]
- 7.Mukherjee R, Wagar D, Stephens TA, Lee-Chan E, Singh B. Identification of CD4+ T cell-specific epitopes of islet-specific glucose-6-phosphatase catalytic subunit-related protein: a novel beta cell autoantigen in type 1 diabetes. J Immunol 2005;174:5306–5315 [DOI] [PubMed] [Google Scholar]
- 8.Yang J, Danke NA, Berger D, et al. Islet-specific glucose-6-phosphatase catalytic subunit-related protein-reactive CD4+ T cells in human subjects. J Immunol 2006;176:2781–2789 [DOI] [PubMed] [Google Scholar]
- 9.Jarchum I, Nichol L, Trucco M, Santamaria P, DiLorenzo TP. Identification of novel IGRP epitopes targeted in type 1 diabetes patients. Clin Immunol 2008;127:359–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takaki T, Marron MP, Mathews CE, et al. HLA-A*0201-restricted T cells from humanized NOD mice recognize autoantigens of potential clinical relevance to type 1 diabetes. J Immunol 2006;176:3257–3265 [DOI] [PubMed] [Google Scholar]
- 11.Han B, Serra P, Amrani A, et al. Prevention of diabetes by manipulation of anti-IGRP autoimmunity: high efficiency of a low-affinity peptide. Nat Med 2005;11:645–652 [DOI] [PubMed] [Google Scholar]
- 12.Serreze DV, Chapman HD, Varnum DS, et al. B lymphocytes are essential for the initiation of T cell-mediated autoimmune diabetes: analysis of a new “speed congenic” stock of NOD.Ig mu null mice. J Exp Med 1996;184:2049–2053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takaki T, Lieberman SM, Holl TM, et al. Requirement for both H-2Db and H-2Kd for the induction of diabetes by the promiscuous CD8+ T cell clonotype AI4. J Immunol 2004;173:2530–2541 [DOI] [PubMed] [Google Scholar]
- 14.Lieberman SM, Takaki T, Han B, Santamaria P, Serreze DV, DiLorenzo TP. Individual nonobese diabetic mice exhibit unique patterns of CD8+ T cell reactivity to three islet antigens, including the newly identified widely expressed dystrophia myotonica kinase. J Immunol 2004;173:6727–6734 [DOI] [PubMed] [Google Scholar]
- 15.Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 2010;464:1293–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol 2005;23:447–485 [DOI] [PubMed] [Google Scholar]
- 17.Atkinson MA, Leiter EH. The NOD mouse model of type 1 diabetes: as good as it gets? Nat Med 1999;5:601–604 [DOI] [PubMed] [Google Scholar]
- 18.Wang J, Tsai S, Shameli A, Yamanouchi J, Alkemade G, Santamaria P. In situ recognition of autoantigen as an essential gatekeeper in autoimmune CD8+ T cell inflammation. Proc Natl Acad Sci USA 2010;107:9317–9322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakayama M, Abiru N, Moriyama H, et al. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature 2005;435:220–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krishnamurthy B, Dudek NL, McKenzie MD, et al. Responses against islet antigens in NOD mice are prevented by tolerance to proinsulin but not IGRP. J Clin Invest 2006;116:3258–3265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krishnamurthy B, Mariana L, Gellert SA, et al. Autoimmunity to both proinsulin and IGRP is required for diabetes in nonobese diabetic 8.3 TCR transgenic mice. J Immunol 2008;180:4458–4464 [DOI] [PubMed] [Google Scholar]
- 22.Itoh N, Hanafusa T, Miyazaki A, et al. Mononuclear cell infiltration and its relation to the expression of major histocompatibility complex antigens and adhesion molecules in pancreas biopsy specimens from newly diagnosed insulin-dependent diabetes mellitus patients. J Clin Invest 1993;92:2313–2322 [DOI] [PMC free article] [PubMed] [Google Scholar]