

Abstract
OBJECTIVE
Autoimmune diseases, including type 1 diabetes, are thought to have a Th17-cell bias and/or a T-regulatory cell (Treg) defect. Understanding whether this is a hallmark of patients with type 1 diabetes is a crucial question that is still unsolved, largely due to the difficulties of accessing tissues targeted by the disease.
RESEARCH DESIGN AND METHODS
We phenotypically and functionally characterized Th17 cells and Tregs residing in the pancreatic-draining lymph nodes (PLNs) of 19 patients with type 1 diabetes and 63 nondiabetic donors and those circulating in the peripheral blood of 14 type 1 diabetic patients and 11 healthy subjects.
RESULTS
We found upregulation of Th17 immunity and functional defects in CD4+CD25bright Tregs in the PLNs of type 1 diabetic subjects but not in their peripheral blood. In addition, the proinsulin-specific Treg-mediated control was altered in the PLNs of diabetic patients. The dysfunctional Tregs isolated from diabetic subjects did not contain contaminant effector T cells and were all epigenetically imprinted to be suppressive, as defined by analysis of the Treg-specific demethylated region within the forkhead box P3 (FOXP3) locus.
CONCLUSIONS
These data provide evidence for an unbalanced immune status in the PLNs of type 1 diabetic subjects, and treatments restoring the immune homeostasis in the target organ of these patients represent a potential therapeutic strategy.
Type 1 diabetes is a chronic autoimmune disease caused by the selective destruction of pancreatic insulin-producing β-cells (1). Autoimmunity commonly occurs when central and/or peripheral tolerance barriers are broken down, allowing the activation of self-reactive T cells (2), which are clearly pivotal for type 1 diabetes development (3). Animal models of diabetes, such as NOD mice, have indicated that Th1 cells that secrete interferon-γ (INF-γ) are key in the destruction of β-cells as activators of CD8+ T cells (4). More recently, Th17 immunity has also been implicated in type 1 diabetes, as supported by the observation that interleukin-17 (IL-17) is expressed in the pancreas of NOD mice, and that inhibition of IL-17 leads to delayed diabetes onset (5,6). In humans, Th17 cells are defined as CD4+ T cells that produce IL-17 and are characterized by the expression of a unique pattern of surface receptors, including CCR4, CCR6, and CD161 (7). Th17 immunity is increased in the blood of patients with type 1 diabetes compared with healthy subjects (8,9), but how these findings are relevant to the organ targeted by autoimmune diabetes is still unknown.
Self-reactive T cells in healthy individuals are controlled by peripheral tolerance mechanisms, in which natural T regulatory cells (Tregs) have emerged as primary mediators (10,11). Natural Tregs are characterized by the high expression of the IL-2Rα chain (CD25) and the transcription factor forkhead box P3 (FOXP3) (12). Importantly, CD25 and FOXP3 are also upregulated in human activated non-Tregs, and their expression can easily be altered by inflammation and/or drug administration (13). We found that natural Tregs differ from other T-cell populations in their DNA methylation status of a defined region within the FOXP3 locus, termed the Treg-specific demethylated region (TSDR); demethylation of this region is observed exclusively in natural Tregs, but not in activated FOXP3+ non-Tregs or in TGF-β–induced Tregs (14–16). The TSDR is currently considered a reliable marker for bona fide Tregs (17). However, it is still uncertain whether T cells completely demethylated at the FOXP3 locus automatically possess a functional regulatory activity.
Recent studies have shown that Tregs in the pancreas of diabetic NOD mice express low levels of CD25 owing to diminished levels of IL-2, which is a critical factor for Treg cell growth (18). These tissue-infiltrating Tregs are prone to apoptosis and thus fail to control autoimmune responses that lead to diabetes (18). In addition, lymphopenic and inflammatory environments can both lead to loss of FOXP3 expression in murine Tregs (19–21). In humans, the data are less comprehensive, and to our knowledge, all of the studies performed so far on Tregs in type 1 diabetes have been conducted on peripheral blood (PB). They demonstrate that the frequency of peripheral Tregs is not altered but that they may have an increased sensitivity to apoptosis (22). Their in vitro suppressive function is slightly reduced in some experimental settings (23–26), but not in others (27,28). In addition, recent data suggest that effector T cells in patients with diabetes are refractory to inhibition by Tregs (29,30) and that aberrant IL-2 signaling in CD4+ T cells contributes to reduced FOXP3 expression (31). Overall, these data corroborate the idea that a defect in Tregs in diabetes may contribute to the loss of self-tolerance, but we do not yet have solid evidence to support this hypothesis.
Self-reactive T cells and Tregs in patients should preferably be considered and assessed in the context of the organ targeted by the autoimmune process rather than in the circulation. We initiated the current study to investigate effector T cells and Tregs in the pancreatic-draining lymph nodes (PLNs) of patients affected by type 1 diabetes compared with those circulating in their PB.
RESEARCH DESIGN AND METHODS
Donors and sample collection.
PB was collected before transplantation from patients with type 1 diabetes undergoing pancreas or pancreas/kidney transplant at San Raffaele Hospital. These patients donated one PLN, which was collected during the surgical procedure using scissors (Supplementary Fig. 1). Five to 10 PLNs were also collected from the pancreata of nondiabetic brain-dead multiorgan donors received at the Islet Isolation Facility of San Raffaele Hospital. Spleens of six nondiabetic donors were also received and were used to generate dendritic cells (DC) for the antigen-specific assays. Finally, PB was collected from healthy individuals. All studies were approved by the local ethics committee (protocols: HSR-TIGET 004, TIGET PERIBLOOD). Diabetic patients and healthy control subjects provided written informed consent before any study procedure.
Cell isolation.
Lymphocytes were extracted from PLNs by mechanical dissociation. PB mononuclear cells were isolated by density-gradient centrifugation on Lymphoprep (Axis-Shield, Oslo, Norway) as previously described (32).
Phenotype analysis.
The phenotype analysis was performed by flow cytometry on freshly isolated lymphocytes. The intracytoplasmic staining was performed upon 12-O-tetradecanoylphorbol-13-acetate/ionomycin activation for 5 h in the presence of GolgiStop (BD Biosciences, San Jose, CA), followed by permeabilization with saponin (Sigma, St. Louis, MO). All monoclonal antibodies (mAbs) were obtained from BD Pharmingen (San Jose, CA). Samples were acquired using the BD FACSCanto II (Becton Dickinson, San Jose, CA).
Analysis of the methylation status of the FOXP3 TSDR.
The assay was performed by Epiontis, as described before (16).
Functional assays.
To perform the polyclonal-suppressive assay, part of the PLN was sorted by flow cytometry (Supplementary Fig. 2). The remaining unfractionated PLN cells were stained with carboxy fluorescein succinimidyl ester (CFSE; Molecular Probes, Eugene, OR) (33) and cultured in 96-well plates (21.000 cells/well) in X-vivo 15 medium and 5% pooled AB human serum (Lonza, Basel, Switzerland). Cells were stimulated with anti-CD3, anti-CD2, and anti-CD28–coated beads (Treg Inspector, Miltenyi Biotech, Bergisch Gladbach, Germany), at a ratio of three beads per cell. Sorted CD4+CD25bright T cells (hereafter referred to as CD25bright) were added at a ratio of one to three responder PLN cells. To perform antigen-specific proliferation and suppression, we followed our previously published protocol (11). On day 6 after activation, 100 μL of supernatant was collected for cytokine analysis by the Luminex bead system (Bioplex, Bio-Rad, Hercules, CA). All of the cells were harvested, stained with anti-CD3 APC mAb (Becton Dickinson) and 7-aminoactinomycin D (BD Biosciences) to exclude dead cells, and analyzed by flow cytometry. The performance of this antigen-specific proliferation test was assessed by using total PB mononuclear cells isolated from healthy subjects. An average of 8% of CD3+ T cells proliferated in response to proinsulin (n = 2), proving the goodness of our assay (data not shown).
RT-PCR.
Total RNA was extracted from 5,000 cells with the RNeasy kit (Qiagen, Valencia, CA), and cDNA was synthesized using the High-Capacity cDNA Archive kit (Applied Biosystems, Foster City, CA). A preamplification step (Assay on Demand, Applied Biosystems) was performed using Taqman PreAmp Master Mix (Applied Biosystems). Levels of human hypoxanthine phosphoribosyltransferase and mRNAs were determined using Assay on Demand, TaqMan Universal PCR Master Mix, and ABI PRISM 7700 Sequencer detector (Applied Biosystems).
Statistical analysis.
Comparisons between groups were performed using the unpaired, two-tailed Student t test or the Mann-Whitney U test. For the analysis of paired samples in the suppression assay, the Wilcoxon test was applied. The linear regression was used to determine the correlation between variables. For all analyses, a two-tailed P value ≤ 0.05 was considered significant. Statistical analyses were performed using Stata 10.1 software (Stata Corp., College Station, TX).
RESULTS
Th17 cells are expanded in the PLNs of type 1 diabetic patients.
PLNs were obtained from 19 subjects with long-term type 1 diabetes and from 63 nondiabetic brain-dead multiorgan donors. PB was also collected from 14 type 1 diabetic patients who each donated a PLN, from 4 type 1 diabetic subjects who did not donate the PLN, and from 11 healthy control subjects. Donor characteristics are summarized in Table 1, and detailed information on each diabetic patient is given in Supplementary Table 1.
TABLE 1.
Characteristics of type 1 diabetic patients and nondiabetic donors
| Donor group and sample |
||||||
|---|---|---|---|---|---|---|
| T1D PLN | ND PLN | T1D PB | HC PB | |||
| Characteristic | n = 19 | n = 63 | P | n = 18 | n = 11 | P |
| Age, years | 41 (24–56) | 49 (11–66) | 0.0015* | 40 (30–56) | 34 (27–39) | 0.0196† |
| Men | 13 (68) | 33 (52) | NS | 12 (67) | 4 (36) | NS‡ |
| Type 1 diabetes duration, years | 27 (7–44) | NA | 26 (7–44) | NA | ||
| EIR, unit/day | 36 (11–52) | NA | 36 (12–68) | NA | ||
| Intensive care unit stay, days | NA | 4 (1–27) | NA | NA | ||
Continuous data are expressed as mean (range), and categoric data as n (%). EIR, exogenous insulin requirements; HC, healthy control subjects; NA, not applicable; ND, nondiabetic donors; T1D, type 1 diabetic patients.
*Mann-Whitney–Wilcoxon test.
†t Test with unequal variances, χ2 test.
‡Fisher exact t test.
The percentages of total CD3+ T cells and of CD3+CD4+ T cells were similar in diabetic patients and nondiabetic donors in the PLNs and PB (Fig. 1A). Conversely, the frequency of CD3+CD8+ T cells was higher in the PLNs of diabetic patients (Fig. 1A), and this led to a lower CD4/CD8 T-cell ratio in diabetic donors compared with nondiabetic subjects (Supplementary Table 2). The antigen-experienced T cells, defined by the expression of CD45RO, were enriched within the lymph node and circulating CD4+ T cells, but not within the CD8+ T cells, of diabetic subjects (Fig. 1B). The expansion of CD4+CD45RO+ T cells observed in diabetic patients was independent of donor age (data not shown).
FIG. 1.
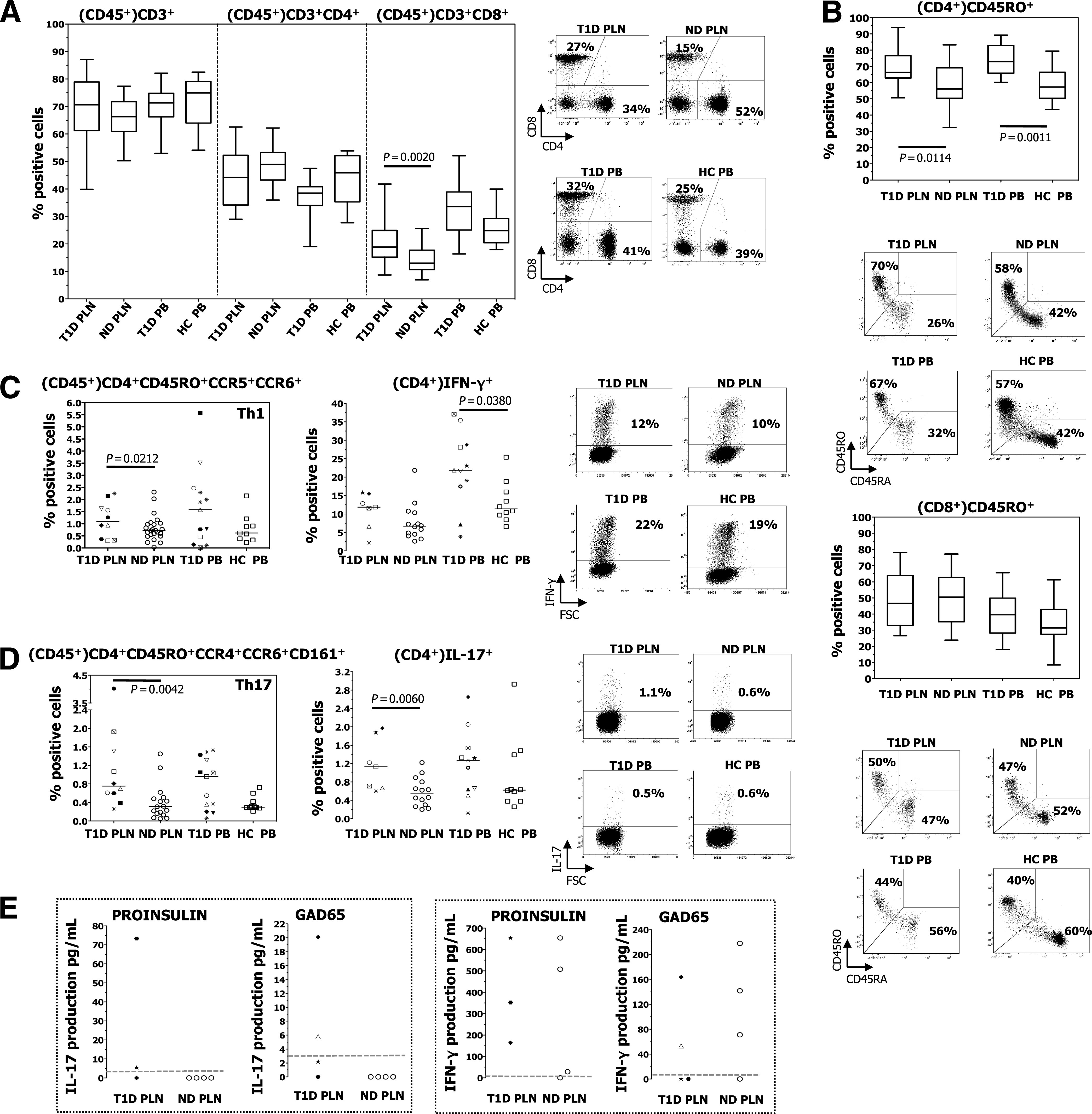
Frequency of T-cell subsets. The PLNs and PB isolated from diabetic (T1D) and nondiabetic donors (ND) had comparable percentages of isolated live cells (all >90%), and the PB contained similar cell numbers in the two donor groups (data not shown). The number of cells within the PLN was difficult to compare due to the manual isolation of the lymphocytes and the variability in the size of the PLNs. A: Box plots represent percentages of CD3+ T cells, CD3+CD4+ T cells, and CD3+CD8+ T cells all within CD45+ cells determined by flow cytometry–based analysis. The number of donors in each group is as follows: T1D PLN, n = 18; ND PLN, n = 58; T1D PB, n = 16; and healthy control subjects (HC) PB, n = 10. The horizontal line in the middle of each box indicates the median; the top and bottom borders of the box mark the 75th and 25th percentiles, respectively; and the whiskers mark the minimum and maximum. One representative dot plot of CD4 and CD8 cells (within CD45+ cells) with relative percentages is shown. B: Box plots represent the frequency of CD45RO+ T cells within CD4+ and CD8+ T cells determined by flow cytometry–based analysis. The number of donors in each group is as follows: T1D PLN, n = 13; ND PLN, n = 37; T1D PB, n = 14; and HC PB, n = 10. One representative dot plot of CD45RO/CD45RA (within CD4+ and CD8+ T cells) with relative percentages is shown. C: The frequency of Th1 cells was measured by flow cytometry (left), and by IFN-γ+–producing CD4+ T cells (right). One representative dot plot of IFN-γ+ cells (within CD4+ T cells) with relative percentages is shown. D: The frequency of Th17 cells was measured by surface phenotype (left) and by IL-17–producing CD4+ T cells (right). One representative dot plot of IL-17+ cells (within CD4+ T cells) with relative percentages is shown. E: The IL-17 (left) and IFN-γ (right) produced by total PLN in response to proinsulin and GAD65 were tested. Dotted lines indicate the detection level of the assay. Each symbol identifies a patient (as described in Supplementary Table 1), and lines indicate median values. The Mann-Whitney–Wilcoxon test and the t test with unequal variances were used for group comparisons.
To determine the frequency of the various T-helper cell subsets within the antigen-experienced CD4+ T cells, we tested the frequency of Th1 and Th17 cells by the expression of specific chemokine-receptors by CD4+CD45RO+ T cells (i.e., CCR5+CCR6+ and CCR4+CCR6+CD161+, respectively) (34,35) (Supplementary Fig. 3), and by cytokine-release (i.e., IFN-γ and IL-17, respectively) upon polyclonal activation of CD4+ T cells. Th1 cells were expanded only in the PLNs of diabetic donors, as determined by chemokine-receptor expression but not by IFN-γ release. However, Th1 cells in the PB of diabetic patients were slightly increased compared with those of healthy control subjects, as determined by IFN-γ production but not by chemokine receptor expression (Fig. 1C). Th2 cells were equally present in all tissues and donors analyzed (data not shown). Th17 cells were significantly expanded only in the PLNs of diabetic patients (Fig. 1D). In addition, IL-17 was produced by the PLNs of diabetic donors upon diabetes-related antigen-specific activation (i.e., proinsulin and GAD65), whereas the same IL-17 was never released by the PLNs of nondiabetic subjects. IFN-γ was secreted at similar levels upon diabetes-related antigen-specific activation of the PLNs isolated from diabetic and nondiabetic individuals (Fig. 1E). The results obtained with the few PLNs of diabetic patients that we could test have thus far eluded attempts to establish any correlation between anti-GAD65 autoAb (reported in the Supplementary Table 1) and in vitro reactivity to GAD65. These data show that Th17 immunity is enhanced in the PLNs isolated from long-term diabetic patients but not in their PB.
FOXP3-expressing CD4+ T cells are reduced, whereas epigenetically imprinted Tregs are similar in the PLNs of diabetic patients compared with those of nondiabetic donors.
The frequency of Tregs was first determined by flow cytometry through analysis of CD25bright within the CD45+ cells, of FOXP3+CD127− within the CD25bright T cells, and of CD25bright FOXP3+CD127− within the CD45+ cells. All three analyses revealed a significant Treg reduction only in the PLNs of patients with type 1 diabetes and not, as already largely demonstrated by us and others (23,24,26–28), in their PB (Fig. 2A). The amount of FOXP3 assessed by median fluorescence intensity (MFI) expressed in CD4+ T cells and in CD25bright T cells was similar within the PLNs of diabetic and nondiabetic donors (Supplementary Table 3). Because CD25 and FOXP3 are markers that can be altered by the environment (13), we performed the Treg-specific demethylated region (TSDR) assay on the total cells isolated from the PLNs and PB. Unlike the phenotype analysis, this readout identifies epigenetically imprinted Tregs (14–16) and demonstrated that the Treg frequency was similar among diabetic and nondiabetic donors, both in the PLNs and PB (Fig. 2B). Thus, the frequency of T cells epigenetically marked to be Tregs is similar in the PLNs and PB of diabetic and nondiabetic individuals, whereas the frequency of T cells that express the FOXP3 protein is reduced only in the target organ of diabetic patients.
FIG. 2.
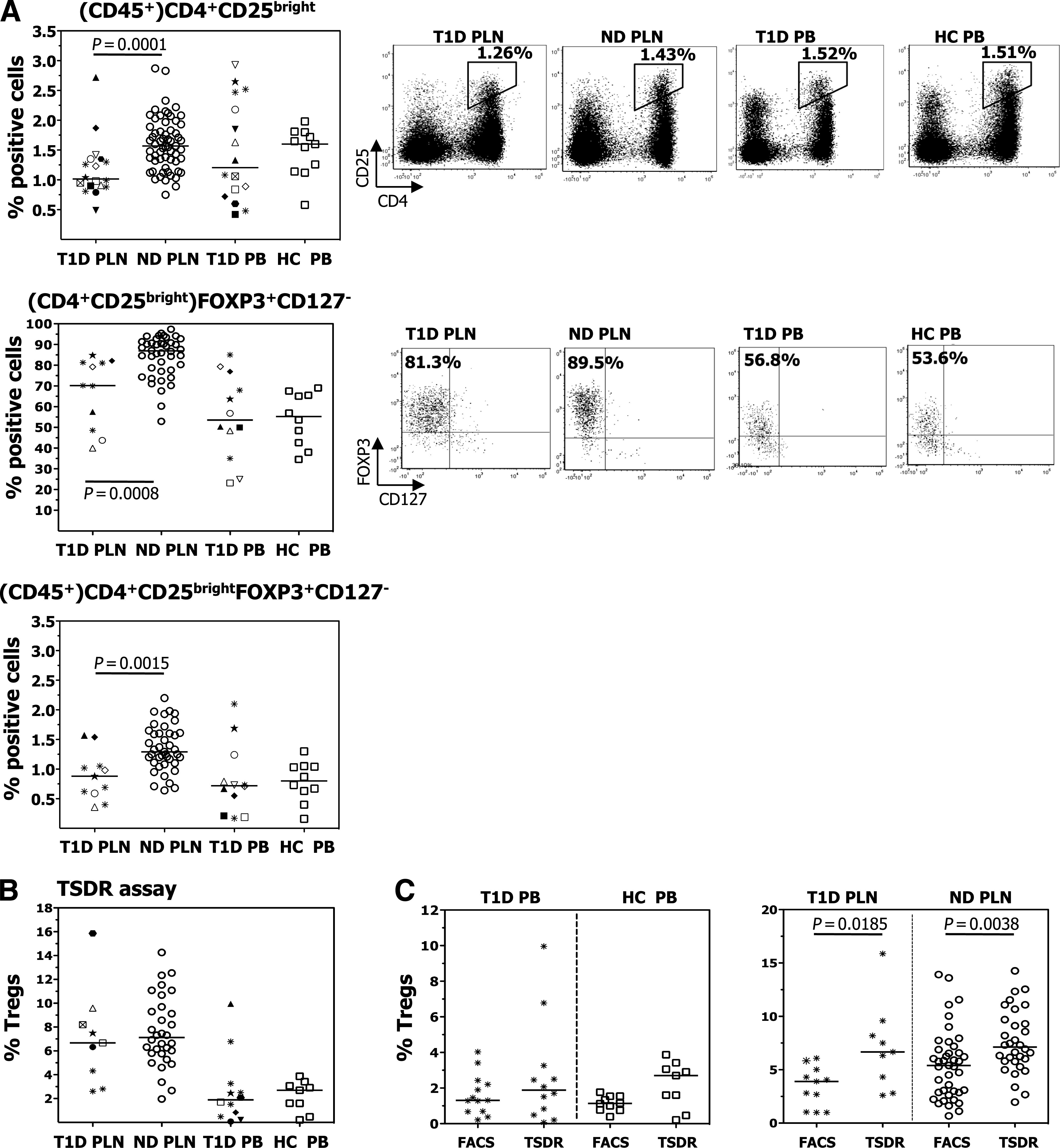
Frequency of Tregs. A: The frequency of CD25bright T cells within CD45+ cells (upper), of FOXP3+CD127− cells within CD25bright T cells (middle), and of CD25brightFOXP3+CD127− T cells within CD45+ cells (lower) were determined by flow cytometry. One representative dot plot for each staining is shown on the right. B: The frequency of Tregs was determined by TSDR analysis within total PLNs and PB. C: The frequency of Tregs, determined by FACS as CD4+FOXP3+ cells and by the TSDR assay, within total PB (left) and total PLN (right) of type 1 diabetic (T1D) patients, healthy control subjects (HC), and nondiabetic donors (ND) is shown. Each symbol identifies a patient (as described in Supplementary Table 1), and lines indicate median values. The Mann-Whitney–Wilcoxon test and the t test with unequal variances were used for group comparisons.
The TSDR is an exclusive marker for thymic-derived FOXP3+ Tregs, and pervious work has demonstrated that it correlates well with flow cytometry data in determining the Treg frequency in the PB of healthy donors (16). We confirmed the presence of this correlation in the PB of diabetic patients and healthy control subjects. Unexpectedly, the Treg frequency in the PLNs of diabetic and nondiabetic donors, as detected by TSDR, was higher than that detected by fluorescence-activated cell sorter (FACS) (Fig. 2C). Thus, some of the cells epigenetically imprinted to be FOXP3+ Tregs lose their FOXP3 expression within the PLN, independently of the disease presence.
Tregs isolated from type 1 diabetic patients lose their suppressive ability once they reside in the PLN.
The most efficient way to isolate viable and pure human Tregs devoid of contaminating effector T cells remains the flow cytometry–based cell sorting. Thus, functional experiments were performed in this study by using FACS to isolate Tregs, and this led to a highly purified population of Tregs (Supplementary Fig. 2). The limited CD25bright T cells that could be sorted from PLNs of diabetic subjects forced us to perform the in vitro functional assay with a low number of Tregs (i.e., 7,000 cells) in the presence of responder PLN cells that were three times higher. At this Treg/responder cell ratio, the CD25bright T cells isolated from nondiabetic subjects had an average suppressive function of 30%, which is consistent with published data (36,37). In addition, the same cells had an average suppressive activity of 80% when the assay was performed at the commonly used 1:1 ratio, confirming the solidity of our assay (Fig. 3A). CD25bright T cells isolated from PLNs of three diabetic patients did not inhibit proliferation of polyclonally activated autologous PLN T cells, but rather, they had a helper function because their presence in the culture led to increased proliferation of the responder T cells. The CD25bright T cells purified from the PLNs of a further three diabetic patients had a reduced suppressive activity that fell into the low range of those isolated from nondiabetic donors (Fig. 3A). The statistical analysis performed on all the samples tested showed that Tregs isolated from the PLNs of diabetic patients have an impaired suppressive capability compared with those of nondiabetic donors (P = 0.0066).
FIG. 3.
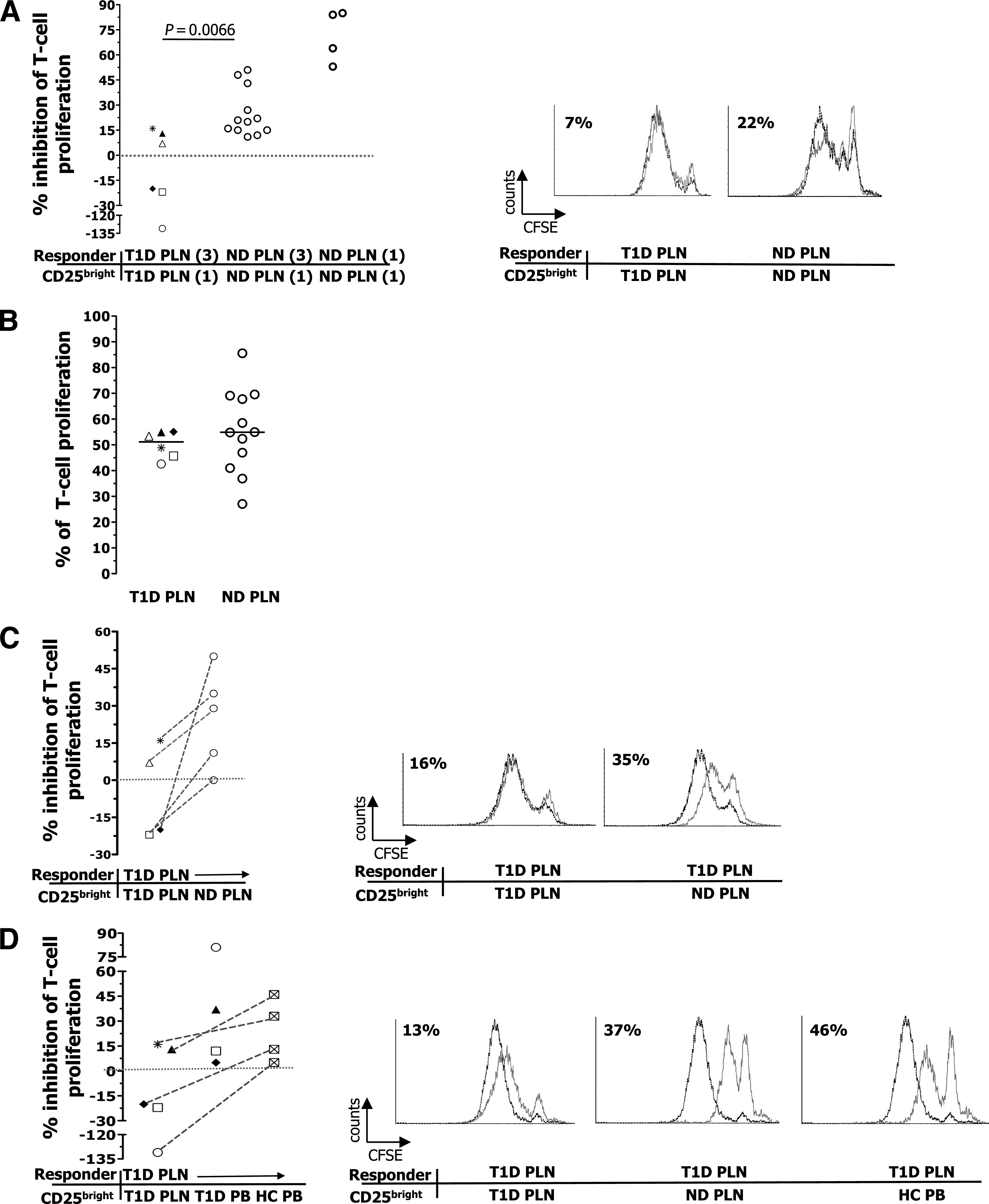
Suppression of polyclonally activated PLN T cells by CD25bright T cells. A: The inhibition of proliferation of PLN responder T cells by autologous CD25bright–sorted T cells at the indicated ratios (3:1 and 1:1) was tested. A negative percentage of inhibition of T-cell proliferation indicates that the presence of Tregs in the culture led to improved proliferation. The Mann-Whitney U test was used for group comparison, and P ≤ 0.05 was considered significant. B: T-cell proliferation after 6 days of culture. C: The inhibition of proliferation of responder PLN T cells of diabetic (T1D) patients by CD25bright T cells sorted from autologous PLNs or allogeneic PLNs of nondiabetic donors (ND) was tested. The dotted lines link experiments in which the same responder T cells were used. D: The inhibition of proliferation of responder PLN T cells of diabetic patients by CD25bright T cells sorted from autologous PLN, autologous PB, or allogeneic PB of healthy control subjects (HC) was tested. The same symbol corresponds to the same diabetic patient. The Wilcoxon test was used for comparison of data shown in C and D, and P ≤ 0.05 was considered significant. Each dot represents one donor, and lines indicate median values. In A, C, and D, one representative histogram for each responder/CD25bright T-cell pairs with relative percentage of suppression is shown on the right. Dotted black lines represent responder PLN cells activated in the absence of CD25bright T cells, and solid gray lines represent responder PLN cells activated in the presence of sorted CD25bright T cells.
This different behavior of the CD25bright T cells isolated from PLNs of diabetic and nondiabetic individuals was not due to different levels of T-cell proliferation (Fig. 3B) or proinflammatory cytokine (i.e., TNF-α) production (Supplementary Fig. 4) by the responder PLN cells between the two groups. In addition, responder PLN T cells of patients with type 1 diabetes were not refractory to CD25bright T-cell–mediated suppression because their proliferation was well inhibited by CD25bright T cells isolated from allogeneic PLNs of nondiabetic donors (Fig. 3C). There was also no defect in the frequency of IL-2–producing PLN T cells among diabetic patients after 5 h of in vitro stimulation, but rather, it appeared to be higher in diabetic compared with nondiabetic donors (Supplementary Fig. 5). Thus, an IL-2–dependent Treg defect in the PLNs of diabetic subjects is unlikely to be present, although a specific IL-2–signaling deficiency (31) cannot at this moment be ruled out.
To determine whether the Treg-defective suppressive activity observed in the PLNs of type 1 diabetic patients was lineage- or tissue-specific, Tregs were isolated from the PB of some of the above-tested diabetic patients, and their ability to suppress proliferation of autologous PLN T cells was investigated. Peripheral Tregs, contrary to their PLN counterpart, exerted a suppressive function as efficient as that of peripheral Tregs isolated from allogeneic healthy control subjects (Fig. 3D). These data did not reach a statistically significant difference, probably because of the limited number of samples that we could test. However, the data all show the same trend; Tregs purified from three patients with long-lasting diabetes retain a suppressive function when circulating in the periphery but lose their regulatory activity once they reside within the site of autoinflammatory drainage.
We then tested the ability of the CD25bright T cells to inhibit diabetes-related antigen-specific T-cell proliferation, maintaining the physiologic CD25bright/responder cell ratio present within the PLN, which was estimated to be ∼1/50. To accomplish this, we compared the frequency of T cells proliferating in response to proinsulin within the lymph nodes depleted of CD25bright T cells by flow cytometry (as shown in Supplementary Fig. 2) with that within the total PLN. The CD25bright T cells present in the PLNs of diabetic patients did not inhibit proinsulin-specific T-cell proliferation but, rather, had a helper activity. By contrast, CD25bright T cells present in the PLNs of nondiabetic subjects exerted an average suppressive function of 38% in response to proinsulin in inhibition of T-cell proliferation and IFN-γ production, and this is in line with the low Treg/responder cell ratio (Fig. 4A). Moreover, we found that CD25bright T cells from PLNs of patients with diabetes did not inhibit proinsulin-specific IFN-γ production, contrary to those isolated from nondiabetic subjects. Removal of CD25bright T cells from PLNs of patients with diabetes led to a dramatic fall in IFN-γ release (Fig. 4B). Thus, depletion of Tregs in the PLNs of diabetic patients leads to a reduced proinsulin-specific cell response rather than to an increased one (as observed in the PLNs of nondiabetic donors). These data further strengthen those shown in the polyclonal suppressive assays and demonstrate that Tregs isolated from the PLNs of diabetic donors have a defective suppressive activity and also acquire a helper phenotype.
FIG. 4.
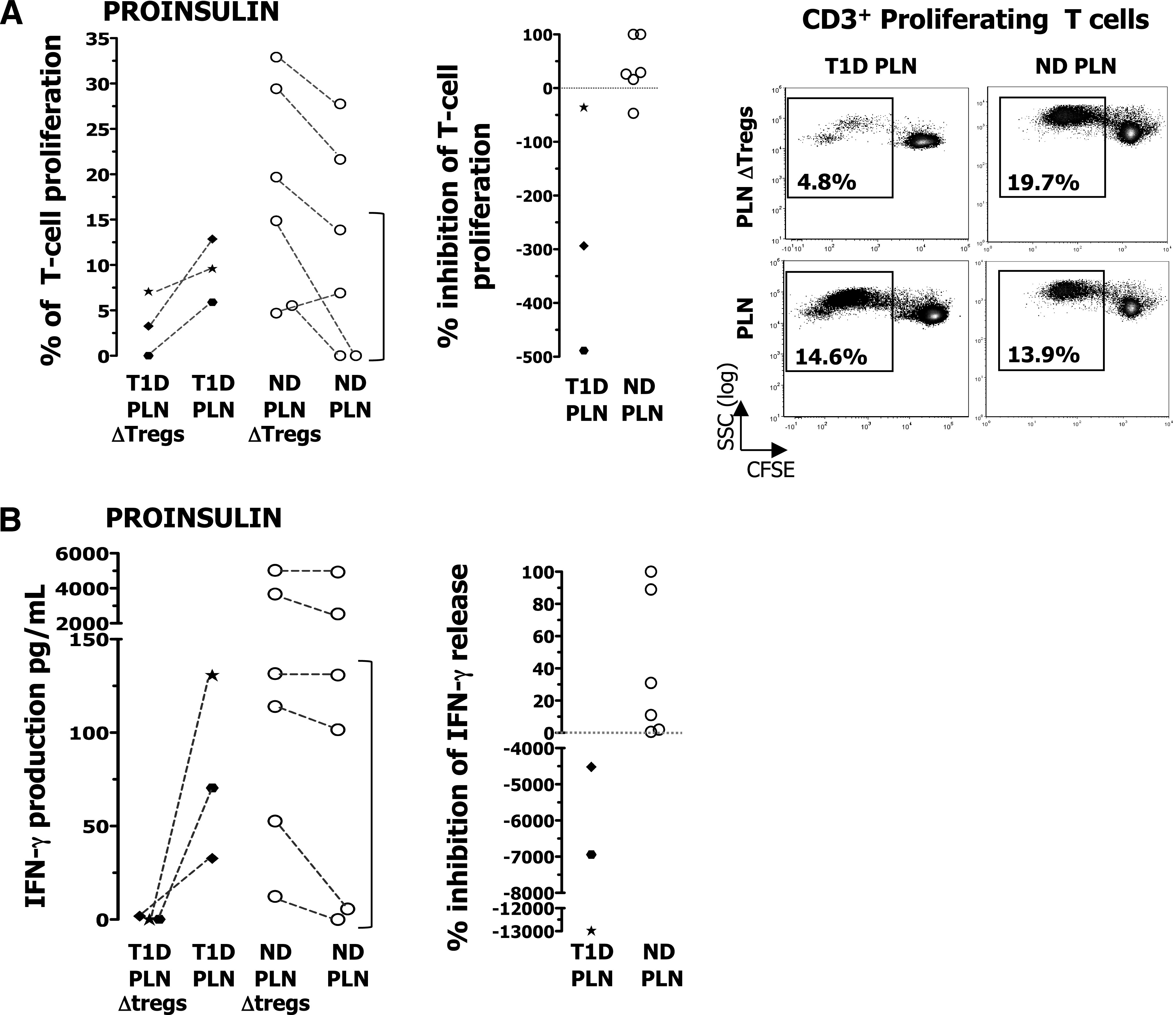
Suppression of proinsulin-activated PLN T cells by CD25bright T cells. CD14+ cells isolated from the spleen of nondiabetic donors (ND) or PB of diabetic (T1D) patients were cultured for 7 days with 10 ng/mL IL-4 (R&D Systems, Minneapolis, MN) and 50 ng/mL granulocyte microphage colony-stimulating factor (R&D Systems) to generate DCs. On day 6, DCs were incubated for 6 h with 10 μg of rhu-proinsulin or rhu-GAD65 or left unpulsed and then activated with 100 units/mL of IFN-γ and 10 ng/mL of lipopolysaccharide overnight. On day 7, PLNs depleted of CD25bright T cells were stained with CFSE and cultured with autologous DC unloaded or loaded with antigen at a ratio of 1:40 (DC/PLN). PLN ΔTregs, lymph-node cells depleted of CD25bright T cells by flow cytometry–based sorting. A: The percentages of CD3+ T-cell proliferation in response to proinsulin upon culture of CD25bright T-cell–depleted PLN and total PLN are shown (left). The same data are presented to illustrate inhibition of T-cell proliferation in response to proinsulin by CD25bright T cells (middle). Representative dot plots of CD25bright T-cell–depleted PLN and total PLN proliferating in response to proinsulin are shown (right). In the dot plots, proliferating T cells are gated on CD3+ T cells and percentages of proinsulin-specific T cells are indicated without background subtraction. SSC, side scatter. B: The IFN-γ produced by CD25bright T-cell–depleted PLN and that produced by total PLN in response to proinsulin was tested (left). The same data are presented to show inhibition of IFN-γ-production in response to proinsulin by CD25bright T cells (right). The bracket highlights the “low-responder” nondiabetic donors. PLN cell proliferation observed in the presence of proinsulin-loaded DC was measured upon subtraction of PLN cell proliferation observed in the presence of unloaded control DC.
Our data seem to suggest that total PLNs of nondiabetic donors respond better to proinsulin in vitro than those of type 1 diabetic donors. However, the PLNs of the three type 1 diabetic donors tested responded to proinsulin at levels similar to those of the “low-range” nondiabetic donors in cell proliferation and IFN-γ production. Thus far, we cannot conclude that the PLNs of nondiabetic donors respond better to proinsulin than those of diabetic patients because of the low number of patients tested.
CD25bright T cells isolated from PLNs of diabetic patients are not contaminated by effector T cells.
Overall, the just-mentioned functional assays posed the question of whether the CD25bright T cells isolated from PLNs of diabetic patients were contaminated by activated cytokine-producing effector T cells. We exclude such a possibility because the CD25bright T cells sorted from PLNs of diabetic and nondiabetic donors contained the same amount of FOXP3 mRNA (Fig. 5A) and expressed the same low transcript levels of the master regulators of Th1 (T-box expressed in T cells [T-bet]), Th2 (GATA-binding protein 3 [GATA3]), and Th17 cells (retinoic acid–related orphan receptor C [RORC]) and of IL-2 and IFN-γ (Fig. 5B). Most importantly, the sorted CD25bright T cells were all demethylated at the FOXP3 locus, proving their epigenetic Treg imprinting (Fig. 5C). These data demonstrate that the sorted CD25bright T cells within the PLNs of diabetic patients do not contain effector T cells, and despite being epigenetically determined to be Tregs, they do not exert consistent in vitro regulatory function.
FIG. 5.
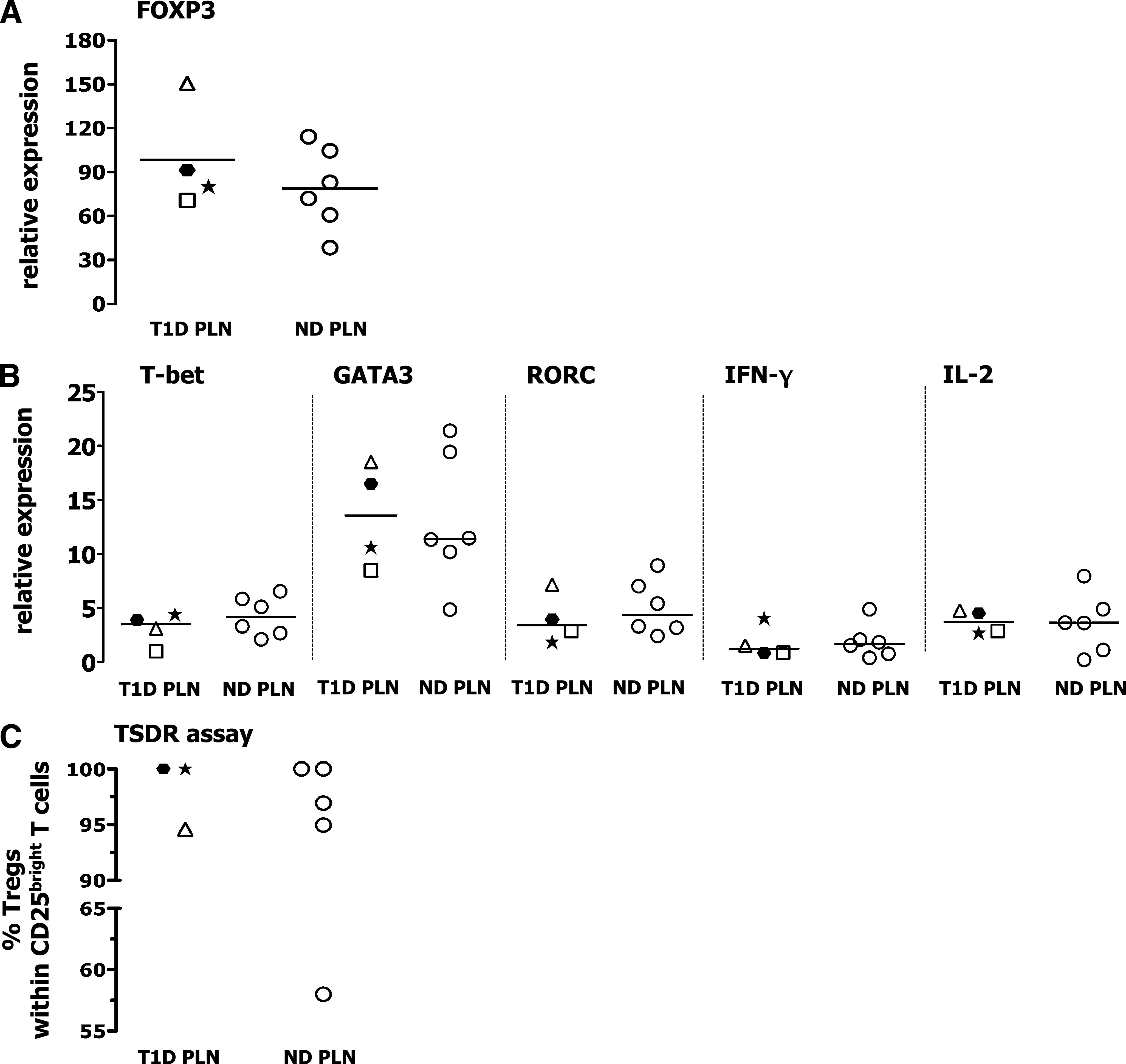
Purity of the flow cytometry–based sorted CD25bright T cells tested in the functional assays. A and B: The amount of transcript expression (relative to an internal control) of the indicated genes in the sorted CD25bright T cells was assessed by RT-PCR. Relative expression of the mRNA of interest was determined by normalizing to hypoxanthine phosphoribosyltransferase expression and calculating the fold-change vs. the expression values measured in total PBMC isolated from normal donors. C: The frequency of Tregs within the sorted CD25bright T cells was measured by the TSDR assay. Each symbol identifies a patient (as described in Supplementary Table 1), and lines indicate median values. ND, nondiabetic donors.
Altered tissue-specific immune status in diabetic patients.
The Treg/Th17 cell balance could be key in controlling autoimmune diseases, type 1 diabetes included (38,39). Our data show that the PLNs of diabetic patients are enriched in Th17 cells while having similar numbers of bona fide epigenetically imprinted Tregs. To determine whether there is a compensatory mechanism by which Tregs expand in those patients in which Th17 cells are more frequent, we measured the Treg/Th17 cell ratio as determined by TSRD analysis and chemokine receptor expression, respectively. The Treg/Th17 cell ratio was about four to five times lower in the PLNs of diabetic patients compared with nondiabetic donors, but was not different in the PB. Such an inverse correlation was not found between Tregs and Th1 cells (Fig. 6A). Thus, the immune balance is shifted toward a Th17-proinflammatory environment only in the target organ of long-lasting diabetic patients and not in their PB.
FIG. 6.
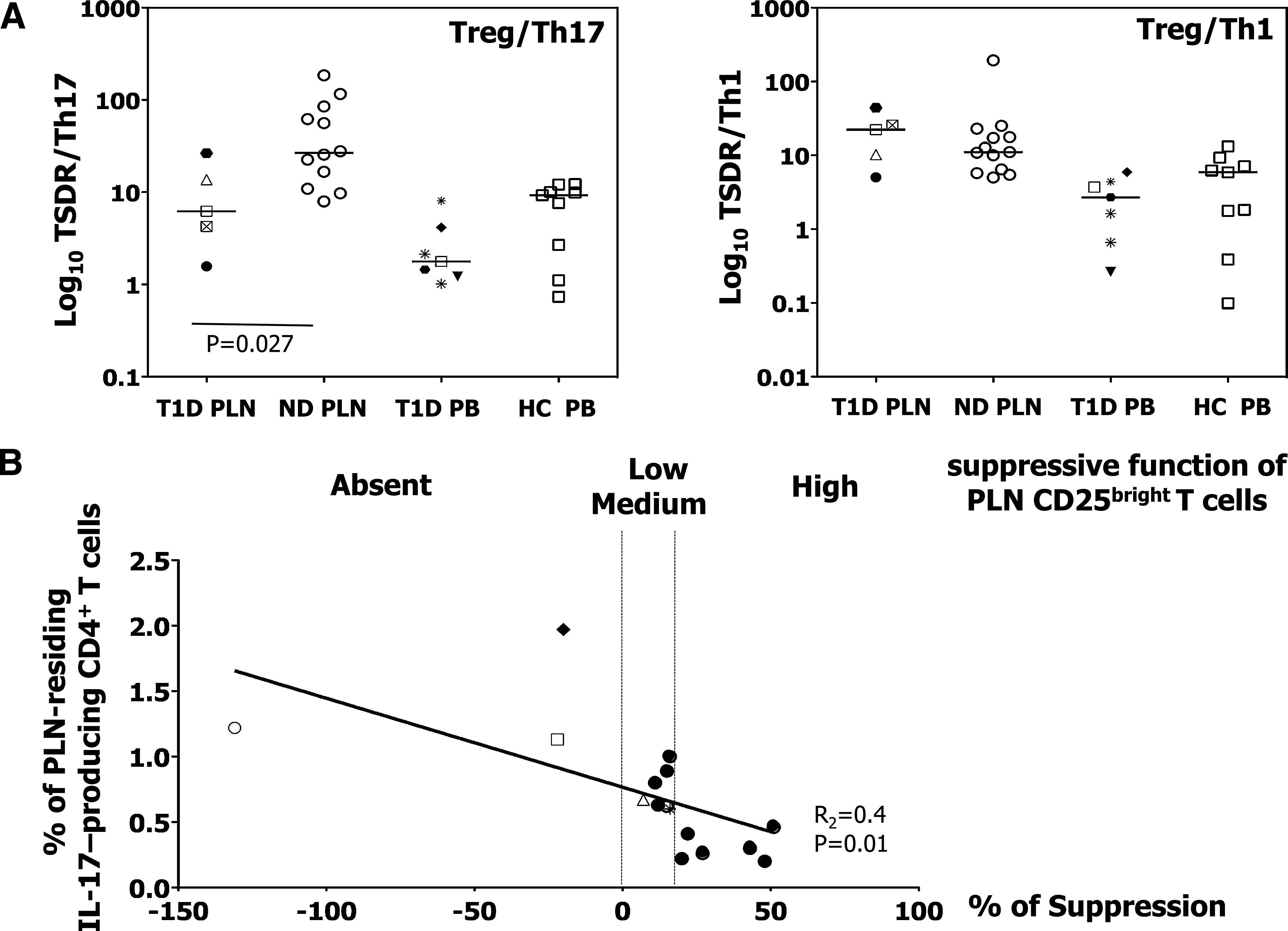
Immune status in the PLN and PB of diabetic patients. A: Treg/Th17 and Treg/Th1 cell ratio was determined by TSRD and chemokine-receptor expression in the indicated donors and samples. HC, healthy control subjects; ND, nondiabetic donors; T1D, type 1 diabetic patients. Each symbol identifies a patient (as described in Supplementary Table 1), and the Mann-Whitney–Wilcoxon test was used for group comparisons. B: Correlation between the frequency of IL-17-producing CD4+ T cells and the suppressive function of CD25bright T cells isolated from PLN of diabetic (light symbols) and nondiabetic (bold filled circles) subjects. The linear regression analysis was used to compare the two measurements.
We then tested whether the presence of IL-17–producing CD4+ T cells in vivo within the PLN correlates with the suppressive capacity of the sorted CD25bright T cells. Linear regression analysis revealed that a higher frequency of IL-17–producing CD4+ T cells residing in the PLN was correlated with a lower/absent Treg suppressive function (R2 = 0.40; P = 0.01) (Fig. 6B). These data are suggestive of an inverse correlation between Th17 immunity and the Treg-mediated regulatory activity in the target organ of long-term diabetic individuals.
DISCUSSION
To what extent proinflammatory Th17 cells and defects in Treg-mediated regulation contribute to the development of type 1 diabetes in humans is highly debated. Here we show that the PLNs of patients with type 1 diabetes, unlike their PB, have an altered immune status due to the expansion of Th17 cells and the presence of CD25bright T cells epigenetically imprinted to have a regulatory activity but which lack a proper function.
Increased Th17 cells in the PB of children with diabetes has been recently reported (8), and Tan and colleagues (9) have demonstrated that these circulating IL-17–producing T cells may reside mainly within the CD4+CD45RA−CD25intFOXP3low cells. Although expressing FOXP3, this latter cell subset does not have suppressive activity, but rather, it has a helper function and contains proinflammatory cytokine-producing cells (40,41). Our data demonstrate that the expansion of Th17 immunity is also present in the target organ of patients with long-term diabetes. However, this was not due to a higher number of CD4+CD45RA−CD25intFOXP3low T cells, which were as frequent as those in nondiabetic donors (Supplementary Fig. 6). Contrary to what has been published (8,9), we did not find signs of excessive Th17 immunity in the circulating CD4+ T cells of type 1 diabetic patients, and this may be because the individuals in our patient population had the disease for an average of 26 years. One could speculate that Th17 cells circulate early after disease manifestation but tend to accumulate in the target organ once the disease is established.
The TSDR is an exclusive marker for thymic-derived FOXP3+ Tregs, and previous work has demonstrated that it correlates well with flow cytometry data in determining the Treg frequency in the PB of healthy donors (16). We confirmed the presence of such a correlation in the PB of healthy control subjects and diabetic patients. Unexpectedly, the Treg frequency in the PLNs of both diabetic and nondiabetic donors, as detected by TSDR, was higher than that detected by FACS. Thus, some of the cells epigenetically imprinted to be FOXP3+ Tregs lose their FOXP3 expression within the PLN, and these new findings are currently under investigation.
Another unexpected finding of our study was that CD25bright T cells completely demethylated at the FOXP3 locus have an impaired regulatory activity only in the PLNs of patients with type 1 diabetes. The crucial question is why cells that are epigenetically imprinted to be regulatory do not function as such, and why this occurs only in diabetic patients and, specifically, in their PLNs. Firstly, post-transcriptional modification of the gene expressing FOXP3 may occur, leading to reduced or unstable FOXP3 expression. Secondly, there may be a FOXP3-stabilizing factor that is reduced in the PLN Tregs. Alternatively, other molecules fundamental for Treg function and signature may be defective within the PLN CD25bright T cells of diabetic patients. Unfortunately, the limited number of cells obtained from our samples was an obstacle to such an extensive analysis. Finally, important environmental factors, such as cytokines and/or chemokines, may also prevent the Tregs from being functionally fit only in diabetic subjects.
We have shown that the key features of the PLN of diabetic subjects are 1) an unbalanced Treg/Th17 cell ratio; 2) increased IL-17–producing CD4+ T cells in response to diabetes-related antigens; and 3) the presence of CD25bright T cells epigenetically imprinted to be Tregs but which overall are reduced in FOXP3 expression and have a defective suppressive activity. We know that 1) IL-17 has an apoptotic activity on human pancreatic β-cells (8) and 2) pathogenic Th17 responses in mice are restrained by Tregs via FOXP3 binding to the phosphorylated form of the signal transducers and activators of transcription 3 (STAT3) (42,43). Thus, one could speculate that certain Tregs in diabetic patients, for still unknown reasons, turn off FOXP3 expression once they migrate to the pancreas, leading to defective control of Th17 cells, which expand and cause the destruction of the pancreas by releasing IL-17.
We recognize that the current study has some limitations. First, the type 1 diabetic patients had an average disease duration of 26 years. However, we believe that this does not reduce the significance of our results because it was previously found that PLNs of long-term type 1 diabetic subjects, and not of type 2 diabetic patients or nondiabetic donors, harbor insulin-specific T cells (44), demonstrating the presence of effector T cells in the target organ of type 1 diabetic individuals years after onset. A recent study showed that insulin-producing β-cells are still present in patients who have had diabetes for more than 50 years (45), and these cells can represent an important antigen-reservoir, drawing inflammatory cells to the pancreas and PLNs even years after disease onset. For instance, the diabetic patient T1D024 included in our study (symbol △) had preserved fasting C-peptide levels 9 years after disease onset, and he was one of those donors whose PLN showed IL-17 production upon GAD65 stimulation and displayed a very limited suppressive ability in polyclonal assays. In addition, studies performed in autoimmune murine models suggest that Tregs are still actively and continuously involved in inflammatory regulation after disease development (46,47).
Second, the age and sex of the various groups of donors were not strictly comparable. Certain recognized (i.e., dialysis for diabetic patients and period in intensive care or cause of death for nondiabetic donors) and nonrecognized characteristics of the donors might also have influenced our results. However, the age and sex of the donors, and their clinical characteristics, had no influence on the Th17-cell and TSDR data (data not shown). In addition, we recently collected PLNs from three living nondiabetic donors who underwent pancreas surgery for chronic pancreatitis or pancreatic pseudocysts. Phenotypical and functional data on Tregs isolated from these PLNs were identical to those generated with PLNs of brain-dead nondiabetic donors, further supporting the fact that our results were not affected by external confounding factors (data not shown). We therefore believe that, given the uniqueness of the patient samples in our hands, this study is informative and provides original data.
Our work puts forward the hypothesis that type 1 diabetic patients have an altered immune balance in their target organ caused by the presence of excessive Th17 immunity and unfit Tregs. It is still unknown whether these findings are related and whether they are a cause or a consequence of the autoimmune disease. However, our data point to the fact that treatments targeted at suppressing Th17 cells and/or refitting Tregs in the target organ may represent a solution for diabetic patients.
ACKNOWLEDGMENTS
This work was supported by the Juvenile Diabetes Research Foundation Grant CDA #2-2008-8 to M.B.
S.O. is the Founder and Chief Executive Officer of Epiontis GmbH. No other potential conflicts of interest relevant to this article were reported.
A.F. processed the samples; performed the experiments; collected, analyzed, and discussed data; and contributed to writing the manuscript. C.So. recruited diabetic patients and collected blood before surgery and the PLNs during surgery. A.St. assisted with sample processing and experiments. A.V. assisted with data analysis and interpretation. P.Mo. assisted with antigen-specific T-cell suppression assays, data analysis, and interpretation. L.P. performed statistical analysis and discussed data. R.N. collected the PLNs of nondiabetic donors. S.O. performed the TSDR analysis. P.Ma. collected the blood of diabetic patients. M.S. performed statistical analysis and contributed to manuscript organization. A.Se. supervised P.Ma. C.St. supervised C.So. E.B. contributed to data analysis and interpretation. M.B. designed and supervised the study and wrote the manuscript.
The authors thank Maria-Grazia Roncarolo (San Raffaele Scientific Institute) and her laboratory, and individuals at M.B.’s laboratory, for helpful and continuous discussion. The authors thank the following individuals from the San Raffaele Scientific Institute: Alessio Palini, Emanuele Canonico, and Chiara Villa for FACS sorting and technical support; Ilaria Santagostino and Federica Mescia for collecting data of diabetic patients; Vito Lampasona for testing autoAbs; and Alessandro Ambrosi for statistical support.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db11-0090/-/DC1.
REFERENCES
- 1.Eisenbarth GS. Update in type 1 diabetes. J Clin Endocrinol Metab 2007;92:2403–2407 [DOI] [PubMed] [Google Scholar]
- 2.von Boehmer H, Melchers F. Checkpoints in lymphocyte development and autoimmune disease. Nat Immunol 2010;11:14–20 [DOI] [PubMed] [Google Scholar]
- 3.Lehuen A, Diana J, Zaccone P, Cooke A. Immune cell crosstalk in type 1 diabetes. Nat Rev Immunol 2010;10:501–513 [DOI] [PubMed] [Google Scholar]
- 4.Wang B, André I, Gonzalez A, et al. Interferon-gamma impacts at multiple points during the progression of autoimmune diabetes. Proc Natl Acad Sci USA 1997;94:13844–13849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jain R, Tartar DM, Gregg RK, et al. Innocuous IFNgamma induced by adjuvant-free antigen restores normoglycemia in NOD mice through inhibition of IL-17 production. J Exp Med 2008;205:207–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Emamaullee JA, Davis J, Merani S, et al. Inhibition of Th17 cells regulates autoimmune diabetes in NOD mice. Diabetes 2009;58:1302–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maggi L, Santarlasci V, Capone M, et al. CD161 is a marker of all human IL-17-producing T-cell subsets and is induced by RORC. Eur J Immunol 2010 ;40:2174–2181 [DOI] [PubMed] [Google Scholar]
- 8.Honkanen J, Nieminen JK, Gao R, et al. IL-17 immunity in human type 1 diabetes. J Immunol 2010;185:1959–1967 [DOI] [PubMed] [Google Scholar]
- 9.Marwaha AK, Crome SQ, Panagiotopoulos C, et al. Cutting edge: increased IL-17-secreting T cells in children with new-onset type 1 diabetes. J Immunol 2010;185:3814–3818 [DOI] [PubMed] [Google Scholar]
- 10.Danke NA, Koelle DM, Yee C, Beheray S, Kwok WW. Autoreactive T cells in healthy individuals. J Immunol 2004;172:5967–5972 [DOI] [PubMed] [Google Scholar]
- 11.Monti P, Scirpoli M, Rigamonti A, et al. Evidence for in vivo primed and expanded autoreactive T cells as a specific feature of patients with type 1 diabetes. J Immunol 2007;179:5785–5792 [DOI] [PubMed] [Google Scholar]
- 12.Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol 2010;11:7–13 [DOI] [PubMed] [Google Scholar]
- 13.Wang J, Ioan-Facsinay A, van der Voort EI, Huizinga TW, Toes RE. Transient expression of FOXP3 in human activated nonregulatory CD4+ T cells. Eur J Immunol 2007;37:129–138 [DOI] [PubMed] [Google Scholar]
- 14.Baron U, Floess S, Wieczorek G, et al. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3(+) conventional T cells. Eur J Immunol 2007;37:2378–2389 [DOI] [PubMed] [Google Scholar]
- 15.Floess S, Freyer J, Siewert C, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol 2007;5:e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wieczorek G, Asemissen A, Model F, et al. Quantitative DNA methylation analysis of FOXP3 as a new method for counting regulatory T cells in peripheral blood and solid tissue. Cancer Res 2009;69:599–608 [DOI] [PubMed] [Google Scholar]
- 17.Huehn J, Polansky JK, Hamann A. Epigenetic control of FOXP3 expression: the key to a stable regulatory T-cell lineage? Nat Rev Immunol 2009;9:83–89 [DOI] [PubMed] [Google Scholar]
- 18.Tang Q, Adams JY, Penaranda C, et al. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity 2008;28:687–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duarte JH, Zelenay S, Bergman ML, Martins AC, Demengeot J. Natural Treg cells spontaneously differentiate into pathogenic helper cells in lymphopenic conditions. Eur J Immunol 2009;39:948–955 [DOI] [PubMed] [Google Scholar]
- 20.Komatsu N, Mariotti-Ferrandiz ME, Wang Y, Malissen B, Waldmann H, Hori S. Heterogeneity of natural Foxp3+ T cells: a committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc Natl Acad Sci U S A 2009;106:1903–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou X, Bailey-Bucktrout SL, Jeker LT, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol 2009;10:1000–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jailwala P, Waukau J, Glisic S, et al. Apoptosis of CD4+ CD25(high) T cells in type 1 diabetes may be partially mediated by IL-2 deprivation. PLoS ONE 2009;4:e6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brusko TM, Wasserfall CH, Clare-Salzler MJ, Schatz DA, Atkinson MA. Functional defects and the influence of age on the frequency of CD4+ CD25+ T-cells in type 1 diabetes. Diabetes 2005;54:1407–1414 [DOI] [PubMed] [Google Scholar]
- 24.Lindley S, Dayan CM, Bishop A, Roep BO, Peakman M, Tree TI. Defective suppressor function in CD4(+)CD25(+) T-cells from patients with type 1 diabetes. Diabetes 2005;54:92–99 [DOI] [PubMed] [Google Scholar]
- 25.Glisic-Milosavljevic S, Waukau J, Jailwala P, et al. At-risk and recent-onset type 1 diabetic subjects have increased apoptosis in the CD4+CD25+ T-cell fraction. PLoS ONE 2007;2:e146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Monti P, Scirpoli M, Maffi P, et al. Rapamycin monotherapy in patients with type 1 diabetes modifies CD4+CD25+FOXP3+ regulatory T-cells. Diabetes 2008;57:2341–2347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu W, Putnam AL, Xu-Yu Z, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med 2006;203:1701–1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Putnam AL, Vendrame F, Dotta F, Gottlieb PA. CD4+CD25high regulatory T cells in human autoimmune diabetes. J Autoimmun 2005;24:55–62 [DOI] [PubMed] [Google Scholar]
- 29.Lawson JM, Tremble J, Dayan C, et al. Increased resistance to CD4+CD25hi regulatory T cell-mediated suppression in patients with type 1 diabetes. Clin Exp Immunol 2008;154:353–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schneider A, Rieck M, Sanda S, Pihoker C, Greenbaum C, Buckner JH. The effector T cells of diabetic subjects are resistant to regulation via CD4+ FOXP3+ regulatory T cells. J Immunol 2008;181:7350–7355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Long SA, Cerosaletti K, Bollyky PL, et al. Defects in IL-2R signaling contribute to diminished maintenance of FOXP3 expression in CD4(+)CD25(+) regulatory T-cells of type 1 diabetic subjects. Diabetes 2010;59:407–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo MG. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J Immunol 2006;177:8338–8347 [DOI] [PubMed] [Google Scholar]
- 33.Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood 2005;105:4743–4748 [DOI] [PubMed] [Google Scholar]
- 34.Sallusto F, Lanzavecchia A. Heterogeneity of CD4+ memory T cells: functional modules for tailored immunity. Eur J Immunol 2009;39:2076–2082 [DOI] [PubMed] [Google Scholar]
- 35.Crome SQ, Wang AY, Levings MK. Translational mini-review series on Th17 cells: function and regulation of human T helper 17 cells in health and disease. Clin Exp Immunol 2010;159:109–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med 2004;199:971–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Häringer B, Lozza L, Steckel B, Geginat J. Identification and characterization of IL-10/IFN-gamma-producing effector-like T cells with regulatory function in human blood. J Exp Med 2009;206:1009–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jäger A, Kuchroo VK. Effector and regulatory T-cell subsets in autoimmunity and tissue inflammation. Scand J Immunol 2010;72:173–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van den Brandt J, Fischer HJ, Walter L, Hünig T, Klöting I, Reichardt HM. Type 1 diabetes in BioBreeding rats is critically linked to an imbalance between Th17 and regulatory T cells and an altered TCR repertoire. J Immunol 2010;185:2285–2294 [DOI] [PubMed] [Google Scholar]
- 40.Battaglia M, Roncarolo MG. The fate of human Treg cells. Immunity 2009;30:763–765 [DOI] [PubMed] [Google Scholar]
- 41.Miyara M, Yoshioka Y, Kitoh A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 2009;30:899–911 [DOI] [PubMed] [Google Scholar]
- 42.Chaudhry A, Rudra D, Treuting P, et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science 2009;326:986–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang XO, Panopoulos AD, Nurieva R, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem 2007;282:9358–9363 [DOI] [PubMed] [Google Scholar]
- 44.Kent SC, Chen Y, Bregoli L, et al. Expanded T cells from pancreatic lymph nodes of type 1 diabetic subjects recognize an insulin epitope. Nature 2005;435:224–228 [DOI] [PubMed] [Google Scholar]
- 45.Keenan HA, Sun JK, Levine J, et al. Residual insulin production and pancreatic ß-cell turnover after 50 years of diabetes: Joslin Medalist Study. Diabetes 2010;59:2846–2853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen Z, Herman AE, Matos M, Mathis D, Benoist C. Where CD4+CD25+ T reg cells impinge on autoimmune diabetes. J Exp Med 2005;202:1387–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stephens LA, Gray D, Anderton SM. CD4+CD25+ regulatory T cells limit the risk of autoimmune disease arising from T cell receptor crossreactivity. Proc Natl Acad Sci U S A 2005;102:17418–17423 [DOI] [PMC free article] [PubMed] [Google Scholar]