

Abstract
OBJECTIVE
Proinflammatory activation of Kupffer cells is implicated in the effect of high-fat feeding to cause liver insulin resistance. We sought to determine whether reduced endothelial nitric oxide (NO) signaling contributes to the effect of high-fat feeding to increase hepatic inflammatory signaling and if so, whether this effect 1) involves activation of Kupffer cells and 2) is ameliorated by increased NO signaling.
RESEARCH DESIGN AND METHODS
Effect of NO/cGMP signaling on hepatic inflammation and on isolated Kupffer cells was examined in C57BL/6 mice, eNos−/− mice, and Vasp−/− mice fed a low-fat or high-fat diet.
RESULTS
We show that high-fat feeding induces proinflammatory activation of Kupffer cells in wild-type mice coincident with reduced liver endothelial nitric oxide synthase activity and NO content while, conversely, enhancement of signaling downstream of endogenous NO by phosphodiesterase-5 inhibition protects against high fat–induced inflammation in Kupffer cells. Furthermore, proinflammatory activation of Kupffer cells is evident in eNos−/− mice even on a low-fat diet. Targeted deletion of vasodilator-stimulated phosphoprotein (VASP), a key downstream target of endothelially derived NO, similarly predisposes to hepatic and Kupffer cell inflammation and abrogates the protective effect of NO signaling in both macrophages and hepatocytes studied in a cell culture model.
CONCLUSIONS
These results collectively imply a physiological role for endothelial NO to limit obesity-associated inflammation and insulin resistance in hepatocytes and support a model in which Kupffer cell activation during high-fat feeding is dependent on reduced NO signaling. Our findings also identify the NO/VASP pathway as a novel potential target for the treatment of obesity-associated liver insulin resistance.
Inflammatory activation of Kupffer cells, the resident macrophages of the liver, is implicated in the pathogenesis of obesity-induced insulin resistance and fatty liver disease (1–3). One potential mechanism whereby nutrient excess can induce hepatic insulin resistance involves a “two-hit” process in which proinflammatory activation of Kupffer cells occurs initially and then, in the “second hit,” release of cytokines from these cells in turn causes hepatocellular inflammation that downregulates insulin signal transduction. Although consumption of a high-fat diet clearly induces proinflammatory activation of Kupffer cells in mice (4), the question of whether reduced endothelial nitric oxide (NO) signaling contributes to this effect has previously not been investigated.
Among early responses observed in association with obesity-induced insulin resistance is a reduction in vascular content of NO, an effect that predisposes to increased endothelial inflammation, thrombosis, and vasoconstriction (5,6). Because reduced vascular NO levels precede the onset of increased liver nuclear factor-κB (NF-κB) signaling and impaired insulin-mediated phosphorylation of Akt in a mouse model of diet-induced obesity (DIO) (7), we sought in the current work to determine whether reduced endothelial NO signaling contributes to the effect of high-fat feeding to induce inflammatory activation of Kupffer cells and associated hepatic insulin resistance.
The liver is a highly vascularized tissue receiving 20% of cardiac output, and hepatic sinusoidal endothelial cells make up 50% of nonparenchymal cells of the liver (8,9). Sinusoidal endothelial cells regulate vascular resistance in the liver through flow-mediated NO production (10) and are also able to scavenge and eliminate soluble waste macromolecules from portal venous blood. In conjunction with sinusoidal endothelial cells, Kupffer cells situated on the luminal side of the endothelium contribute to this scavenging function as well as to host immunity and immune tolerance (8,11). Although endothelial NO exerts anti-inflammatory effects in endothelial cells and vascular smooth muscle cells (12), whether it regulates Kupffer cell activation has yet to be tested directly.
NO is produced by three distinct NO synthase (NOS) isoforms: endothelial (eNOS), inducible (iNOS), and neuronal. eNOS is expressed primarily in endothelial cells, and NO production by this enzyme is regulated via multiple mechanisms (calcium, phosphorylation, cellular localization), whereas iNOS is expressed predominantly in immune cells and is induced by NF-κB–dependent pathways, and it produces much higher concentrations of NO than does eNOS. At these higher levels, iNOS-generated NO exerts cytoxic effects that trigger cellular inflammation and are important in pathogen killing, whereas at far lower levels (from eNOS), NO activates soluble guanylate cyclase, which activates cGMP-dependent protein kinase (PKG) by increasing cytoplasmic cGMP levels. Among downstream targets of PKG is vasodilator-stimulated phosphoprotein (VASP), a protein implicated in the control of cytoskeletal dynamics and cell migration (13). Although PKG-mediated VASP activation is believed to increase endothelial cell permeability and may underlie growth inhibitory effects of NO on smooth muscle cells (14), its role in the anti-inflammatory action of NO/cGMP/PKG signaling remains unexplored.
RESEARCH DESIGN AND METHODS
Adult male C57BL/6 wild-type mice and eNos−/− mice were purchased from The Jackson Laboratory, and Vasp −/− mice on a C57BL/6 background were obtained from Dr. Guenter Daum (University of Washington, Seattle, WA) (14). Age-matched groups were maintained in a temperature-controlled facility with a 12-h light-dark cycle and were fed an equicaloric diet that was either low (10% saturated fat) or high (60% saturated fat) in fat content (cat. nos. D12492 and D12450B; Research Diet). Whole-body insulin signaling was assessed following intraperitoneal insulin injection (0.06 units/g body wt) after an overnight fast. Fifteen minutes later, mice were killed with an overdose of CO2, followed by cervical dislocation. Littermate mice were used as controls for Vasp −/− mice. All procedures were approved by the University of Washington Institutional Animal Care and Use Committee.
Quantitative RT-PCR analyses.
RNA was extracted using RNAase kit (Quiagen). For gene expression analysis, real-time RT-PCR reactions were conducted as previously described (7) using TaqMan Gene Expression Analysis (Applied Biosystems).
Western blotting and immunoprecipitation.
Cell lysis and tissue extract was performed as described previously (15). All Western blots and immunoprecipitation used equal amounts of total protein for each condition from individual experiments and were performed as previously described (16). Quantification of Western blots was performed using ImageJ Processing and Analysis (National Institutes of Health).
Materials.
Antiserine 473 Akt, anti-Akt, antiphosphorylated eNOS (Ser 1177), antiphosphorylated inhibitor of κB (IκB)-α, anti-PKG, anti-VASP, anti–insulin receptor substrate (IRS)-1 rabbit polyclonal antibody, and phospho-tyrosine monoclonal antibody were obtained from Cell Signaling. Anti–IRS-2 rabbit polyclonal antibody was obtained from Millipore. Anti-eNOS mouse polyclonal antibody was obtained from BD Bioscience. Anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH) rabbit polyclonal antibody was obtained from Santa Cruz Biotechnology. Total Akt and phospho-Akt (Ser 473) ELISA kits were obtained from Biosource. (Z)-1-[N-(2-aminoethyl)-N-(2-ammonioethyl)amino] diazen-1-IM1,2-diolate (DETA-NO) and 8-bromoguanosine-3′,5′ (8Br)-cGMP were purchased from Enzo Life Sciences. DETA-NO was used within 24 h after the reconstitution.
NO measurement and hepatic triglyceride measurement.
Tissue NO content was measured by using electron spin resonance spectroscopy technique as previously described (7,17). Hepatic triglyceride content was measured in liver lysates as previously described (18).
Kupffer cell isolation and cell culture.
Kupffer cell isolation and primary hepatocyte cell culture were performed as described previously (3) with minor modifications. After hepatic perfusion through the portal vein with liver digest medium (GIBCO) containing 0.05 mg/mL collagenase type 4 (Worthington), the liver was minced and transferred into a 50-mL conical tube through a 70-μm cell strainer. After centrifugation (5 min at 50g), primary hepatocytes (the cell pellet) were collected. Supernatants after the first centrifugation of 50g were spun for 10 min at 1000g, the resultant pellets were resuspended with rat anti-F4/80 antibody (AbD Serotec), and magnetic beads were conjugated with sheep anti-rat IgG (Invitrogen) at 4°C for 30 min. RAW264.7 cells were purchased from American Type Culture Collection and cultured in Dulbecco’s modified Eagle’s medium (cat. no. 10-013-CV; Cellgro) with 10% FBS. AML12 cells were purchased from American Type Culture Collection and cultured in Dulbecco’s modified Eagle’s medium/F-12 50/50 (cat. no. 10-090-CV; Cellgro) with 0.005 mg/mL insulin, 0.005 mg/mL transferrin, 5 ng/mL selenium, 40 ng/mL dexamethasone, and 10% FBS. Peritoneal macrophages were isolated and cultured as previously described (19).
Statistical analysis.
In all experiments, densitometry measurements were normalized to controls incubated with vehicle and fold increase above the control condition was calculated. Analysis of the results was performed using the GraphPad statistical package. Data are expressed as means ± SEM, and values of P < 0.05 were considered statistically significant. A two-tailed t test was used to compare mean values in two-group comparisons. To compare responses between sildenafil and vehicle-treated mice that also received either vehicle or insulin, data were analyzed by two-way ANOVA, and the Bonferroni post hoc comparison test was used to compare mean values between groups.
RESULTS
Effect of high-fat feeding on liver NO production, hepatic inflammation, and Kupffer cell activation.
Based on published evidence that during high-fat feeding in mice, reduced NO content in thoracic aorta precedes the onset of both hepatic inflammation (increased NF-κB signaling) and hepatic insulin resistance (reduced insulin-stimulated activation of IRS-1/ phosphatidylinositol 3-kinase/Akt) (7), we first asked whether liver NO content declines prior to the onset of increased NF-κB signaling and Kupffer cell activation in this setting.
We found that compared with low fat–fed control mice, liver NO content and phosphorylation of eNOS were reduced after 4 weeks of high-fat feeding (Fig. 1A and B). Markers of liver NF-κB activation—hepatic IκB-α phosphorylation (Fig. 1C), interleukin (IL)-6 (Fig. 1D) and tumor necrosis factor (TNF)-α mRNA expression, TNF-α (Fig. 1E), and intracellular adhesion molecule (ICAM) (Fig. 1F)—were also increased by high-fat feeding, but this did not occur until 8 weeks on the high-fat diet. During high-fat feeding, liver NO content falls prior to the onset of high fat–induced NF-κB signaling.
FIG. 1.
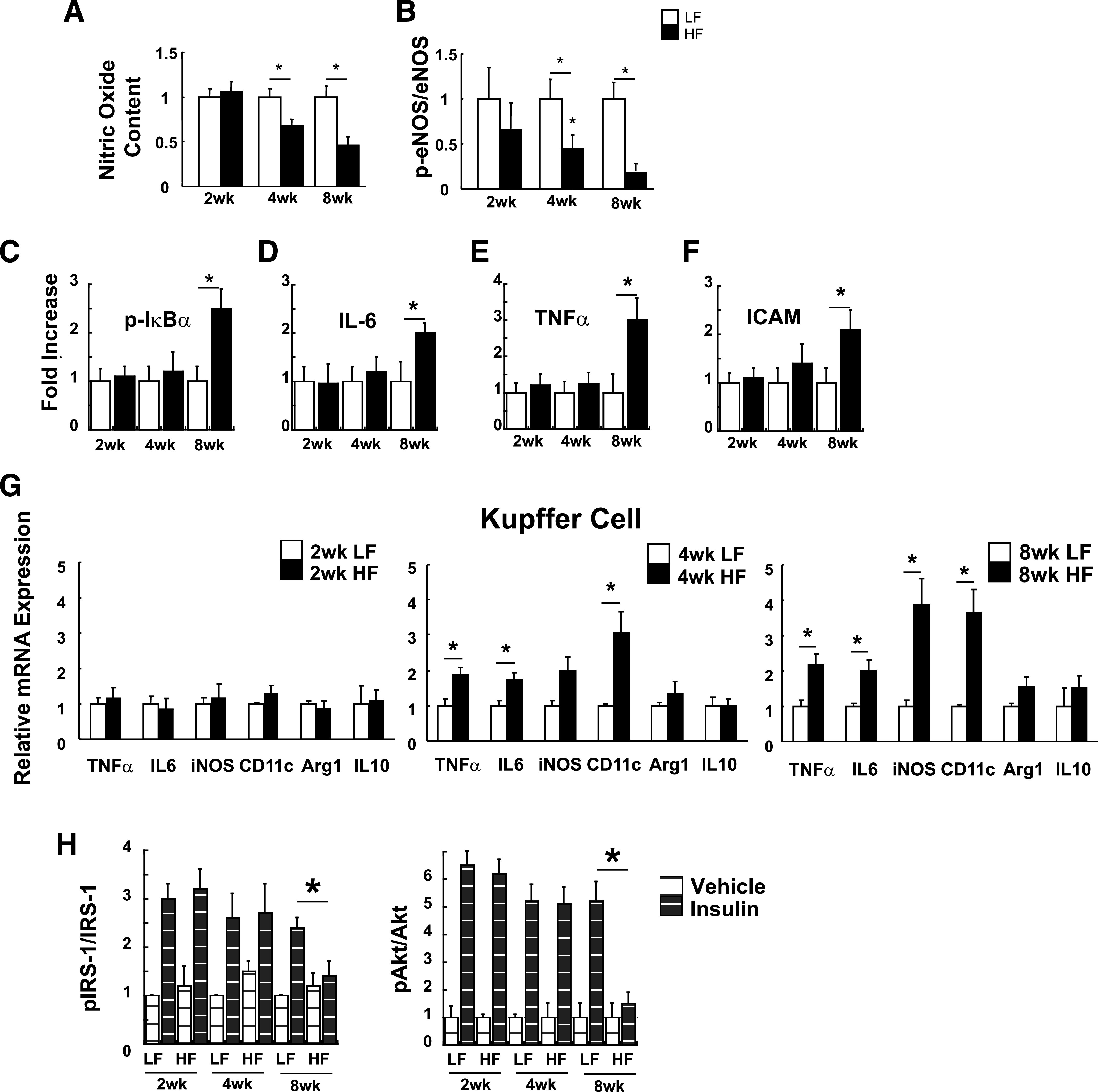
The effect of high-fat (HF) feeding on liver NO, hepatic inflammation, and Kupffer cell activation. Wild-type (WT) mice were fed either a low-fat (LF) or HF diet for 2, 4, or 8 weeks. A: Hepatic NO content during HF and LF feeding as measured by electron spin resonance spectroscopy (n = 9). B: Liver eNOS phosphorylation of Ser 1177 as measured by Western blot (n = 9). C: Liver phospho–IκB-α as measured by Western blot, normalized to total GAPDH levels (n = 9). D: Liver IL-6. E: TNF-α mRNA (n = 9). F: ICAM mRNA levels (n = 9). G: Relative mRNA levels from isolated Kupffer cells as measured by RT-PCR after 2, 4, and 8 weeks of LF or HF diet (n = 5 mice). H: In parallel experiments in WT mice, hepatic insulin signaling was assessed following intraperitoneal injection of insulin or saline vehicle (n = 5 per group). Liver protein lysates were analyzed for IRS-1 tyrosine phosphorylation (p–IRS-1), IRS-1, pAkt, and Akt levels by enzyme-linked immunosorbent assay. *P < 0.05.
Because Kupffer cells play an important role in the development of hepatic insulin resistance during high-fat feeding (2,20), we next sought to investigate the time course of Kupffer cell activation during high-fat feeding in normal mice using a validated Kupffer cell isolation method (3). Compared with those in low fat–fed controls, levels of TNF-α, IL-6, and CD11c mRNA were significantly increased in Kupffer cells after 4 weeks of high-fat feeding, and this effect persisted throughout the 8-week study period (Fig. 1G), although induction of iNOS mRNA in these cells was not detected until the 8-week time point. Impairment of insulin-mediated p–IRS-1 and p-Akt in liver lysates was evident at 8 weeks of high-fat feeding (Fig. 1H) after the onset of reduction of liver NO content and activation of Kupffer cells.
These data collectively suggest that high-fat feeding reduces liver NO signaling/content and triggers Kupffer cell inflammatory activation prior to the onset of generalized liver inflammation and biochemical insulin resistance at the level of IRS-1/Akt signaling.
Hepatic inflammation and insulin resistance in eNos−/− mice.
To test whether genetic absence of vascular eNOS reproduces the effect of high-fat feeding to induce liver inflammation and insulin resistance, we studied adult male eNos−/− mice and wild-type control mice fed either a low-fat or high-fat diet for a relatively short period (4 weeks). As expected, liver IκB-α phosphorylation and IL-6 mRNA expression in liver tissues of wild-type mice were not increased after only 4 weeks on either diet, whereas both markers of NF-κB activation were significantly increased in the liver of eNos−/− mice, regardless of the fat content of their diet (Fig. 2A and B). Similarly, inflammatory activation was observed in Kupffer cells in the eNos−/− mice, and mRNA expression encoding TNF-α, iNOS, IL-6, and CD11c were each increased significantly (Fig. 2C).
FIG. 2.
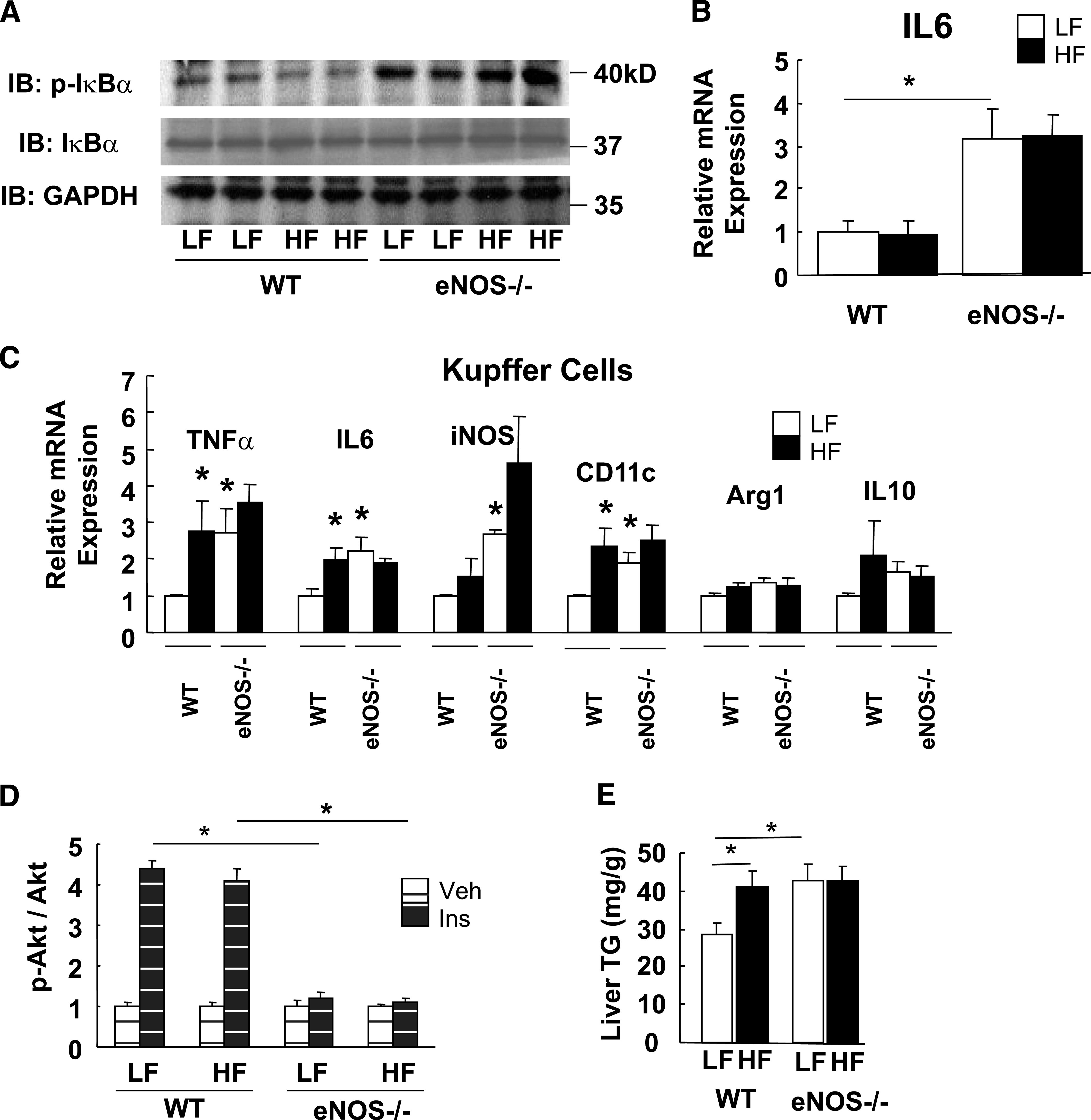
Hepatic inflammation and insulin resistance in eNos−/− mice. eNos−/− mice and littermate control mice were fed a low-fat (LF) or a high-fat (HF) diet for 4 weeks from age 8 weeks. A and B: Liver lysates were analyzed for IκB-α phosphorylation by Western blot and IL-6 mRNA expression by RT-PCR (n = 7). *P < 0.05. C: Inflammatory markers from isolated Kupffer cells by RT-PCR (n = 5). *P < 0.05. D: Hepatic insulin (Ins) signaling at the level of Akt phosphorylation (n = 5). *P < 0.05. E: Hepatic triglyceride (TG) content (n = 7). *P < 0.05. Veh, vehicle; WT, wild type. IB, immunoblot; kD, kilodalton.
Consistent with previous evidence from a glucose clamp study that eNOS-deficient mice have hepatic insulin resistance (21), we next found that insulin stimulation of hepatic IRS-1 and IRS-2 tyrosine phosphorylation (data not shown) and Akt 473 phosphorylation were attenuated in eNos−/− mice compared with wild-type control mice fed a low-fat diet (Fig. 2D), whereas deficiency of eNOS was associated with increased liver triglyceride content (Fig. 2E). These data suggest that loss of eNOS-derived NO is sufficient to reproduce the effect of prolonged high-fat feeding to cause liver inflammation and insulin resistance.
Effect of sildenafil in liver inflammation and activation of Kupffer cells during high-fat feeding.
Because increased NO/cGMP levels are associated with reduced NF-κB signaling, we next sought to determine whether increased NO/cGMP signaling would restore hepatic insulin signaling in an in vivo model. Adult male C57BL/6 mice were fed either a low-fat or a high-fat diet for 8 weeks to cause obesity, liver inflammation, and insulin resistance (15). During the final 2 weeks of the high-fat feeding protocol, mice on each diet received daily oral administration of either vehicle or the phosphodiesterase-5 (PDE-5) inhibitor sildenafil at a dose (30 mg/kg per day) that did not affect weight gain on either diet compared with the vehicle control group (15). In response to 8 weeks of high-fat feeding, liver IκB-α phosphorylation and IL-6 mRNA content in vehicle-treated mice were increased compared with those in low fat–fed mice, whereas in sildenafil-treated mice these inflammatory responses to high-fat feeding were not observed (Fig. 3A and B). In addition, a similar anti-inflammatory effect was seen in an analysis of Kupffer cells, in which expression of mRNA encoding TNF-α, iNOS, and CD11c was each reduced compared with Kupffer cells isolated from vehicle-treated controls (Fig. 3C).
FIG. 3.
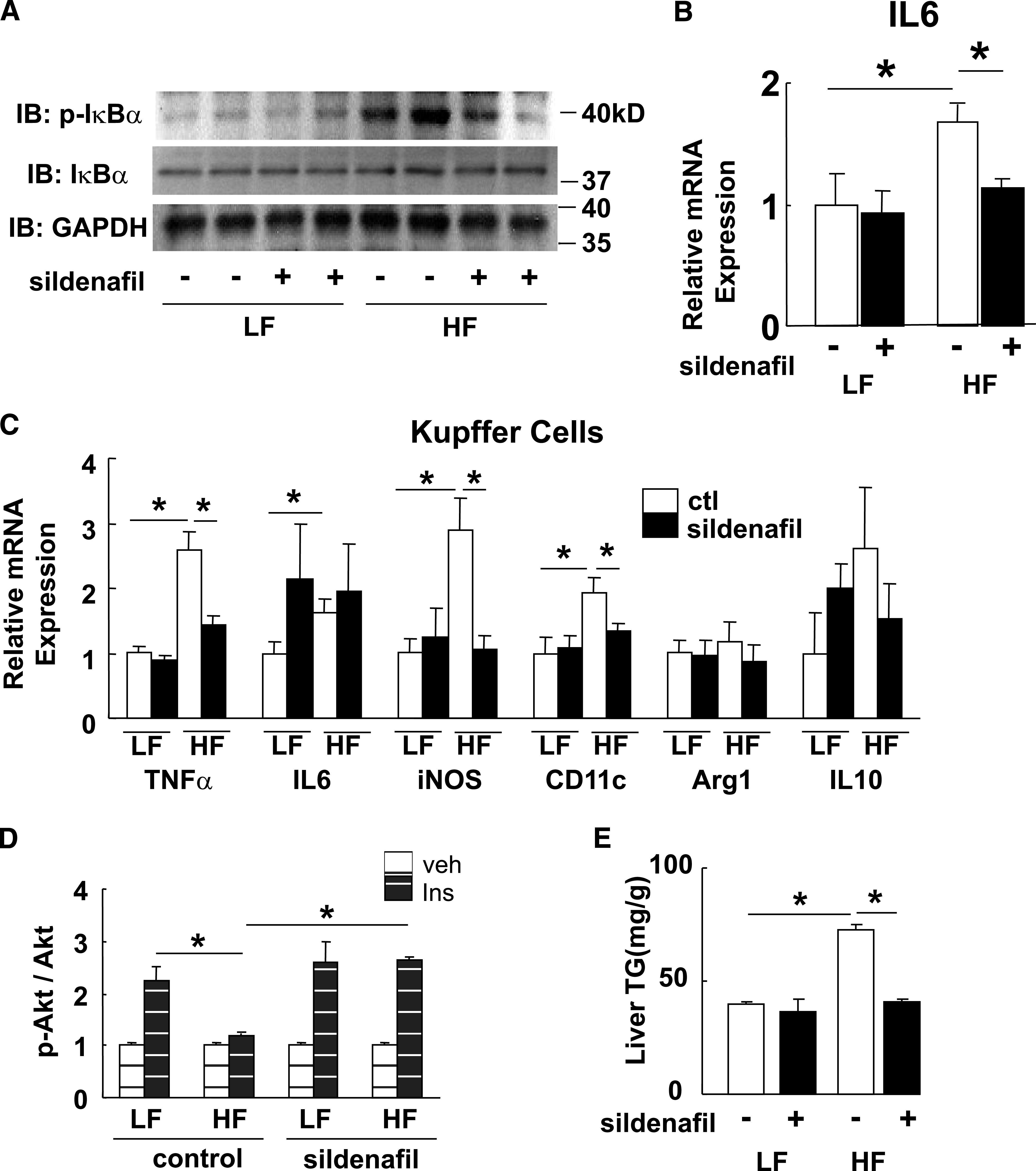
Effect of daily sildenafil during high-fat (HF) feeding on liver NF-κB and hepatic insulin signaling. Six-week-old male C57BL/6 mice were maintained on either a low-fat (LF; 10% saturated fat) or HF (60% saturated fat) diet for 8 weeks, and for the last 2 weeks of the diet, study mice received 30 mg/kg per day oral sildenafil (n = 8) or vehicle (n = 8). A: IκB-α phosphorylation in liver lysates. B: IL-6 mRNA levels. C: Inflammatory markers from isolated Kupffer cells by quantitative RT-PCR (n = 5). *P < 0.05. D: Hepatic insulin (Ins) signaling at the level of Akt phosphorylation (n = 5). *P < 0.05. E: Hepatic triglyceride (TG) content (n = 5). *P < 0.05. ctl, control; veh, vehicle. IB, immunoblot; kD, kilodalton.
Finally, sildenafil was associated with attenuation of the deleterious effects of high-fat feeding on insulin-mediated IRS-1 and IRS-2 tyrosine phosphorylation (data not shown) as well as on Ser 473 phosphorylation of Akt (Fig. 3D) and on hepatic triglyceride content (Fig. 3E). In wild-type mice, therefore, hepatic inflammation and insulin resistance induced by high-fat feeding can be fully blocked by pharmacological inhibition of PDE-5, which increases intracellular signaling via cGMP.
Role of VASP in liver inflammation and activation of Kupffer cells in vivo.
In tissues such as vascular smooth muscle cells and platelets, NO/cGMP signaling activates PKG, which in turn phosphorylates VASP on Ser 239. To determine whether high-fat feeding is associated with changes in PKG or phospho-VASP (Ser 239), we performed Western blot analysis on liver lysates from wild-type mice fed either a low-fat or high-fat diet for 8 weeks. As expected, we found that high-fat feeding is associated with reduced liver phospho-VASP levels, whereas total PKG protein levels were not significantly altered (Fig. 4A). This finding suggests that in mouse liver, the effect of high-fat feeding to reduce endothelial NO signaling decreases the basal level of VASP activation—an effect that in turn could contribute to the proinflammatory response to reduced endothelial NO signaling.
FIG. 4.
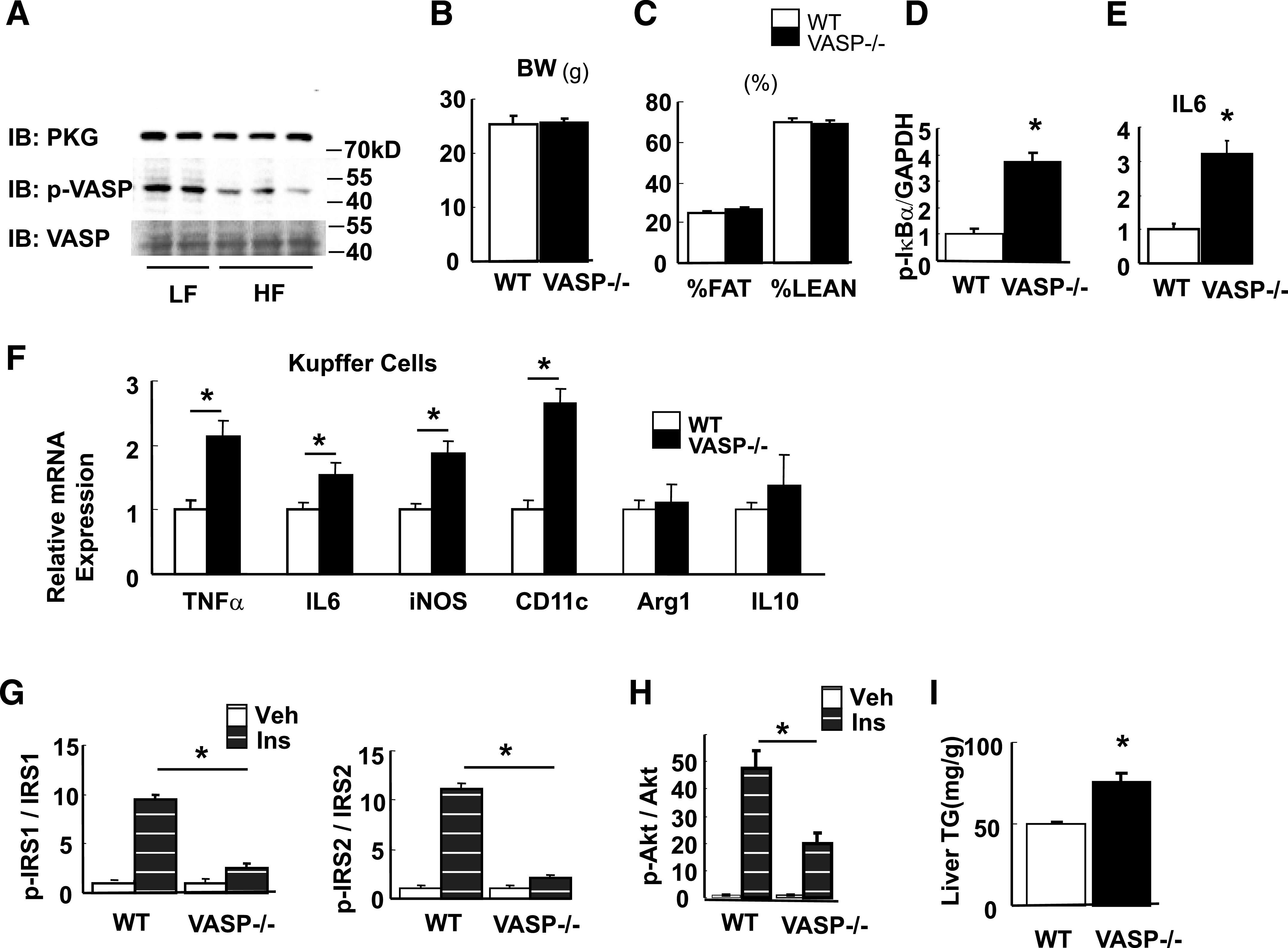
Effect of VASP deficiency on hepatic and Kupffer cell inflammation and hepatic insulin signaling. A: Hepatic PKG expression and phosphorylation of VASP Ser 239 from wild-type (WT) mice fed either a low-fat (LF) or high-fat (HF) diet for 4 weeks from age 8 weeks (n = 5). B–E: Vasp−/− mice and littermate control mice were fed an LF diet for 4 weeks. B: Body weight (BW) (g). C: Body composition (%). Liver lysates were analyzed for IκB-α phosphorylation by Western blot (D), and IL-6 mRNA expression was measured by RT-PCR (E) (n = 8). *P < 0.05. F: Kupffer cells were isolated from Vasp−/− mice or WT control mice on an LF diet for 4 weeks. Inflammatory markers as measured by RT-PCR (n = 5). *P < 0.05. G and H: In parallel experiments in Vasp−/− (n = 7) and littermate control (n = 9) mice, hepatic insulin (Ins) signaling was assessed following intraperitoneal injection of insulin. IRS-1 and IRS-2 tyrosine phosphorylation and Akt serine phosphorylation. *P < 0.05. I: Hepatic triglyceride (TG) from Vasp−/− and control (n = 5). *P < 0.05. Veh, vehicle. IB, immunoblot; kD, kilodalton.
For further investigation of this hypothesis, studies were performed on Vasp−/− mice and their littermate controls, which were fed a low-fat diet for 4 weeks: a diet and time period known not to induce hepatic inflammation or impair insulin signaling (7). Wild-type and Vasp−/− mice did not exhibit significant differences in body weight or body composition in response to 4 weeks of low-fat feeding (Fig. 4B and C). Nevertheless, hepatic IκB-α phosphorylation and IL-6 gene expression were increased in Vasp−/− compared with wild-type mice (Fig. 4D and E). In Kupffer cells, TNF-α, IL-6, iNos, and CD11c mRNA were also significantly higher in Vasp−/− than in control mice (Fig. 4F). Furthermore, insulin-stimulated phosphorylation of IRS-1 and IRS-2 (Fig. 4G) and Akt (Fig. 4H) were each attenuated in the liver of Vasp−/− mice compared with littermate controls after 4 weeks of low-fat feeding, whereas deficiency of Vasp is associated with increased liver triglyceride content (Fig. 4I). These results collectively suggest that the absence of VASP induces inflammation in both hepatocytes and Kupffer cells—responses that implicate endogenous VASP signaling as a mediator of the effect of NO to attenuate both hepatic inflammation and insulin resistance.
Effect of NO/cGMP/VASP signaling on macrophage activation in vitro.
To more directly investigate whether VASP is a determinant of macrophage activation phenotype, we next asked whether signaling via this protein plays a tonic role to limit inflammatory responses of macrophages in a cell culture system. First, we overexpressed VASP in RAW cells by retroviral gene transfer (Fig. 5A) and observed that VASP overexpression is associated with an attenuation of lipopolysaccharide (LPS)-dependent induction of TNF-α, IL-6, and iNOS mRNA (Fig. 5B) compared with control RAW cells transduced with empty vector. Thus, increased VASP signaling appears to attenuate inflammatory signaling in macrophages.
FIG. 5.
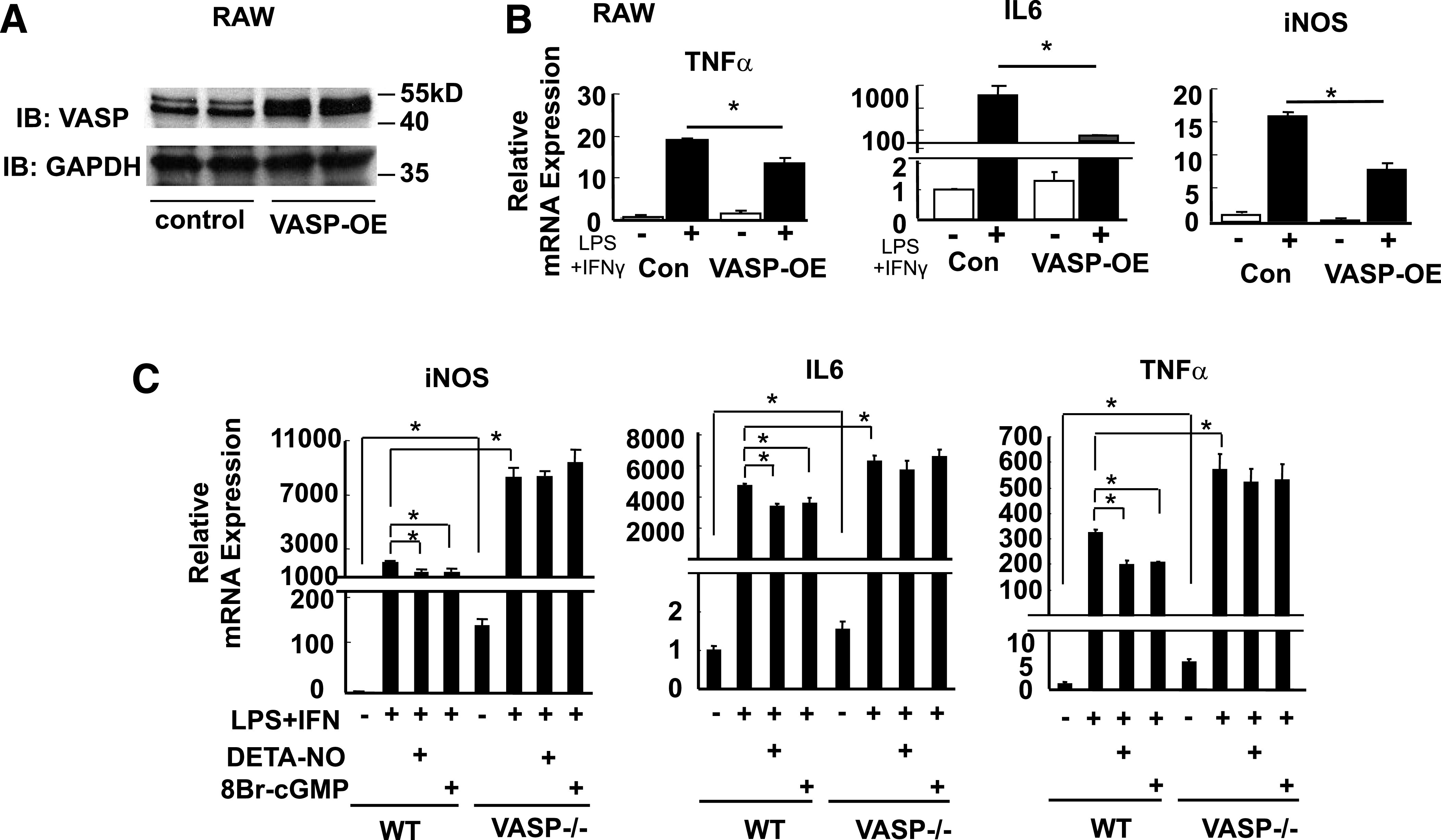
The effect of VASP signaling on RAW cell inflammatory responses to LPS. RAW cells were transduced with VASP (VASP-OE) or control (Con) vector. A: VASP Western blot. B: RAW cells were stimulated with LPS (5 ng/mL) and IFN-γ (12 ng/mL) for 4 h and then analyzed for TNF-α, IL-6, and iNOS mRNA expression (n = 3). *P < 0.05. C: Peritoneal macrophages from VASP−/− and wild-type (WT) control mice were stimulated with LPS (5 ng/mL) and IFN-γ (12 ng/mL) for 4 h in the presence or absence of DETA-NO (10 μmol/L for 4 h) or 8Br-cGMP (10 μmol/L for 4 h). *P < 0.05. IB, immunoblot; kD, kilodalton.
For determination of whether VASP is required for the anti-inflammatory effects of NO in macrophages, peritoneal macrophages were collected from Vasp−/− and wild-type control mice and were subsequently stimulated with the combination of LPS and γ-interferon (IFN-γ) to increase NF-κB signaling. Even in the absence of inflammatory stimuli, VASP deficiency is associated with increased NF-κB activation compared with wild-type macrophages, suggesting that the absence of VASP is sufficient to increase inflammatory signaling (Fig. 5C). When LPS stimulation was performed in the presence of DETA-NO or 8Br-cGMP, agents known to increase VASP signaling, NF-κB signaling (e.g., TNFα, IL6, and iNos gene expression) was strongly diminished in peritoneal macrophages from control littermate mice. However, in Vasp-deficient macrophages, DETA-NO and 8Br-cGMP no longer exhibited anti-inflammatory effects (Fig. 5C). Thus, intact VASP signaling appears to be necessary for the anti-inflammatory effects of NO/cGMP signal transduction in macrophages, and furthermore, the absence of VASP signaling is sufficient to increase NF-κB signaling.
Effect of VASP signaling in hepatocytes in vitro.
We next investigated whether VASP signaling is a determinant in hepatic responses to inflammatory stimuli in vitro. VASP was overexpressed in AML12 hepatocytes using retroviral transduction (Fig. 6A). Overexpression of VASP was associated with reduced palmitate-mediated increases in phospho–IκB-α levels (Fig. 6B) and restoration of insulin-mediated p-Akt signaling even in the presence of palmitate (Fig. 6C). Thus, increased VASP signaling in hepatocytes is sufficient to attenuate palmitate-dependent NF-κB activation. Next, primary hepatocytes were isolated from Vasp−/− and littermate control mice. The absence of Vasp was sufficient to increase phospho–IκB-α levels, even in the absence of palmitate (Fig. 6D), and the addition of palmitate did not further increase phospho–IκB-α levels in Vasp-deficient hepatocytes. These results suggest that the absence of Vasp is sufficient in reproducing the inflammatory effects of palmitate. In addition, the anti-inflammatory effects of DETA-NO or 8Br-cGMP were no longer apparent in Vasp-deficient hepatocytes (Fig. 6D).
FIG. 6.
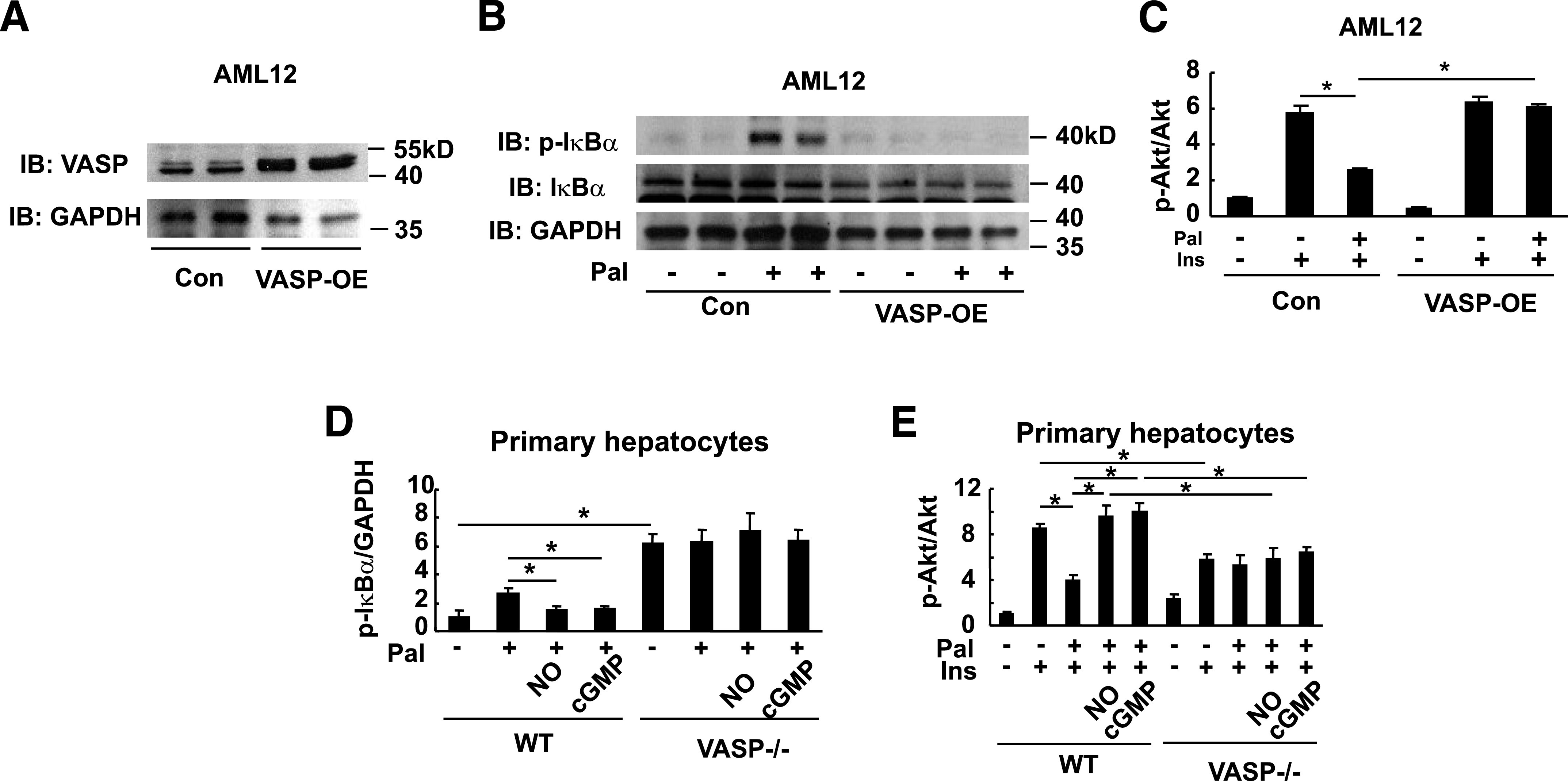
The effect of VASP signaling on hepatocyte inflammatory responses to palmitate. AML12 hepatocytes were transduced with VASP (VASP-OE) or control (Con) vector. A: VASP Western blot. B: Hepatocytes were treated with 100 μmol/L palmitate (Pal) or vehicle for 4 h, and a representative IκB-α phosphorylation Western blot from one of three independent experiments is shown. C: Insulin (Ins)-mediated pAkt signaling (10 nmol/L insulin for 15 min) in transduced hepatocytes, following 4 h palmitate (100 μmol/L). Ser 473 Akt phosphorylation was assessed by Western blot analysis, and fold increase over control condition was calculated (n = 3). *P < 0.05. D: Primary hepatocytes were isolated and cultured from Vasp−/− mice or wild-type (WT) control mice. Isolated hepatocytes were treated with palmitate (100 μmol/L) for 4 h following 4-h pretreatment with 10 μmol/L DETA-NO or 10 μmol/L 8Br-cGMP. IκB-α phosphorylation was assessed by Western blot analysis. *P < 0.05. E: Insulin-mediated pAkt (10 nmol/L for 15 min) following treatment with palmitate (100 μmol/L for 4 h), DETA-NO (10 μmol/L for 4 h), or 8BrcGMP (10 μmol/L for 4 h). Akt Ser 473 phosphorylation assessed by Western blot (n = 3). *P < 0.05. IB, immunoblot; kD, kilodalton.
In parallel experiments, hepatic Vasp deficiency is associated with impaired insulin signaling at the level of p-Akt, even in the absence of palmitate. These results are similar to what was seen in vivo in Vasp−/− mice fed a low-fat diet. As expected, the effect of DETA-NO or 8Br-cGMP to attenuate palmitate-induced insulin resistance was not apparent in Vasp-deficient hepatocytes (Fig. 6E).
These in vitro results collectively suggest that the absence of VASP induces inflammation in hepatocyte responses that implicate endogenous VASP signaling as a mediator of the effect of NO to attenuate both hepatic inflammation and insulin resistance.
DISCUSSION
Growing evidence suggests that inflammatory signaling pathways mediate insulin resistance induced by nutrient excess in both cultured hepatocytes and in hepatic tissue in vivo (2,4,7,20). Because endothelial NO signaling has anti-inflammatory properties and because vascular NO levels decline early in the course of DIO (7), we hypothesized an etiological role for reduced NO/cGMP signaling in the pathogenesis of hepatic inflammation and impaired hepatic insulin signaling in this setting. Consistent with this hypothesis, we found that during high-fat feeding, reduced liver NO content precedes the onset of hepatic inflammation (at the level of NF-κB) and impairment of insulin signaling at the level of IRS-1 and phospho-Akt. High-fat feeding–dependent hepatic inflammation and insulin resistance were prevented by daily oral dosing of sildenafil, a PDE-5 inhibitor that increases signaling downstream of endothelial NO. Furthermore, we showed that deficiency of VASP, a key downstream target of the NO/cGMP signaling pathway, causes hepatic inflammation and insulin resistance comparable with that induced by high-fat feeding. Together, these results provide direct evidence in support of the hypothesis that vascular NO/cGMP/VASP signaling plays a physiological role to protect against hepatic inflammation induced during DIO. By extension, reduction of phospho-eNOS by high-fat feeding may be an early mediator of these deleterious hepatic responses.
In the context of ischemia reperfusion or exposure to liver toxins, decreased production of NO from eNOS has been linked to liver pathology via dysregulation of blood flow and oxygen delivery (22)—effects that may arise in part from NF-κB–dependent inflammation. In support of this model, both NO donor administration and eNOS overexpression are protective against liver injury in animal models of hepatoxicity (23,24). Similarly, inflammatory activation of Kupffer cells is implicated in obesity-induced liver insulin resistance based on evidence that during high-fat feeding, these cells undergo proinflammatory activation while, conversely, deletion of IκB-α in myeloid cells reduces macrophage-mediated inflammation and improves hepatic insulin sensitivity in mice with DIO (2).
Depletion of Kupffer cells using gadolinium chloride attenuated the development of hepatic steatosis and hepatic insulin resistance, suggesting an important early role for Kupffer cells in diet-induced alterations in hepatic insulin resistance (1). Our current studies support and extend this model by offering evidence that endothelial NO exerts a tonic anti-inflammatory effect on Kupffer cells in vivo, as well as in a macrophage cell line in culture, and that during high-fat feeding reduced NO production in liver contributes to the proinflammatory activation of these cells that clearly occurs in mice with DIO.
In the vessel wall, the anti-inflammatory role of NO is well established, especially in the early development of atherosclerosis. Endothelial NO actively regulates a key and early immune response implicated in atherogenesis by regulating macrophage and lymphocyte capture and vessel wall migration via adhesion molecules (25,26). Mice deficient in eNOS consequently exhibit an increase in adhesion molecule expression resulting in increased neutrophil rolling, adherence, and migration in response to inflammatory stimuli, all of which are important early steps in the development of atherosclerosis. Our studies extend these observations to both Kupffer cells and hepatocytes and suggest that NO anti-inflammatory effects extend beyond the vessel wall.
Previous investigators have demonstrated that deletion of eNOS results in increased systemic insulin resistance (21,27). Our data suggest a role for liver eNOS and Kupffer cells in the development of hepatic insulin resistance; however, the lack of vascular eNOS in other metabolically important tissues such as adipose, muscle, or central nervous system might be responsible for the observed alterations in liver inflammation, and this alternative explanation needs to be considered.
In rodent models of diabetes, PKG and VASP signaling are both reduced in vascular tissues (28) resulting in actin cytoskeleton rearrangements (29), but whether VASP signaling mediates inhibitory effects of NO/PKG on NF-κB signaling has not previously been investigated. Our finding that deficiency of VASP reproduces the effect of high-fat feeding in the activation of Kupffer cells while also causing liver inflammation and insulin resistance suggests that this protein plays a key role in mediating the anti-inflammatory effects of vascular NO on these cells and is required for maintenance of normal liver insulin sensitivity. The extent of VASP expression in hepatocytes and Kupffer cells and their relative contribution to the anti-inflammatory effects of NO signaling in vivo await further study; however, our finding that macrophages isolated from VASP-deficient mice exhibit enhanced basal and stimulated inflammation points to a direct role of this pathway to control macrophage activation status. VASP deficiency is associated with impaired insulin-mediated IRS and pAkt signaling and evidence for increased hepatic steatosis suggesting a novel metabolic role for this protein. Additional studies on VASP’s role in hepatic gluconeogenesis, lipid metabolism, and systemic insulin sensitivity are currently under investigation.
The relationship between NO and cellular inflammation is complex because at high concentrations (such as the micromolar levels produced by iNOS), NO is cytotoxic and exerts PKG-independent proinflammatory effects that can lead to cell death and are functionally opposite those induced by the much lower (nanomolar) concentrations of NO generated by eNOS that engage the soluble guanylate cyclase/cGMP/PKG pathway. Intracellular cGMP levels are governed not only by guanylyl cyclase activity but also by phosphodiesterase conversion of cGMP back to GMP. Sildendafil is a PDE-5 inhibitor that attenuates intracellular catabolism of cGMP. A link between cGMP signaling and insulin action was identified in a recent study in which 12-week treatment with sildenafil improved energy balance and insulin sensitivity in mice fed a high-fat diet (30), and this drug also ameliorates diabetes-induced endothelial dysfunction in both a rat model and in humans (31,32). We used a much shorter period of sildenafil treatment, 2 weeks, to avoid the effect of sildenafil on weight change or energy balance. Combined with evidence that liver NO content falls early in the course of DIO in mice, our finding that 2 wk sildenafil treatment prevents high-fat diet-induced liver inflammation offers further evidence that reduced NO/cGMP signaling is a trigger for the deleterious hepatic effects of high-fat feeding. Moreover, we report that sildenafil treatment also blocks M1 activation of Kupffer cells induced by high-fat feeding and further studies are warranted to determine whether this effect can explain the prevention of liver inflammation and insulin resistance observed in this model.
Sildenafil improves insulin sensitivity during high-fat feeding (30), and our data demonstrate improvements in hepatatic steatosis and Kupffer cell inflammation. The target of sildenafil action, however, remains an important unanswered question. In vitro studies suggest that increased cGMP (using 8Br-cGMP) reduces NF-κB signaling in both hepatocytes and macrophage, and previous studies have demonstrated an inhibitory effect of increased cGMP in endothelial cells (33). Collectively, these data would imply that sildenafil’s anti-inflammatory effect is “global”; yet, whether the observed metabolic effects are the result of action on hepatocytes or Kupffer cells remains to be determined. Furthermore, it is also possible that sildenafil’s beneficial metabolic and anti-inflammatory effects do not even require direct action at the liver but may involve action on the adipose tissue or adipose tissue macrophage.
In summary, our findings suggest a key role for vascular NO to limit the effects of high-fat feeding to induce Kupffer cell activation, hepatic inflammation, and impairment of hepatic insulin signaling at the level of IκB-α signaling and phospho-Akt by insulin, respectively. Our additional findings that targeted deletion of VASP predisposes Kupffer cell inflammation, and that VASP knockout attenuates the anti-inflammatory effects of NO, identify VASP as a critical downstream mediator of the anti-inflammatory effects induced by the NO/cGMP/PKG pathway. These results imply a physiologic role for endothelial NO signaling to limit obesity-associated inflammation and impairment of insulin signaling in hepatocytes and Kupffer cells and identify the NO/PKG/VASP pathway as a potential target for the treatment of obesity-associated metabolic impairment.
ACKNOWLEDGMENTS
This study was supported by a grant from the Manpei Suzuki Diabetes Foundation (to S.T.), by National Institutes of Health (NIH) Grant HL-52459 (to A.W.C.), by NIH grants DK-083042 and DK-052989 (to M.W.S.), by NIH Grant DK-073878 (to F.K.), and by a grant from the John L. Locke Jr. Charitable Trust and from the Kenneth H. Cooper Endowed Professorship in Preventive Cardiology (to F.K.).
No potential conflicts of interest relevant to this article were reported.
S.T. designed and performed experiments, provided data analysis, wrote the manuscript, contributed to discussion, and reviewed and edited the manuscript. N.O.R. designed and performed experiments. P.H. performed experiments and contributed to discussion. A.M.C. designed and performed experiments and contributed to discussion. V.M.-S. performed experiments. G.D. interpreted data and contributed to discussion. G.J.M. performed data analysis and contributed to discussion. A.W.C. contributed to discussion and reviewed and edited the manuscript. M.W.S. contributed to discussion and wrote, reviewed, and edited the manuscript. F.K. interpreted data and wrote, reviewed, and edited the manuscript.
Parts of this study were presented in abstract form at the 70th Scientific Sessions of the American Diabetes Association, Orlando, Florida, 25–29 June 2010.
REFERENCES
- 1.Huang W, Metlakunta A, Dedousis N, et al. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes 2010;59:347–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arkan MC, Hevener AL, Greten FR, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med 2005;11:191–198 [DOI] [PubMed] [Google Scholar]
- 3.Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, et al. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab 2008;7:496–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 2010;72:219–246 [DOI] [PubMed] [Google Scholar]
- 5.Verma S, Anderson TJ. Fundamentals of endothelial function for the clinical cardiologist. Circulation 2002;105:546–549 [DOI] [PubMed] [Google Scholar]
- 6.Liu VW, Huang PL. Cardiovascular roles of nitric oxide: a review of insights from nitric oxide synthase gene disrupted mice. Cardiovasc Res 2008;77:19–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim F, Pham M, Maloney E, et al. Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arterioscler Thromb Vasc Biol 2008;28:1982–1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ishibashi H, Nakamura M, Komori A, Migita K, Shimoda S. Liver architecture, cell function, and disease. Semin Immunopathol 2009;31:399–409 [DOI] [PubMed] [Google Scholar]
- 9.Braet F, Wisse E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: a review. Comp Hepatol 2002;1:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shah V, Haddad FG, Garcia-Cardena G, et al. Liver sinusoidal endothelial cells are responsible for nitric oxide modulation of resistance in the hepatic sinusoids. J Clin Invest 1997;100:2923–2930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheung O, Sanyal AJ. Recent advances in nonalcoholic fatty liver disease. Curr Opin Gastroenterol 2010;26:202–208 [DOI] [PubMed] [Google Scholar]
- 12.Grumbach IM, Chen W, Mertens SA, Harrison DG. A negative feedback mechanism involving nitric oxide and nuclear factor kappa-B modulates endothelial nitric oxide synthase transcription. J Mol Cell Cardiol 2005;39:595–603 [DOI] [PubMed] [Google Scholar]
- 13.Trichet L, Sykes C, Plastino J. Relaxing the actin cytoskeleton for adhesion and movement with Ena/VASP. J Cell Biol 2008;181:19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen L, Daum G, Chitaley K, et al. Vasodilator-stimulated phosphoprotein regulates proliferation and growth inhibition by nitric oxide in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 2004;24:1403–1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim F, Pham M, Luttrell I, et al. Toll-like receptor-4 mediates vascular inflammation and insulin resistance in diet-induced obesity. Circ Res 2007;100:1589–1596 [DOI] [PubMed] [Google Scholar]
- 16.Kim F, Tysseling KA, Rice J, et al. Free fatty acid impairment of nitric oxide production in endothelial cells is mediated by IKKbeta. Arterioscler Thromb Vasc Biol 2005;25:989–994 [DOI] [PubMed] [Google Scholar]
- 17.Khoo JP, Alp NJ, Bendall JK, et al. EPR quantification of vascular nitric oxide production in genetically modified mouse models. Nitric Oxide 2004;10:156–161 [DOI] [PubMed] [Google Scholar]
- 18.Kanda H, Tateya S, Tamori Y, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest 2006;116:1494–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Askari B, Kanter JE, Sherrid AM, et al. Rosiglitazone inhibits acyl-CoA synthetase activity and fatty acid partitioning to diacylglycerol and triacylglycerol via a peroxisome proliferator-activated receptor-gamma-independent mechanism in human arterial smooth muscle cells and macrophages. Diabetes 2007;56:1143–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Odegaard JI, Chawla A. Mechanisms of macrophage activation in obesity-induced insulin resistance. Nat Clin Pract Endocrinol Metab 2008;4:619–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shankar RR, Wu Y, Shen HQ, Zhu JS, Baron AD. Mice with gene disruption of both endothelial and neuronal nitric oxide synthase exhibit insulin resistance. Diabetes 2000;49:684–687 [DOI] [PubMed] [Google Scholar]
- 22.Liu J, Waalkes MP. Nitric oxide and chemically induced hepatotoxicity: beneficial effects of the liver-selective nitric oxide donor, V-PYRRO/NO. Toxicology 2005;208:289–297 [DOI] [PubMed] [Google Scholar]
- 23.Rivera-Chavez FA, Toledo-Pereyra LH, Dean RE, Crouch L, Ward PA. Exogenous and endogenous nitric oxide but not iNOS inhibition improves function and survival of ischemically injured livers. J Invest Surg 2001;14:267–273 [DOI] [PubMed] [Google Scholar]
- 24.Duranski MR, Elrod JW, Calvert JW, Bryan NS, Feelisch M, Lefer DJ. Genetic overexpression of eNOS attenuates hepatic ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 2006;291:H2980–H2986 [DOI] [PubMed] [Google Scholar]
- 25.Giordano D, Magaletti DM, Clark EA. Nitric oxide and cGMP protein kinase (cGK) regulate dendritic-cell migration toward the lymph-node-directing chemokine CCL19. Blood 2006;107:1537–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinelli R, Gegg M, Longbottom R, Adamson P, Turowski P, Greenwood J. ICAM-1-mediated endothelial nitric oxide synthase activation via calcium and AMP-activated protein kinase is required for transendothelial lymphocyte migration. Mol Biol Cell 2009;20:995–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duplain H, Burcelin R, Sartori C, et al. Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation 2001;104:342–345 [DOI] [PubMed] [Google Scholar]
- 28.Russo I, Del Mese P, Doronzo G, et al. Resistance to the nitric oxide/cyclic guanosine 5′-monophosphate/protein kinase G pathway in vascular smooth muscle cells from the obese Zucker rat, a classical animal model of insulin resistance: role of oxidative stress. Endocrinology 2008;149:1480–1489 [DOI] [PubMed] [Google Scholar]
- 29.Blume C, Benz PM, Walter U, Ha J, Kemp BE, Renné T. AMP-activated protein kinase impairs endothelial actin cytoskeleton assembly by phosphorylating vasodilator-stimulated phosphoprotein. J Biol Chem 2007;282:4601–4612 [DOI] [PubMed] [Google Scholar]
- 30.Ayala JE, Bracy DP, Julien BM, Rottman JN, Fueger PT, Wasserman DH. Chronic treatment with sildenafil improves energy balance and insulin action in high fat-fed conscious mice. Diabetes 2007;56:1025–1033 [DOI] [PubMed] [Google Scholar]
- 31.Aversa A, Vitale C, Volterrani M, et al. Chronic administration of Sildenafil improves markers of endothelial function in men with Type 2 diabetes. Diabet Med 2008;25:37–44 [DOI] [PubMed] [Google Scholar]
- 32.Schäfer A, Fraccarollo D, Pförtsch S, et al. Improvement of vascular function by acute and chronic treatment with the PDE-5 inhibitor sildenafil in experimental diabetes mellitus. Br J Pharmacol 2008;153:886–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rizzo NO, Maloney E, Pham M, et al. Reduced NO-cGMP signaling contributes to vascular inflammation and insulin resistance induced by high-fat feeding. Arterioscler Thromb Vasc Biol 2010;30:758–765 [DOI] [PMC free article] [PubMed] [Google Scholar]