

Abstract
OBJECTIVE
Consistent with studies in NOD mice, early clinical trials addressing whether depletion of B cells by the Rituximab CD20-specific antibody provides an effective means for type 1 diabetes reversal have produced promising results. However, to improve therapeutic efficacy, additional B-cell–depleting agents, as well as attempts seeking diabetes prevention, are being considered.
RESEARCH DESIGN AND METHODS
Autoantibodies, including those against insulin (IAAs), are used to identify at-risk subjects for inclusion in diabetes prevention trials. Therefore, we tested the ability of anti-CD20 to prevent diabetes in NOD mice when administered either before or after IAA onset.
RESULTS
The murine CD20-specific 18B12 antibody that like Rituximab, depletes the follicular (FO) but not marginal zone subset of B cells, efficiently inhibited diabetes development in NOD mice in a likely regulatory T-cell–dependent manner only when treatment was initiated before IAA detection. One implication of these results is that the FO subset of B cells preferentially contributes to early diabetes initiation events. However, most important, the inefficient ability of anti-CD20 treatment to exert late-stage diabetes prevention was found to be attributable to downregulation of CD20 expression upon B cell entry into pancreatic islets.
CONCLUSIONS
These findings provide important guidance for designing strategies targeting B cells as a potential means of diabetes intervention.
While the autoimmune destruction of pancreatic β-cells that results in type 1 diabetes is ultimately mediated by both CD4 and CD8 T cells, in the NOD mouse model and potentially in humans, disease pathogenesis also requires contributions from B cells (reviewed in Silveira et al. [1]). Studies in NOD mice indicate B cells likely contribute to diabetes by serving as a subset of antigen presenting cells (APCs) that most efficiently support the expansion of pathogenic CD4 T-cell responses (2–4). This is because unlike other APC subsets, B cells express plasma membrane-bound Ig molecules, allowing for their specific and efficient capture of pancreatic β-cell proteins (5,6). Indeed, some diabetes susceptibility genes in NOD mice mechanistically contribute to disease pathogenesis by impairing immunological tolerance induction mechanisms normally deleting or inactivating B cells expressing autoreactive Ig specificities (7–9). Secreted autoreactive Ig molecules may also contribute to diabetes pathogenesis in NOD mice (10,11). In addition, B cells may contribute to diabetes in NOD mice by supporting development in the vicinity of pancreatic islets of tertiary lymphoid structures where pathogenic T cells might be activated (12).
Eliminating B cells from birth by either genetic or antibody-mediated approaches inhibits diabetes development in NOD mice (13,14). Partly on the basis of these findings, early phase clinical trials were initiated to determine whether depletion of B cells using the human CD20-specific Rituximab antibody provided beneficial effects, including preservation of C-peptide production, for recent-onset diabetes patients (15,16). Hope for these trials was bolstered by several reports suggesting that in addition to a capacity to block progression to overt diabetes when initiated at an early prodromal stage of disease development, anti-CD20–mediated B-cell depletion (and in one case, using anti-CD22) can also reverse recently established hyperglycemia in at least a subset of NOD mice (17–19). However, it is unclear if CD20- and CD22-specific antibodies with a reported ability to reverse recent-onset diabetes in NOD mice exert the same pattern of B-cell subset deletion as Rituximab. In this regard, it should be noted that Rituximab efficiently depletes the follicular (FO) but not the marginal zone (MZ) subset of mature B cells (20). Such a characteristic is of potential importance given reports that MZ subset B cells can exert potent APC activity and may preferentially contribute to diabetes development in NOD mice (21,22). Furthermore, the capacity of anti-CD20 treatment to eliminate B cells that become activated within pancreatic insulitic infiltrates during diabetes development is also unknown.
Another factor to consider is the short time frame after onset of overt hyperglycemia in which anti-CD20–mediated B-cell depletion can reportedly exert a disease reversal effect in NOD mice (18). It is unclear how frequently anti-CD20 treatment could be undertaken in an analogous time frame after diabetes onset in humans. Furthermore, the first reports from human diabetes intervention trials indicate Rituximab treatment retards the rate but does not eliminate the further erosion of residual pancreatic β-cell mass in recent disease onset patients (23). With this result, while promising, it has been questioned whether anti-CD20 treatment might prove more effective in preventing the progression to overt diabetes when initiated in individuals at late prodromal stages of disease development. Here, such trials would take advantage of a continual refinement of genetic and immunological susceptibility markers (24,25).
One key marker considered predictive for future diabetes development in humans is the appearance of insulin autoantibodies (IAAs) (26). The presence of IAAs also reportedly marks individual NOD mice that will first develop overt diabetes (27). Hence, to model a potential clinical use setting, we determined if when first initiated in already IAA-positive NOD mice, treatment with a murine CD20-specific antibody sharing B-cell deletional characteristics similar to Rituximab retained a capacity to inhibit diabetes development. We also assessed whether during progression of diabetes development, anti-CD20 treatment could eliminate B cells within pancreatic islet leukocytic infiltrates. Diabetes resistance elicited in NOD mice treated with a B-cell activating factor (BAFF)-blocking reagent depleting all mature B cells reportedly results from an enhanced ability of residual remaining myeloid-type APCs to support a regulatory T-cell (Treg) expansion (28). Hence, we also evaluated if diabetes protection resulting from the possible partial pattern of anti-CD20–mediated B-cell depletion was due to a Treg expansion.
RESEARCH DESIGN AND METHODS
Mice.
NOD/LtDvs mice are maintained by sibling matings at The Jackson Laboratory. B-cell–deficient NOD.Igµnull and totally lymphocyte-deficient NOD-scid mice have been previously described (14,29,30).
Anti-CD20–mediated B-cell depletion.
IgG1 and IgG2a isotypes of the B-cell–depleting mouse CD20-specific 18B12 monoclonal antibody have been described (31,32). Both antibodies were injected intraperitoneally at a 10 mg/kg body wt dose at indicated time points. Control mice received equivalent doses of the isotype-matched non–B-cell depleting 2B8 antibody. In one experiment, at the initiation of anti-CD20 or control antibody treatment, NOD mice also received 250 μg i.p. injections at 2-week intervals of the Treg-depleting PC61 CD25-specific antibody.
Flow cytometry.
Splenic and/or pancreatic lymph node (PLN)-derived leukocytes were assessed for levels of various B-cell subsets by flow cytometry using FACSCalibur instrumentation (BD Biosciences, San Jose, CA) and FlowJo data analysis software (Tree Star Inc., Palo Alto, CA). Fluorochrome conjugated monoclonal antibodies specific for the CD45R/B220 (RA36B2), CD21/CD35 (7G6), CD23 (B3B4), and CD138 (281–2) cell surface molecules were obtained from BD Biosciences. Total B cells were identified based on B220 expression. Among total B cells, expression patterns of CD21 and CD23 defined the T1 (CD21− CD23−), T2/pre-MZ (CD21hi CD23int), MZ (CD21hi CD23−), and FO (CD21int CD23hi) subsets. Plasma cells were identified by CD138 expression. Some studies used the IgG2a CD20-specific 18B12 antibody conjugated to allophycocyanin using the Alexa Fluor Protein Kit (Invitrogen, Carlsbad, CA). Other studies evaluated pancreatic islet–associated B-cell populations in NOD mice. Islets were isolated as previously described (33) and cultured overnight on an individual donor basis in previously described tissue culture medium (34), allowing for egress of associated leukocytes that were harvested for flow cytometric analyses of B-cell content and CD20 expression. Tregs were enumerated based on a CD4+CD25+FoxP3+ phenotype using the eBioscience (San Diego, CA) FoxP3 intracellular staining kit. Functional activity of magnetic bead–purified Tregs (CD4+ CD25+ phenotype) was evaluated by the previously described flow cytometric approach (35), assessing their ability to suppress anti-CD3–stimulated proliferation of carboxyfluorescein succinimidyl ester–labeled CD4+ CD25− responder T cells. A previously described phycoerythrin-conjugated H2-Ag7 MHC class II tetramer supplied by Dr. Luc Teyton (Scripps Research Institute, La Jolla, CA) was used to identify CD4 T cells sharing antigenic specificity with the diabetogenic BDC2.5 clonotype (36).
Diabetes development.
Mice were monitored for glycosuria with Ames Diastix (Bayer Diagnostics Division, Elkhart, IN), with diabetes onset diagnosed as two consecutive values of ≥3.
Antibody response.
Generation and measurement by enzyme-linked immunosorbent assay of antibodies produced in response to priming with hen egg lysozyme (HEL) were carried out by previously described methods (7–9).
IAA assay.
IAAs were detected using the previously described radioimmunoassay methodology (37).
RESULTS
The 18B12 CD20-specific antibody depletes B cells in NOD mice but with the IgG2a isotype inducing anaphylaxis.
The IgG1 isotype of the 18B12 murine CD20-specific antibody rapidly depleted peripheral blood B cells in NOD mice, with minimal rebounding observed at day 21 posttreatment (Fig. 1). NOD mice could be maintained in a state where peripheral blood B cells remained virtually absent by continuous treatments with the IgG1 anti-CD20 reagent at 21-day intervals (Fig. 1). NOD mice receiving a second treatment at 14 days after the first with the IgG2a isotype of 18B12 all succumbed to an anaphylaxis response within 24 h. Hence, the IgG1 anti-murine CD20 reagent was used for all subsequent experiments.
FIG. 1.

Peripheral blood B cells in NOD mice are efficiently deleted by the IgG1 isotype of the 18B12 murine CD20-specific antibody. NOD female mice were initially injected intraperitoneally at 8 weeks of age with 10 mg/kg body wt of the IgG1 anti-CD20 isotype, with controls treated with a similar dose of the irrelevant 2B8 antibody (n = 3 per group). Proportions of B cells among peripheral blood leukocytes were then monitored over time. Depletion of peripheral blood B cells was safely maintained by repeated injection of the IgG1 18B12 antibody at 21-day intervals. PBL, peripheral blood.
Anti-CD20 treatment initiated in IAA-positive NOD mice does not significantly block diabetes development.
There is only a short time frame after onset of overt hyperglycemia in which treatment with a Rituximab-like antibody (2H7) reportedly exerts diabetes reversal effects in a subset of NOD mice transgenically expressing human CD20 (18). Thus, we tested whether anti-CD20 treatment more effectively induces late-stage diabetes prevention effects when initiated in NOD mice that have already developed a significant level of pancreatic β-cell autoimmunity marked by the presence of IAAs. Starting at 10 weeks of age, NOD female mice were treated at 21-day intervals with the 18B12 mouse CD20-specific antibody. Other NOD females were treated at the same intervals with the isotype-matched 2B8 control antibody. Serum samples were obtained from all mice prior to the initiation of anti-CD20 or control antibody treatment and tested for IAAs. This allowed us to retrospectively determine if the efficacy of anti-CD20 treatment in blocking diabetes development potentially differed when initiated before or after IAA onset.
As expected, when initiated in pre-IAA onset NOD mice, anti-CD20 treatment significantly inhibited diabetes development (Fig. 2A). Among NOD mice treated with the irrelevant control antibody, the presence of IAAs marked a significantly increased propensity for progression to overt diabetes (100 vs. 64%) (Fig. 2A). Conversely, while anti-CD20 treatment may have a marginal capacity to inhibit diabetes development when initiated in already IAA-positive NOD mice, this effect did not achieve statistical significance (Fig. 2A). We cannot exclude the possibility that the minority subset of NOD mice typed as IAA negative at 10 weeks of age, and subsequently not protected from diabetes development by anti-CD20 treatment, was transiently positive at an earlier time point. However, overall, these results indicate B cells targetable by the 18B12 CD20-specific antibody appear to more prominently contribute to earlier rather than later stages of autoimmune diabetes development in NOD mice.
FIG. 2.

Anti-CD20–mediated B-cell depletion strongly inhibits progression to overt diabetes development only when initiated in NOD mice that have not yet become IAA positive. A: Serum was collected from 10-week-old NOD female mice that then immediately began to receive treatments at 21-day intervals with the IgG1 CD20-specific or control antibody at a 10 mg/kg body wt dose. Serum samples were retrospectively assessed for the presence of IAAs and the mice monitored for diabetes development. Mice used for the analyses had a spread of eight separate birth dates and, thus, were entered into the treatment groups in a staggered fashion. Anti-CD20 treatment significantly suppressed diabetes development in the IAA-negative (P = 0.04) but not IAA-positive recipients (P = 0.14, Kaplan-Meier analyses). B: IAA-negative NOD female mice receiving a single anti-CD20 treatment at 10 weeks of age were not protected from subsequent diabetes development. Aby, antibody.
NOD B cells rebounding after transient anti-CD20–mediated depletion retain diabetogenic activity.
NOD B cells rebounding after transient depletion by anti-CD22 treatment are reportedly characterized by a reprogrammed gene expression profile inhibiting their diabetogenic capacity (17). Thus, we assessed if B cells rebounding in NOD mice receiving only one 18B12 anti-CD20 treatment also had suppressed diabetogenic activity. However, transiently depleting B cells by a single anti-CD20 treatment at 10 weeks of age in originally IAA-negative NOD females did not prevent subsequent diabetes development (Fig. 2B). Hence, B cells rebounding after anti-CD20–mediated transient depletion in NOD mice are not reprogrammed to a diabetes protective state.
Anti-CD20–mediated diabetes protection does not require depletion of MZ B cells.
A strain-specific and age-dependent expansion of the MZ B-cell subset has been hypothesized to be a diabetogenic component in NOD mice (22). MZ B cells are absent from peripheral blood that was monitored in the experiments described above. Thus, it was possible the poor ability of IAA-positive NOD mice to be protected from diabetes development by anti-CD20 treatment resulted from MZ B cells undergoing an age-dependent expansion, becoming refractive to deletion. To initially assess this possibility, we determined whether compared with the nonautoimmune-prone C57BL/6 (B6) strain, NOD mice housed at The Jackson Laboratory were also characterized by an age-associated expansion of splenic MZ B cells. A greater age- associated expansion of splenic MZ B cells was indeed observed in NOD than B6 mice (Fig. 3A). Conversely, mature FO subset B cells exhibited a greater age-associated expansion in B6 than NOD mice (Fig. 3B).
FIG. 3.

Selective depletion of the FO subset of mature B cells limits the initiation but does efficiently abrogate already established diabetogenic autoimmune responses in NOD mice. A: Splenic MZ B cells (CD21hi CD23−) undergo a greater age-dependent expansion in NOD than B6 female mice (n = 5 per strain at each time point; P = 0.02 by ANOVA). B: Splenic FO B cells (CD21int CD23hi) undergo a greater age-dependent expansion in B6 than NOD mice (P = 0.001 by ANOVA). C and D: Treatment with the IgG1 isotype of the 18B12 murine CD20-specific antibody deletes splenic FO but not MZ B cells in NOD mice. Treatment at 21-day intervals with the CD20-specific or control antibody was initiated at 5 weeks of age in a cohort of NOD female mice. At the indicated time points, a subset of mice in each group was then assessed for numbers of splenic MZ (C) and FO (D) B cells. E: Antibody responses to exogenous antigens are marginally suppressed in NOD mice selectively depleted of FO B cells. NOD mice were treated at 10 weeks of age with the control or IgG1 CD20-specific antibody (n = 5 per group) and then primed 9 days later with HEL. Serum was collected 11 days after antigen priming and assessed by enzyme-linked immunosorbent assay for relative levels of HEL-specific antibodies. Data are represented as mean OD405 ± SEM at the indicated serum dilutions (control vs. FOB depleted: P = 0.0001 by ANOVA). MZB, MZ B cells; FOB, FO B cells; Aby, antibody.
We next examined the effect of anti-CD20 treatment on MZ B cells in NOD mice at various ages. Starting at 5 weeks of age, NOD females were treated at 21-day intervals with the 18B12 CD20-specific or irrelevant control antibody. Splenic MZ B-cell numbers were assessed in a subset of mice in each group at various time points. At all ages analyzed, MZ B cells in NOD mice were resistant to anti-CD20–mediated deletion (Fig. 3C). Such deletion resistance was not due to an absence of CD20 expression by NOD MZ B cells (Supplementary Fig. 1). Thus, anatomical sequestration likely explains the resistance of MZ B cells to anti-CD20–mediated deletion. These collective results indicate the ability of the 18B12 CD20-specific antibody to inhibit early stages of diabetes development in NOD mice does not result from elimination of MZ B cells.
Initial but not established pancreatic β-cell autoimmunity is efficiently inhibited by FO B-cell depletion.
Unlike what was observed for the MZ subset, at all ages studied, splenic FO B-cell numbers were significantly lower in anti-CD20–treated than control NOD mice (Fig. 3D). Anti-CD20 treatment also significantly depleted the early developmental T1 and T2 subsets of immature B cells in NOD mice (Supplementary Fig. 2). FO B cells were also significantly deleted in the IAA-positive NOD mice depicted in Fig. 2A that were not protected from diabetes by anti-CD20 treatment (Supplementary Fig. 3). Hence, anti-CD20–mediated FO B-cell depletion strongly suppresses diabetes initiation mechanisms, but their absence does not significantly impair the progression of pancreatic β-cell autoimmunity established in IAA-positive NOD mice. As previously noted, Rituximab treatment also selectively depletes FO but not MZ subset B cells in humans {reviewed in Lund and Randall (20)}. Thus, use of the IgG1 18B12 anti-CD20 reagent in NOD mice robustly mimics the pattern of B-cell subset depletion elicited by Rituximab treatment in humans.
An IgG2c isotype of the MB20–11 CD20-specific antibody that inhibits diabetes development in NOD mice depletes both MZ and FO B cells (19). Thus, use of the IgG1 isotype of the 18B12 CD20-specific antibody that depletes FO, but not MZ, B cells now implicates the former subset as preferentially contributing to the initiation of diabetogenic autoimmunity in NOD mice. Furthermore, albeit at lower levels than in controls, NOD mice selectively depleted of FO B cells still generated a strong antibody response after priming with the exogenous antigen HEL (Fig. 3E). Thus, provided treatment could be initiated early enough, a further potentially important advantage of inhibiting diabetes development by selective depletion of the FO subset rather than total B cells would be induction of a less severe state of generalized immunosuppression.
B cells entering pancreatic islets become CD20 negative.
The ability of anti-CD20 treatment to block early initiating but not late stages of diabetes development in NOD mice raised the question of what effects this agent may have on B cells present in pancreatic islet leukocytic infiltrates at various points of disease pathogenesis. We initially tested whether the proportion of B cells differed within islet-associated leukocytes from 7- or 13-week-old NOD female mice at, respectively, early and late stages of diabetes development. The level of B cells was significantly greater in insulitic infiltrates of 13- than 7-week-old untreated NOD female mice (Fig. 4A). We then assessed what effect anti-CD20 treatment initiated at early or late stages of diabetes development may have on proportions of B cells within insulitic infiltrates. NOD female mice were treated with anti-CD20 at 5 or 11 weeks of age, and levels of B cells in islet-associated leukocytes evaluated 2 weeks later. While both less than in age-matched controls, levels of B cells were significantly lower in islet-associated leukocytes from 7- than 13-week-old NOD female mice treated with anti-CD20 2 weeks earlier (Fig. 4B). It is interesting that islet-associated B cells in untreated NOD mice at both 13 (Fig. 4C) and 7 weeks of age (data not shown) developed a CD20-negative phenotype, in contrast to those from PLNs. The CD20-negative phenotype was associated with islet-derived B cells in untreated NOD mice converting to a CD138-positive plasma cell–like phenotype (Fig. 4D).
FIG. 4.

B cells entering pancreatic islets in NOD mice become CD20-negative plasma cells. A: Proportion of total B cells in pancreatic islet–associated leukocytes from 7- or 13-week-old untreated NOD female mice. B: Proportion of total B cells in pancreatic islet–associated leukocytes from 7- or 13-week-old NOD female mice treated 2 weeks earlier with anti-CD20. Noted statistical significance values in A and B were determined by ANOVA analyses. C: Pancreatic islet–infiltrating B cells in 13-week-old untreated NOD control mice acquire a CD20-negative phenotype (right panel). B cells in PLNs were assessed as a CD20-staining positive control (left panel). Flow cytometric profiles are shown for B220 gated cells. D: CD20-negative pancreatic islet–associated B cells in untreated NOD mice convert to a CD138-positive plasma cell phenotype (right panel). B cells in PLNs were analyzed as a control (left panel). Flow cytometric profiles are shown for B220 gated cells.
These collective results indicate that regardless of the stage of disease development when it is initiated, anti-CD20 treatment of NOD mice eliminates diabetogenic FO subset B cells only prior to their entry into pancreatic islets since subsequent to doing so, they downregulate CD20 expression while converting to a plasma cell–like phenotype. Plasma cells that have also been previously shown by others to convert to a CD20-negative phenotype (38) would by definition be the source of IAAs. Hence, the presence of IAAs would appear to mark the accumulation within pancreatic islets of a highly pathogenic level of diabetogenic B cells that have become refractive to anti-CD20–mediated deletion. However, while not assessed in the current study, anti-CD20 treatment could ultimately diminish IAA levels in NOD mice or humans by eliminating diabetogenic B cells before their conversion to plasma cells.
FO B-cell depletion results in a Treg expansion.
Diabetes resistance elicited in NOD mice treated with a global B-cell–depleting BAFF-blocking reagent reportedly results from an enhanced ability of residual remaining myeloid-type APCs to support a Treg expansion (28). Hence, we assessed whether preferential depletion of FO B cells in NOD mice also supported a Treg expansion and/or altered numbers of BDC2.5-like diabetogenic effector CD4 T cells. These possibilities were assessed in PLNs, given this is reportedly the site for pathogenic expansion of diabetogenic T cells (39). Totally B-cell–deficient NOD.Igµnull mice served as an additional control.
Compared with controls, total CD4 T cells were significantly increased in the PLNs of NOD mice selectively depleted of FO B cells by anti-CD20 treatment (Fig. 5A). There was an even greater significant increase of total CD4 T cells within PLNs of totally B-cell–deficient NOD.Igµnull mice (Fig. 5A). In contrast, numbers of BDC2.5-like diabetogenic CD4 T cells detected by tetramer staining did not differ in PLNs from NOD control or NOD.Igµnull mice (Fig. 5B). Compared with controls, numbers of BDC2.5-like CD4 T cells were somewhat higher in PLNs of anti-CD20–treated NOD mice, but this difference achieved only marginal statistical significance (Fig. 5B). However, the ability to inhibit diabetes development in NOD mice by genetically ablating all B cells or selectively eliminating the FO subset was also associated with significantly increased numbers of phenotypic Tregs (CD4+ CD25+ FoxP3+) within PLNs (Fig. 5C). Splenic Treg numbers were not increased in either anti-CD20–treated NOD mice or the NOD.Igµnull stock (data not shown). Thus, the intra-PLN expansion of Tregs in anti-CD20–treated NOD mice and the NOD.Igµnull stock is most likely an antigen-dependent process.
FIG. 5.
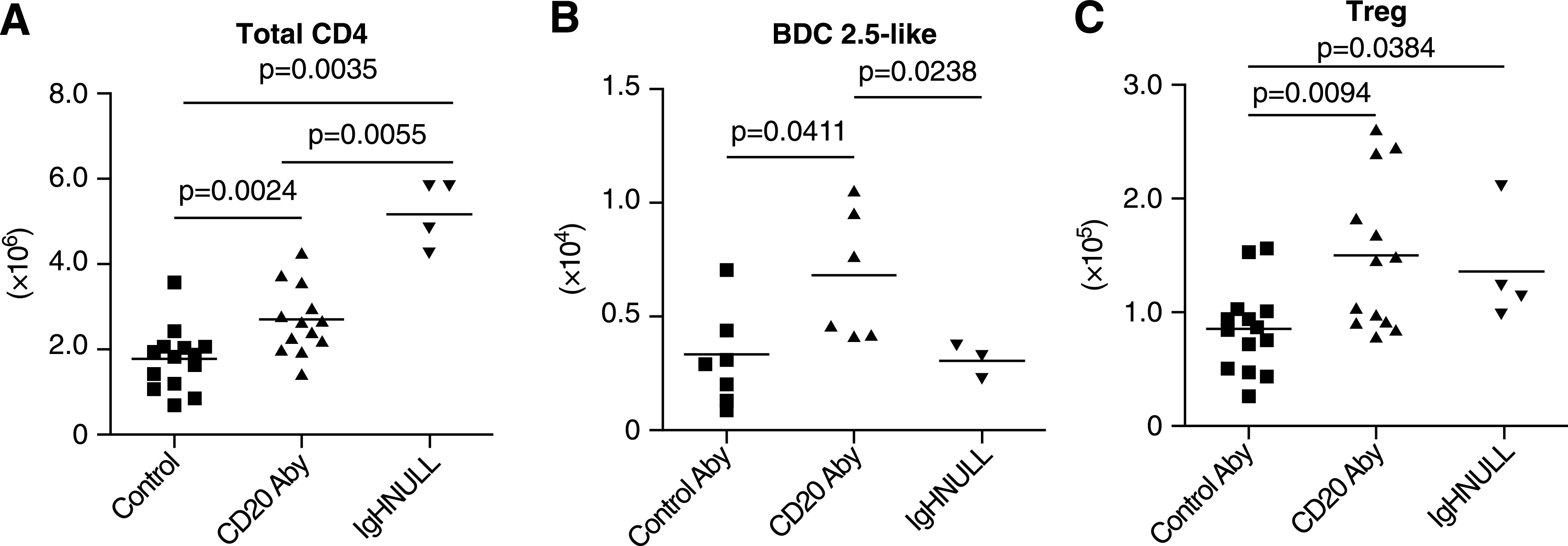
Both Tregs and diabetogenic BDC2.5 clonotypic CD4 T cells expand within PLNs of NOD mice depleted of FO B cells by anti-CD20 treatment. Beginning at 4 weeks of age, NOD females received two injections at a 21-day interval of the CD20-specific or control antibody and at 4–7 days after the second treatment, were assessed for numbers of (A) total CD4 T cells, (B) BDC2.5 clonotypic cells by specific tetramer staining, and (C) Tregs within PLNs. Noted statistical significance values were determined by ANOVA analyses. Aby, antibody.
Diabetes protective effects of eliminating FO B cells prior to IAA onset are dependent on CD25+ cells.
While numerically increased, on a per cell basis, Treg functional activity in anti-CD20–treated NOD mice was equivalent to that in controls (Supplementary Fig. 4). We then tested if the elicited numerical increase in Tregs might contribute to the ability of anti-CD20 treatment to inhibit diabetes development when initiated in pre-IAA onset NOD mice. As expected, compared with controls, diabetes development was significantly reduced in NOD mice in which anti-CD20 treatment was initiated before IAA onset (Fig. 6). Unlike other T-cell types, Tregs constitutively express CD25 (40). It is significant that the ability of anti-CD20 treatment to inhibit diabetes development in pre-IAA onset NOD mice was abrogated by coinfusions with a depleting CD25-specific antibody to a disease rate indistinguishable from controls (Fig. 6). Hence, anti-CD25 coinfusions did not abrogate the diabetes protective effects of anti-CD20 treatment by eliciting alterations in pathogenic effector T-cell activity differing from in NOD control mice. Further supporting this conclusion is the finding that while Tregs were reduced, there was no loss in conventional CD4 and CD8 T cells in coanti-CD20/CD25–treated NOD mice (Supplementary Fig. 5). Instead, these results support a possible Treg-dependent component for the mechanism by which selective depletion of FO subset B cells inhibits diabetes development in pre-IAA onset NOD mice.
FIG. 6.

Coablation of Tregs abrogates the diabetes protective effects of selectively eliminating FO B cells in pre-IAA onset NOD mice. Anti-CD20 or control antibody treatment was initiated in 10-week-old NOD female mice typed to be IAA negative, with a subset also injected at 2-week intervals with a CD25-specific antibody to deplete Tregs. Compared with controls, diabetes development was significantly decreased only in NOD mice treated with anti-CD20 alone (P = 0.0175, Kaplan-Meier analyses). T1D, type 1 diabetes.
DISCUSSION
Our results indicate treatment with the murine CD20-specific 18B12 antibody that like the clinically used Rituximab reagent, selectively depletes the FO but not the MZ subset of B cells, strongly inhibits the early initiating events of autoimmune diabetes development in NOD mice. Preventing the early stages of diabetes development with an anti-CD20 reagent selectively depleting the FO subset, rather than total B cells, could have the important advantage of inducing a less severe state of generalized immunosuppression. However, individuals included in possible diabetes prevention trials are currently selected on the basis of markers of already established high levels of ongoing pancreatic β-cell autoimmunity, such as the presence of autoantibodies. Of potential importance in this regard, our current results indicate that when first initiated in NOD mice that are already IAA positive, the ability of selective FO B-cell depletion by anti-CD20 treatment to block progression to overt diabetes is largely lost. This likely results from B cells entering pancreatic islets of NOD mice rapidly converting to a CD20-negative plasma cell–like phenotype. If this is also true in humans, our results could have potential negative implications for use of the anti-CD20 Rituximab agent as a monotherapeutic clinical approach for either the late-stage prevention of diabetes or to reverse recent-onset disease.
Our current findings differ from a previous report that anti-CD20 treatment could reverse recent-onset diabetes in a subset of NOD mice (18). This previous study used a Rituximab-like antibody to target NOD B cells transgenically expressing human CD20 molecules. Thus, one potential explanation for the possible discrepancy between this previous study and our currently reported results is differential expression regulation of the transgene-encoded human versus endogenous murine CD20 molecules on NOD B cells entering pancreatic islets.
A strain-specific, age-dependent expansion of MZ B cells has been proposed to contribute to diabetes development in NOD mice (22). Our results suggest that while an age-dependent expansion of MZ B cells is an NOD strain characteristic, in the absence of the FO subset, this phenotype does not contribute to diabetes development. However, it remains possible that with increasing age, NOD mice shift from reliance on the FO to the MZ subset of B cells to support diabetogenic T-cell responses. Furthermore, agents such as BAFF inhibitors that eliminate MZ as well as FO B cells might have a stronger capacity than anti-CD20 treatment to prevent progression to overt diabetes after significant levels of pancreatic β-cell autoimmunity have already developed, but this prevention could come at the price of greater generalized immunosuppression. Such possibilities will be assessed in future studies.
It has been reported that in NOD mice, the ability of B cells to mediate the expansion of diabetogenic T cells may normally outpace the capacity of other APC subtypes to support Treg responses that functionally suppress such autoreactive effectors (28). This conclusion was based on a finding that diabetes protection elicited in NOD mice treated with a global B-cell–depleting BAFF-blocking agent was abrogated by anti-CD25–targeting of Tregs. The current study found selective depletion of FO B cells is sufficient to elicit a Treg expansion specifically within the PLNs of NOD mice. Furthermore, anti-CD25–treatment studies support the possibility that at least in part, the capacity of selective FO B-cell depletion to inhibit diabetes development when initiated in pre-IAA onset NOD mice is a Treg-dependent process. By possibly preventing the egress of pathogenic clonotypes into islets, the expansion of Tregs may account for why total CD4 T cells were also increased in the PLNs of FO B-cell–depleted NOD mice. This possibility is supported by a previous finding that pathogenic T cells were also increased in the PLNs of NOD mice protected from diabetes by the activation of NKT cells (41), reportedly to also involve a Treg induction component (42).
The current results also suggest a previously unconsidered, and potentially clinically relevant, means of diabetes intervention, this being a potential combination therapy that both limits the ability of B cells to mediate pathogenic T-cell responses and enhances the capacity of myeloid APC to support Treg activity. Such a combination therapy could entail using Rituximab to block the further entry of diabetogenic B cells into pancreatic islets in conjunction with granulocyte-colony stimulating factor (GCSF; Neupogen), another U.S. Food and Drug Administration–approved reagent that can increase recruitment of dendritic cells with a capacity to enhance Treg activity (43). Further support for this possibility includes the previous finding that while ineffective when used alone, cotreatment with GCSF significantly synergized the ability of antithymocyte globulin to reverse recent-onset diabetes in NOD mice (44). One potential concern regarding this previously described combination therapy is the generalized immunosuppression effects resulting from global antithymocyte globulin–mediated T-cell depletion. Thus, we propose a more benign yet still effective diabetes intervention may be achieved through use of GCSF in combination with Rituximab that selectively depletes FO B cells. However, regardless of their ultimate nature, B-cell targeting approaches continue to represent a potentially attractive means of diabetes intervention. Because of their potential significance as disease intervention targets, it will be important to continue gaining an increased understanding of the pathogenic contributions of various B-cell subsets to differing stages of autoimmune diabetes development. Our current results also indicate it will be important to determine if upon entry into pancreatic islets, diabetogenic B cells change expression levels of molecules that may be targets of agents hoped to attenuate such pathogenic effectors.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants DK-46266 and DK-51090, Cancer Center Support Grant CA-34196, the Brehm Coalition, and by grants from the Juvenile Diabetes Research Foundation International. R.D. and M.R.K. were former employees of Biogen Idec, which supplied the CD20 antibody used in these studies. No other potential conflicts of interest relevant to this article were reported.
D.V.S. directed research and wrote the manuscript. H.D.C. and M.N. researched data. R.D. produced the CD20 antibody and contributed to discussion. M.R.K. and M.H. contributed to discussion. J.P.D. contributed to discussion. C.W. researched data. M.A.A. contributed to discussion and reviewed and edited the manuscript.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db11-0705/-/DC1.
REFERENCES
- 1.Silveira PA, Serreze DV, Grey ST. Invasion of the killer B’s in type 1 diabetes. Front Biosci 2007;12:2183–2193 [DOI] [PubMed] [Google Scholar]
- 2.Falcone M, Lee J, Patstone G, Yeung B, Sarvetnick N. B lymphocytes are crucial antigen-presenting cells in the pathogenic autoimmune response to GAD65 antigen in nonobese diabetic mice. J Immunol 1998;161:1163–1168 [PubMed] [Google Scholar]
- 3.Noorchashm H, Lieu YK, Noorchashm N, et al. I-Ag7-mediated antigen presentation by B lymphocytes is critical in overcoming a checkpoint in T cell tolerance to islet β cells of nonobese diabetic mice. J Immunol 1999;163:743–750 [PubMed] [Google Scholar]
- 4.Serreze DV, Fleming SA, Chapman HD, Richard SD, Leiter EH, Tisch RM. B lymphocytes are critical antigen-presenting cells for the initiation of T cell-mediated autoimmune diabetes in nonobese diabetic mice. J Immunol 1998;161:3912–3918 [PubMed] [Google Scholar]
- 5.Hulbert C, Riseili B, Rojas M, Thomas JW. B cell specificity contributes to the outcome of diabetes in nonobese diabetic mice. J Immunol 2001;167:5535–5538 [DOI] [PubMed] [Google Scholar]
- 6.Silveira PA, Johnson EA, Chapman HD, Bui T, Tisch RM, Serreze DV. The preferential ability of B lymphocytes to act as diabetogenic APC in NOD mice depends on expression of self-antigen-specific immunoglobulin receptors. Eur J Immunol 2002;32:3657–3666 [DOI] [PubMed] [Google Scholar]
- 7.Silveira PA, Dombrowsky J, Johnson E, Chapman HD, Nemazee D, Serreze DV. B cell selection defects underlie the development of diabetogenic APCs in nonobese diabetic mice. J Immunol 2004;172:5086–5094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Silveira PA, Chapman HD, Stolp J, et al. Genes within the Idd5 and Idd9/11 diabetes susceptibility loci affect the pathogenic activity of B cells in nonobese diabetic mice. J Immunol 2006;177:7033–7041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zekavat G, Rostami SY, Badkerhanian A, et al. In vivo BLyS/BAFF neutralization ameliorates islet-directed autoimmunity in nonobese diabetic mice. J Immunol 2008;181:8133–8144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greeley SA, Katsumata M, Yu L, et al. Elimination of maternally transmitted autoantibodies prevents diabetes in nonobese diabetic mice. Nat Med 2002;8:399–402 [DOI] [PubMed] [Google Scholar]
- 11.Inoue Y, Kaifu T, Sugahara-Tobinai A, Nakamura A, Miyazaki J, Takai T. Activating Fc gamma receptors participate in the development of autoimmune diabetes in NOD mice. J Immunol 2007;179:764–774 [DOI] [PubMed] [Google Scholar]
- 12.Kendall PL, Yu G, Woodward EJ, Thomas JW. Tertiary lymphoid structures in the pancreas promote selection of B lymphocytes in autoimmune diabetes. J Immunol 2007;178:5643–5651 [DOI] [PubMed] [Google Scholar]
- 13.Noorchashm H, Noorchashm N, Kern J, Rostami SY, Barker CF, Naji A. B-cells are required for the initiation of insulitis and sialitis in nonobese diabetic mice. Diabetes 1997;46:941–946 [DOI] [PubMed] [Google Scholar]
- 14.Serreze DV, Chapman HD, Varnum DS, et al. B lymphocytes are essential for the initiation of T cell-mediated autoimmune diabetes: analysis of a new “speed congenic” stock of NOD.Ig mu null mice. J Exp Med 1996;184:2049–2053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skyler JS. Prediction and prevention of type 1 diabetes: progress, problems, and prospects. Clin Pharmacol Ther 2007;81:768–771 [DOI] [PubMed] [Google Scholar]
- 16.TrialNet Study Group. Further research to prevent and treat type 1 diabetes. Diabetes Forecast 2004;57:77–79 [PubMed] [Google Scholar]
- 17.Fiorina P, Vergani A, Dada S, et al. Targeting CD22 reprograms B-cells and reverses autoimmune diabetes. Diabetes 2008;57:3013–3024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu CY, Rodriguez-Pinto D, Du W, et al. Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J Clin Invest 2007;117:3857–3867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiu Y, Wong CP, Bouaziz JD, et al. B lymphocyte depletion by CD20 monoclonal antibody prevents diabetes in nonobese diabetic mice despite isotype-specific differences in Fc gamma R effector functions. J Immunol 2008;180:2863–2875 [DOI] [PubMed] [Google Scholar]
- 20.Lund FE, Randall TD. Effector and regulatory B cells: modulators of CD4(+) T cell immunity. Nat Rev Immunol 2010;10:236–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pillai S, Cariappa A, Moran ST. Marginal zone B cells. Annu Rev Immunol 2005;23:161–196 [DOI] [PubMed] [Google Scholar]
- 22.Mariño E, Batten M, Groom J, et al. Marginal-zone B-cells of nonobese diabetic mice expand with diabetes onset, invade the pancreatic lymph nodes, and present autoantigen to diabetogenic T-cells. Diabetes 2008;57:395–404 [DOI] [PubMed] [Google Scholar]
- 23.Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, et al. ; Type 1 Diabetes TrialNet Anti-CD20 Study Group. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med 2009;361:2143–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pociot F, Akolkar B, Concannon P, et al. Genetics of type 1 diabetes: what’s next? Diabetes 2010;59:1561–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pihoker C, Gilliam LK, Hampe CS, Lernmark A. Autoantibodies in diabetes. Diabetes 2005;54(Suppl. 2):S52–S61 [DOI] [PubMed] [Google Scholar]
- 26.Yu L, Eisenbarth GS. Humoral autoimmunity. In Immunology of Type 1 Diabetes. Eisenbarth GS, Ed. Georgetown, TX, Landes Biosciences Publishers, 2004, p. 247–267 [Google Scholar]
- 27.Melanitou E, Devendra D, Liu E, Miao D, Eisenbarth GS. Early and quantal (by litter) expression of insulin autoantibodies in the nonobese diabetic mice predict early diabetes onset. J Immunol 2004;173:6603–6610 [DOI] [PubMed] [Google Scholar]
- 28.Mariño E, Villanueva J, Walters S, Liuwantara D, Mackay F, Grey ST. CD4(+)CD25(+) T-cells control autoimmunity in the absence of B-cells. Diabetes 2009;58:1568–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prochazka M, Gaskins HR, Shultz LD, Leiter EH. The nonobese diabetic scid mouse: model for spontaneous thymomagenesis associated with immunodeficiency. Proc Natl Acad Sci USA 1992;89:3290–3294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Serreze DV, Leiter EH, Hanson MS, et al. Emv30null NOD-scid mice. An improved host for adoptive transfer of autoimmune diabetes and growth of human lymphohematopoietic cells. Diabetes 1995;44:1392–1398 [DOI] [PubMed] [Google Scholar]
- 31.Ahuja A, Shupe J, Dunn R, Kashgarian M, Kehry MR, Shlomchik MJ. Depletion of B cells in murine lupus: efficacy and resistance. J Immunol 2007;179:3351–3361 [DOI] [PubMed] [Google Scholar]
- 32.Hamel K, Doodes P, Cao Y, et al. Suppression of proteoglycan-induced arthritis by anti-CD20 B Cell depletion therapy is mediated by reduction in autoantibodies and CD4+ T cell reactivity. J Immunol 2008;180:4994–5003 [DOI] [PubMed] [Google Scholar]
- 33.Gerling IC, Serreze DV, Christianson SW, Leiter EH. Intrathymic islet cell transplantation reduces beta-cell autoimmunity and prevents diabetes in NOD/Lt mice. Diabetes 1992;41:1672–1676 [DOI] [PubMed] [Google Scholar]
- 34.Serreze DV, Leiter EH. Defective activation of T suppressor cell function in nonobese diabetic mice. Potential relation to cytokine deficiencies. J Immunol 1988;140:3801–3807 [PubMed] [Google Scholar]
- 35.Chen YG, Scheuplein F, Osborne MA, Tsaih SW, Chapman HD, Serreze DV. Idd9/11 genetic locus regulates diabetogenic activity of CD4 T-cells in nonobese diabetic (NOD) mice. Diabetes 2008;57:3273–3280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stratmann T, Martin-Orozco N, Mallet-Designe V, et al. Susceptible MHC alleles, not background genes, select an autoimmune T cell reactivity. J Clin Invest 2003;112:902–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bonifacio E, Atkinson MA, Eisenbarth GS, et al. International Workshop on Lessons From Animal Models for Human Type 1 Diabetes: identification of insulin but not glutamic acid decarboxylase or IA-2 as specific autoantigens of humoral autoimmunity in nonobese diabetic mice. Diabetes 2001;50:2451–2458 [DOI] [PubMed] [Google Scholar]
- 38.DiLillo DJ, Hamaguchi Y, Ueda Y, et al. Maintenance of long-lived plasma cells and serological memory despite mature and memory B cell depletion during CD20 immunotherapy in mice. J Immunol 2008;180:361–371 [DOI] [PubMed] [Google Scholar]
- 39.Melli K, Friedman RS, Martin AE, et al. Amplification of autoimmune response through induction of dendritic cell maturation in inflamed tissues. J Immunol 2009;182:2590–2600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sakaguchi S, Powrie F. Emerging challenges in regulatory T cell function and biology. Science 2007;317:627–629 [DOI] [PubMed] [Google Scholar]
- 41.Chen Y-G, Choisy-Rossi C-M, Holl TM, et al. Activated NKT cells inhibit autoimmune diabetes through tolerogenic recruitment of dendritic cells to pancreatic lymph nodes. J Immunol 2005;174:1196–1204 [DOI] [PubMed] [Google Scholar]
- 42.Ly D, Mi Q-S, Hussain S, Delovitch TL. Protection from type 1 diabetes by invariant NK T cells requires the activity of CD4+CD25+ regulatory T cells. J Immunol 2006;177:3695–3704 [DOI] [PubMed] [Google Scholar]
- 43.Kared H, Masson A, Adle-Biassette H, Bach JF, Chatenoud L, Zavala F. Treatment with granulocyte colony-stimulating factor prevents diabetes in NOD mice by recruiting plasmacytoid dendritic cells and functional CD4(+)CD25(+) regulatory T-cells. Diabetes 2005;54:78–84 [DOI] [PubMed] [Google Scholar]
- 44.Parker MJ, Xue S, Alexander JJ, et al. Immune depletion with cellular mobilization imparts immunoregulation and reverses autoimmune diabetes in nonobese diabetic mice. Diabetes 2009;58:2277–2284 [DOI] [PMC free article] [PubMed] [Google Scholar]