

Abstract
OBJECTIVE
Class IIa histone deacetylases (HDACs) belong to a large family of enzymes involved in protein deacetylation and play a role in regulating gene expression and cell differentiation. Previously, we showed that HDAC inhibitors modify the timing and determination of pancreatic cell fate. The aim of this study was to determine the role of class IIa HDACs in pancreas development.
RESEARCH DESIGN AND METHODS
We took a genetic approach and analyzed the pancreatic phenotype of mice lacking HDAC4, -5, and -9. We also developed a novel method of lentiviral infection of pancreatic explants and performed gain-of-function experiments.
RESULTS
We show that class IIa HDAC4, -5, and -9 have an unexpected restricted expression in the endocrine β- and δ-cells of the pancreas. Analyses of the pancreas of class IIa HDAC mutant mice revealed an increased pool of insulin-producing β-cells in Hdac5−/− and Hdac9−/− mice and an increased pool of somatostatin-producing δ-cells in Hdac4−/− and Hdac5−/− mice. Conversely, HDAC4 and HDAC5 overexpression showed a decreased pool of insulin-producing β-cells and somatostatin-producing δ-cells. Finally, treatment of pancreatic explants with the selective class IIa HDAC inhibitor MC1568 enhances expression of Pax4, a key factor required for proper β-and δ-cell differentiation and amplifies endocrine β- and δ-cells.
CONCLUSIONS
We conclude that HDAC4, -5, and -9 are key regulators to control the pancreatic β/δ-cell lineage. These results highlight the epigenetic mechanisms underlying the regulation of endocrine cell development and suggest new strategies for β-cell differentiation-based therapies.
The mature mammalian pancreas is a gland composed mainly of acinar cells, which belong to the exocrine pancreas and secrete digestive enzymes into the intestine, and the islets of Langerhans, which produce hormones that underlie the endocrine functions of the pancreas. Endocrine islets consist of four different cell types that produce hormones (β-cells [representing 80% of the islet cells in rodents], α-cells, δ-cells, and pancreatic polypeptide cells) and regulate blood glucose homeostasis.
During embryogenesis, the pancreas originates from the dorsal and ventral regions of the foregut endoderm, which expresses the homeodomain transcription factor pancreatic and duodenal homeobox 1 (PDX1) (1). The endocrine differentiation program is initiated with expression of the transcription factor neurogenin 3 (NGN3) (2). Subsequent expression of additional transcription factors, such as NEUROD1, IA1, NKX6.1, NKX2.2, MAFA, ARX, and PAX4, determine the specific endocrine cell fate (3). PAX4 and ARX exert opposing effects on endocrine cell differentiation. PAX4 is required for β/δ-cell differentiation, and ARX is involved in α-cell differentiation (4,5). In mice, a major peak of mature insulin- and glucagon-expressing cells occurs around embryonic day 14 (E14), and somatostatin-expressing cells differentiate around E15 and pancreatic polypeptide cells around E18. Then, endocrine cells aggregate and form well-organized islets of Langerhans (6). Defining how pancreatic development is regulated and the signals that enhance β-cells remains a challenge.
Gene transcription is modulated by acetylation and deacetylation of histones. Acetylation of lysine residues of histones by histone acetyltransferases serves to neutralize the positive charge of histones, relaxing chromatin structure and promoting transcription. Deacetylation by histone deacetylases (HDACs) enables chromatin to compact causing transcription repression. Of note, recent evidence indicates that HDACs also regulate several biological processes by targeting nonhistone proteins (7). HDACs belong to three classes on the basis of their phylogenetic conservation: class I includes HDAC1–3 and -8; class II includes HDAC4–7, -9, and -10; and class IV includes HDAC11. Class II HDACs are further divided into the subclass IIa (HDAC4, -5, -7, and -9) and subclass IIb (HDAC6 and -10) (8). In contrast to other HDACs, class IIa HDACs show restricted expression patterns. For example, HDAC5 and -9 are enriched in skeletal muscle, heart, and brain (9,10). Class IIa HDACs mediate multiple biological processes by interactions with various transcription factors, including myocyte enhancer factor (MEF)-2 (7,11). Moreover, class IIa HDACs undergo nucleo-cytoplasmic shuttle in response to extracellular signals such as phosphorylation by calcium/calmodulin-dependent protein kinase (7). The phenotype of HDAC mutant mice revealed that HDACs play specific roles during embryogenesis and postnatal life (12).
HDAC inhibitors are powerful tools to modulate cell differentiation programs in a broad range of cell types (13). We previously compared the effects of different HDAC inhibitors that either preferentially inhibit class I HDACs or inhibit both class I and class II HDACs. We showed that treatment with different HDAC inhibitors modified the timing and determination of pancreatic cell fate and proposed distinct roles for class I and class II HDACs in pancreatic differentiation, with a specific role of class II HDACs in endocrine cell subtype development (14).
In the current study, we investigated the role of class IIa HDACs in pancreatic cell differentiation. We found that class IIa HDACs display a restricted expression pattern in the pancreas, with HDAC4, -5, and -9 specifically expressed in β- and/or δ-cells. Analysis of mutant mice revealed increased δ-cell mass in the pancreas of Hdac4−/− and Hdac5−/− mice and increased β-cell mass in the pancreas of Hdac5−/− and Hdac9−/− mice. Overexpression of HDAC4 or HDAC5 in the pancreas caused a decrease in β- and δ-cells, further supporting the role of class IIa HDACs in the control of the β/δ-cell lineage. We also found that MEF2A and MEF2D, which form complexes with class IIa HDACs (11), are expressed in the fetal pancreas. Treatment of pancreatic explants with a selective class IIa HDAC inhibitor, MC1568, which modulates the stability of class IIa HDAC-MEF2 complexes (15), amplified the pool of β- and δ-cells. Our data demonstrate the major role of HDAC4, -5, and -9 in the control of the β/δ-cell development. These results provide new insight into the mechanisms that control pancreatic endocrine cell development and the number of β-cells.
RESEARCH DESIGN AND METHODS
Pregnant Wistar rats were purchased from CERJ (Le Genest, France). Hdac4, Hdac5, and Hdac9 mutant mice were described previously (9,10,16). The first day postcoitum was designated embryonic day 0.5 (E0.5). Pregnant rats and mice were killed by CO2 asphyxiation, and postnatal day 1 (P1) and P7 pups were killed by CO2 asphyxiation followed by decapitation, in compliance with the guidelines of the French Animal Care Committee. Dorsal pancreatic buds from E13.5 rat embryos were dissected, as described previously (17).
Organ culture and treatment with MC1568.
E13.5 rat dorsal pancreatic buds were laid on 0.45-μm filters (Millipore, Billerica, MA) at the air-medium interface in Petri dishes containing culture medium as previously described (14). MC1568 (from A.M.) was used at 10 μmol/L.
Lentiviral vector construction and production.
The backbone of the lentiviral construct, pTRIP ΔU3, was previously described (18). New lentiviral vectors were generated through Gateway (Invitrogen, Carlsbad, CA) in vitro recombination using HDAC4 and HDAC5 entry clones (pENTR) and the pTRIP ΔU3 cytomegalovirus (CMV)-Gateway destination vector (19). The 3,660-bp fragment containing the HDAC4 coding sequence was obtained by HindIII and EcoRI restriction from the commercial plasmid pYX-Asc-HDAC4 (Invitrogen) and subcloned into pENTR polylinker to generate the HDAC4 entry clone. The HDAC5-myc fusion was amplified by PCR from the commercial vector using the following primers: HDAC5 sense 5′ caccatgaactctcccaacgagtc 3′ and HDAC5-myc stop antisense 5′ ttacagatcctcttctgagatgagtttc 3′. The resulting 3,342-pb PCR fragment was cloned into the pENTR/D Topo vector (Invitrogen) to generate an HDAC5-myc entry clone. Both cDNAs were cloned into the pTRIP ΔU3 CMV-Gateway destination vector by LR Clonase II recombination according to the manufacturer’s recommendations (Invitrogen) to generate pTRIP ΔU3 CMV-HDAC4 and pTRIP ΔU3 CMV-HDAC5 lentiviral vectors. pTRIP ΔU3 CMV-green fluorescent protein (GFP) vector was described previously (20). Lentiviral vector stocks were produced as previously described (21). The amount of p24 capsid protein was quantified by the HIV-1 p24 ELISA antigen assay (Beckman Coulter, Villepinte, France). All infections were normalized relative to p24 capsid protein quantification.
Lentiviral infection of E13.5 rat pancreata.
E13.5 pancreata were incubated for 15 min at 37°C with collagenase IV (250 units/mL; Worthington, Lakewood, NJ) and mechanically dispersed through 23-G and 25-G needles to obtain a single cell suspension. Lentivirus (1 μL) was preincubated in 10 μL culture medium supplemented with diethylaminoethyl-dextran (20 μg/mL) for 15 min at 37°C and added to 100,000 dispersed pancreatic cells in 10 μL culture medium. Cells were cultured overnight in hanging drops to form pancreatic spheres that were next laid on 0.45-μm filters (Millipore) and grown for 6 days on a filter at the air/medium interface in Petri dishes containing culture medium.
Immunohistochemistry.
Pancreata were immersed in 10% formalin and embedded in paraffin. Sections (4 μm thick) were processed for immunohistochemistry using a previously described protocol (17,22). Primary antibodies were used at the following dilutions: mouse anti-insulin (1:2,000; Sigma-Aldrich, St. Louis, MO), mouse antiglucagon (1:2,000; Sigma-Aldrich), rabbit antiglucagon (1:1,000; Euromedex, Souffelweyerrsheim, France), mouse antisomatostatin (1:500; SOMO18 β-Cell Biology Consortium), rabbit antisomatostatin (1:500; Dako, Glostrup, Denmark), rabbit anti-amylase (1:300; Sigma-Aldrich), rabbit anti-PDX1 (1:1,000 [22]), rabbit anti-HDAC4 (1:250; Abcam, Cambridge, U.K.), rabbit anti-HDAC5 (1:100; Cell Signaling, Beverly, MA), rabbit anti-HDAC9 (1:100; Abgent), mouse anti-Ki67 (1:20; BD Pharmingen, San Diego, CA), and mouse anti-smooth muscle actin (SMA) (1:1,000; Sigma-Aldrich). MEF2A immunohistochemistry was performed using Tyramide Signal Amplification (TSA) according to the manufacturer’s protocol (Cyanine5 TSA kit; Perkin Elmer, Waltham, MA), with rabbit anti-MEF2A (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA). Secondary antibodies were used at the following dilutions: anti-rabbit/mouse fluorescein (1:200; Jackson Immunoresearch, West Grove, PA), anti-rabbit/mouse Texas red (1:200; Jackson Immunoresearch), anti-rabbit/mouse biotinylated conjugated (1:200; Vector Laboratories, Burlingame,CA), and anti-rabbit/mouse horseradish peroxidase conjugated (1:200; Vector Laboratories). For fluorescent immunohistochemistry, the nuclei were stained using the Hoechst 33342 fluorescent stain (0.3 μg/mL; Invitrogen). For colorimetric immunohistochemistry, revelations were performed using 3-3′ diaminobenzidine-tetrahydrochloride substrate.
Quantification and morphometric analysis.
Pancreata at E18.5, P1, and P7 were embedded in paraffin and entirely sectioned (4 μm thick). For each staining, eight sections (separated by at least 150 μm) were selected at regular intervals to cover the whole pancreas. Sections were processed for insulin, somatostatin, or glucagon immunohistochemistry with a 3-3′ diaminobenzidine-tetrahydrochloride substrate and lightly counterstained with hematoxylin. The surfaces of insulin, somatostatin, and glucagon stainings as well as the total surface of the pancreatic tissue were measured on each section. These surfaces were determined by computer-assisted measurements using a DMRB microscope and a color video camera coupled to Q500IW software (Leica, Weitzlar, Germany), as previously described (23). The percentage of α-, β-, or δ-cell fraction was calculated as the ratio of the sum of the glucagon-, insulin-, or somatostatin-positive cell areas to the sum of the total pancreatic tissue area, respectively. For cultured fetal pancreata, surface quantification was performed as previously described (14). The results are expressed as means ± SEM. Statistical significance was determined using the Student t test.
RNA extraction and real-time PCR.
Total RNA extraction and real-time PCR was performed as previously described (14). The oligonucleotide sequences are available upon request. Cyclophilin A was used as an internal reference control. Data are presented as the fold-change in gene expression. The results are expressed as means ± SEM, and statistical significance was determined using a Student t test.
RESULTS
Selective expression of HDAC4, -5, and -9 in endocrine β- and δ-cells.
During mouse pancreas development at E15.5 and in the adult pancreas, immunohistochemistry showed that nuclear HDAC1 (Supplementary Fig. 1) and HDAC2 (data not shown) were detected in all pancreatic cell types, consistent with reports of ubiquitous expression of class I HDACs in many tissues (24). By contrast, class IIa HDAC expression was cell type specific. At E15.5, Hdac4, Hdac5, and Hdac9 were expressed in the developing pancreas, as determined by quantitative PCR (qPCR) (Figs. 1A, 2A, and 3A). In adult pancreas, Hdac4, Hdac5, and Hdac9 expression was restricted to endocrine islets and was not detected in exocrine tissue (Figs. 1A, 2A, and 3A). Purity of endocrine versus exocrine fractions was validated by insulin and amylase mRNA expression, respectively (Supplementary Fig. 2). To determine which endocrine cell type expresses class IIa HDACs, we performed immunohistochemistry and showed that HDAC4 was detected at E15.5 and E18.5 in insulin-positive cells (Fig. 1B and data not shown). At P7 and in the adult pancreas, we observed two different expression levels of HDAC4. Low expression of HDAC4 was seen in cells stained positive for insulin (Fig. 1B and data not shown), whereas greater expression of HDAC4 was observed in cells expressing somatostatin (Fig. 1C). HDAC5 was specifically detected in β-cells at E15.5 and E18.5 (Fig. 2B and data not shown). At P7 and in the adult pancreas, HDAC5 was detected in both insulin-expressing cells and in somatostatin-expressing cells (Fig. 2C and D and data not shown). As was seen with HDAC5 expression, HDAC9 was selectively detected in insulin-positive cells at E15.5, E18.5, and P7 and in the adult pancreas (Fig. 3B and data not shown). In contrast, HDAC9 was not detected in somatostatin-expressing cells (Fig. 3C). Strikingly, we found no expression of HDAC4, -5, and -9 in glucagon-expressing cells or in the acinar pancreatic tissue (Figs. 1D, 2B and C, and 3D). Thus, HDAC4 is highly enriched in δ-cells and at a low level in β-cells, HDAC5 expression is highly enriched in β- and δ-cells, and HDAC9 is highly enriched in β-cells. Finally, immunohistochemical analysis showed that the fourth member of the class IIa HDACs (HDAC7) was expressed in vascular endothelial cells and absent from endocrine cells (Supplementary Fig. 3). Altogether, these results reveal specific expression of three class IIa HDACs (HDAC4, -5, and -9) in pancreatic endocrine β- and δ-cells.
FIG. 1.

HDAC4 is highly enriched in endocrine cells at a high level in δ-cells and at a low level in β-cells. A: qPCR analysis of Hdac4 mRNA expression in embryonic pancreas, adult islets, and adult exocrine tissue. B–D: Immunohistological analysis of HDAC4 (green) in E15.5 and adult pancreata. B: β-Cells were detected with insulin (INS) staining (red), and cells coexpressing HDAC4 and insulin (orange) are shown in the merge. C: δ-Cells were detected with somatostatin (SST) staining (red), and cells coexpressing HDAC4 and somatostatin (yellow) are shown in the merge. D: α-Cells were detected with glucagon (GLU) staining (red). No cells coexpressing HDAC4 and glucagon were observed in the merge. Nuclei were stained with Hoechst stain (blue). Scale bar, 50 μm. Endocrine islets are circled. A higher magnification of selected cells is shown in the insets. (A high-quality digital representation of this figure is available in the online issue.)
FIG. 2.
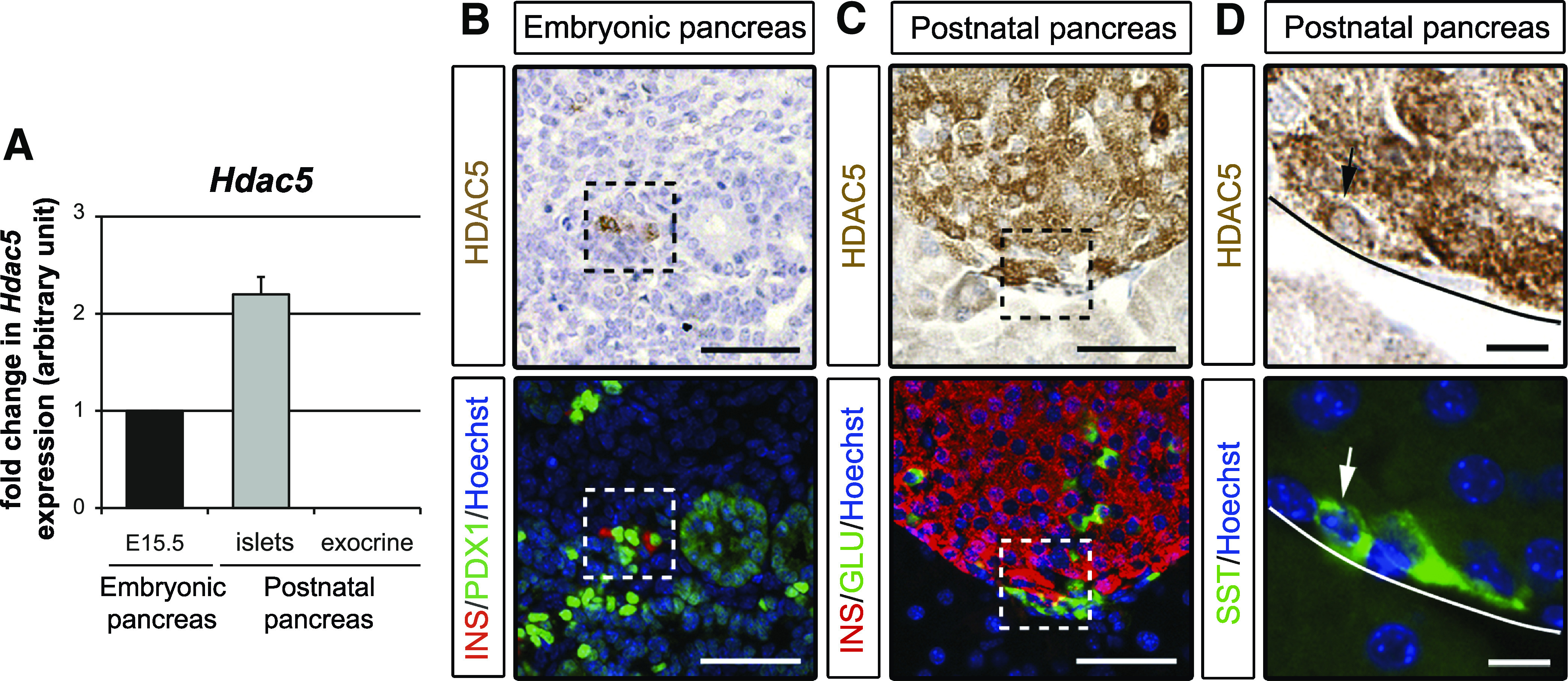
HDAC5 is highly enriched in β- and δ-cells. A: qPCR analysis of Hdac5 mRNA expression in embryonic pancreas, adult islets, and adult exocrine tissue. B–D: Immunohistological analysis of HDAC5 (brown) in E15.5 and adult pancreas. B: β-Cells were detected with insulin (INS) staining (red) and PDX1 staining (green) was used to detect pancreatic epithelium (PDX1low) and β-cells (PDX1high). Some β-cell PDX1high+/insulin+ expressing HDAC5 are framed. C: β- and α-cells were detected using insulin (red) and glucagon (GLU) (green) staining, respectively. Some β-cell insulin+/HDAC5+ and some α-cell glucagon+/HDAC5– are framed. D: δ-Cells were detected with somatostatin (SST) staining (green), and the arrow shows a δ-cell somatostatin+/HDAC5+. Nuclei were stained with Hoechst stain (blue). Scale bar, 50 μm (B and C) and 10 μm (D). (A high-quality digital representation of this figure is available in the online issue.)
FIG. 3.

HDAC9 is highly enriched in β-cells. A: qPCR analysis of Hdac9 mRNA expression in embryonic pancreas, adult islets, and adult exocrine tissue. B–D: Immunohistological analysis of HDAC9 (green) in E15.5 and P7 pancreata. B: β-Cells were detected with insulin (INS) staining (red), and cells coexpressing HDAC9 and insulin (yellow) are shown in the merge. C: δ-Cells were detected with somatostatin (SST) staining (red). No cells coexpressing HDAC9 (green) and somatostatin (red) are observed in the merge. D: α-Cells were detected with glucagon (GLU) staining (red). No cells coexpressing HDAC9 (green) and glucagon (red) are observed in the merge. Nuclei were stained with Hoechst stain (blue). Scale bar, 50 μm. Endocrine islets are circled. A higher magnification of selected cells is shown in the insets. (A high-quality digital representation of this figure is available in the online issue.)
HDAC4 inhibits β- and δ-cell development.
HDAC4 is specifically expressed in chondrocytes, skeletal muscle, heart, brain, and retina (25–27) and plays a central role in the formation of the skeleton (16). As Hdac4−/− mice die by P7 because of bone abnormalities (16), we assessed the pancreas of Hdac4−/− mice at P1. No difference was detected in pancreatic weight between Hdac4−/− and wild-type mice (data not shown). Quantification of insulin staining indicated that β-cell mass of Hdac4−/− and wild-type mice was similar (Fig. 4A and B), as was the case for the α-cell mass quantified by glucagon staining (data not shown). Quantification of somatostatin staining indicated that δ-cell mass was 1.46 ± 0.09 higher in Hdac4−/− mice than in wild-type mice (Fig. 4A and B).
FIG. 4.

HDAC4 loss-of-function enhances δ-cell mass, whereas HDAC4 gain-of-function represses β- and δ-cell mass. A: Immunohistological analyses of wild-type and Hdac4−/− pancreas at P1. β-Cells and δ-cells were detected with insulin (INS) (brown, left panels) and somatostatin (SST) (brown, right panels) stainings. B: Morphometric analysis of β- and δ-cell surfaces by quantification of areas occupied by insulin- and somatostatin-positive cells. β-Cell and δ-cell surfaces were normalized to wild-type (WT) values (100%). Data are shown as means ± SEM. Four pancreata were analyzed for each genotype. C: qPCR analysis of Hdac4,insulin, somatostatin, glucagon, and amylase mRNA expression in pancreatic spheres transduced with CMV-GFP or CMV-HDAC4 lentivirus, followed by a 7-day culture period. D: qPCR analysis of NeuroD1, Pdx1, MafA, Nkx2.2, Znt8, and Ia1 mRNA expression in pancreatic spheres transduced with CMV-GFP or CMV-HDAC4 lentivirus, followed by a 7-day culture period. E: Immunohistological analyses of pancreatic spheres transduced with a lentivirus expressing enhanced GFP or HDAC4 followed by a 7-day culture period. β-Cells and δ-cells were detected with insulin (red) and antibody somatostatin (green) stainings. Nuclei were stained with Hoechst stain (blue). The absolute areas that were occupied by the insulin- and somatostatin-positive cells were quantified. β- and δ-Cell areas are presented as a percentage of the total tissue area. qPCR data and immunohistological analyses are the means ± SEM of five and six independent experiments, respectively. *P < 0.05; **P < 0.005; ***P < 0.001. Scale bar, 100 μm (A) and 50 μm (E). (A high-quality digital representation of this figure is available in the online issue.)
To further investigate the role of HDAC4 in β- and δ-cell development, we overexpressed HDAC4 in E13.5 pancreas by using a novel lentivirus-mediated gene transfer method we developed (Supplementary Fig. 4). Cells were dissociated from E13.5 rat pancreas, transduced by lentivirus-mediated gene transfer, cultured overnight in hanging drops to form pancreatic spheres, and then transferred on a filter at the air/medium interface and cultured for 6 days. Acinar and endocrine cells developed as previously shown for undissociated pancreatic explants (28). Infection of pancreatic spheres with recombinant lentivirus was efficient with >70% of GFP-positive cells after infection with a CMV-GFP lentiviral vector (Supplementary Fig. 4). In spheres infected with a lentiviral vector expressing HDAC4 (CMV-Hdac4), we observed by qPCR a fourfold increase in Hdac4 expression (Fig. 4C). This result was associated with a 31.4 ± 8.3% decrease in insulin expression and a 54.7 ± 5.5% decrease in somatostatin expression (Fig. 4C). Interestingly, the expression of NeuroD1, Pdx1, MafA, Nkx2.2, Znt8, and Ia1, which are required for proper β-cell differentiation or function, was significantly lower in spheres overexpressing HDAC4 (Fig. 4D). In contrast, we did not observe any change in glucagon or amylase expression, markers of differentiated endocrine α-cells and acinar cells, respectively (Fig. 4C). Finally, immunohistochemical analysis confirmed a decrease in both β-cells (a 44.5 ± 10.1% decrease) and δ-cells (a 57.9 ± 5.2% decrease) in spheres overexpressing HDAC4 (Fig. 4E). Taken together, these results demonstrate that HDAC4 is involved in the control of β- and δ-cell development.
HDAC5 inhibits β- and δ-cell development.
HDAC5 is expressed in skeletal muscle, heart, and brain (11,29), and mice lacking Hdac5 are hypersensitive to cardiac stress (10). Because we found HDAC5 expression in β- and δ-cells, we examined the pancreas of Hdac5−/− mice during embryogenesis (E18.5) and after birth (P7) to determine if HDAC5 plays a role in the differentiation of these endocrine cell types. Body weight and pancreatic weight did not differ between Hdac5−/− and wild-type mice (data not shown). Quantification of insulin staining indicated that, at E18.5 and P7, β-cell mass was 2.91 ± 0.148 and 1.56 ± 0.092 higher in Hdac5−/− mice than in wild-type mice (Fig. 5A and B). At E18.5, β-cell proliferation measured by Ki67 immunostaining did not differ between Hdac5−/− and wild-type pancreata (Supplementary Fig. 5C). Quantification of somatostatin staining at P7 revealed a 1.32 ± 0.85-fold increase in δ-cell mass in the pancreas of Hdac5−/− mice (Fig. 5A and B). No change in α-cell mass was observed (Supplementary Fig. 5A and B). Thus, mice lacking HDAC5 in the pancreas have enhanced β- and δ-cell mass.
FIG. 5.

HDAC5 loss-of-function enhances β- and δ-cell mass. A: Immunohistological analyses of wild-type and Hdac5−/− pancreas at E18.5 and P7. On the left panel, β-cells were detected with insulin (INS) staining (brown) at E18.5. On the middle panel, β-cells were detected with insulin staining (brown) at P7. On the right panel, δ-cells were detected with somatostatin (SST) staining (brown) at P7. B: Morphometric analysis of β- and δ-cell surfaces by quantification of areas occupied by insulin- and somatostatin-positive cells. β-Cell surface is shown at E18.5 on the left panel and at P7 on the middle panel. δ-Cell surface is shown at P7 on the right panel. β-Cell and δ-cell surfaces were normalized to wild-type (WT) values (100%). Data are shown as means ± SEM from at least three pancreata per genotype. At E18.5, we analyzed three WT and five Hdac5−/− pancreata. At P7, we analyzed four pancreata of each genotype. *P < 0.05; **P < 0.005; ***P < 0.001. Scale bar, 100 μm. (A high-quality digital representation of this figure is available in the online issue.)
To further investigate the role of HDAC5 in β- and δ-cell development, we used lentivirus-mediated gene transfer to overexpress HDAC5 in E13.5 pancreas. In spheres infected with a lentiviral vector expressing HDAC5 (CMV-Hdac5), we observed by qPCR a 10-fold increase in Hdac5 expression (Fig. 6A). This result was associated with a 21.2 ± 1.9% decrease in insulin expression and a 42.5 ± 12.9% decrease in somatostatin expression (Fig. 6A). Interestingly, the expression of NeuroD1, Pdx1, Nkx2.2, MafA, Znt8, and Ia1 was significantly lower in spheres overexpressing HDAC5 (Fig. 6B). In contrast, we did not observe any change in glucagon or amylase expression (Fig. 6A). Finally, immunohistochemical analysis confirmed a decrease in both β-cells (a 28.9 ± 11% decrease) and δ-cells (a 61.3 ± 6.9% decrease) in spheres overexpressing HDAC5 (Fig. 6C), whereas no change in α-cells was observed (data not shown). Taken together, these results demonstrate that HDAC5 is involved in the control of β- and δ-cell development.
FIG. 6.

HDAC5 gain-of-function represses β- and δ-cell mass. A: qPCR analysis of Hdac5,insulin, somatostatin, glucagon, and amylase mRNAs expression in pancreatic spheres transduced with CMV-GFP or CMV-HDAC4 lentivirus, followed by a 7-day culture period. B: qPCR analysis of NeuroD1, Pdx1, MafA, Nkx2.2, Znt8, and Ia1 mRNA expression in pancreatic spheres transduced with CMV-GFP or CMV-HDAC5 lentivirus, followed by a 7-day culture period. C: Immunohistological analyses of pancreatic spheres transduced with a lentivirus expressing eGFP or HDAC5 followed by a 7-day culture period. β-Cells and δ-cells were detected with insulin (INS) (red) and antibody somatostatin (SST) (green) stainings. Nuclei were stained with Hoechst stain (blue). The absolute areas that were occupied by the insulin- and somatostatin-positive cells were quantified. β-Cell and δ-cell areas are presented as a percentage of the total tissue area. qPCR data and immunohistological analyses are the means ± SEM of three and four independent experiments, respectively. *P < 0.05; **P < 0.005; ***P < 0.001. Scale bar, 50 μm. (A high-quality digital representation of this figure is available in the online issue.)
HDAC9 inhibits β-cell development.
HDAC9 is expressed in skeletal muscle, heart, brain, lymphocytes, and erythrocytes (25,29–31), and mice lacking Hdac9 are hypersensitive to cardiac stress (9). There was no difference detected in the body weight or pancreatic weight between Hdac9−/− and wild-type mice (data not shown). We examined insulin-positive cells in Hdac9−/− mice, since we detected HDAC9 only in β-cells. We observed that at E18.5 and P7, β-cell mass was 1.38 ± 0.08 higher in Hdac9−/− mice than in wild-type mice (Fig. 7A and B). At E18.5, β-cell proliferation measured by Ki67 immunostaining did not differ between Hdac9−/− and wild-type pancreata (Supplementary Fig. 6C). No change in α-cell mass was observed (Supplementary Fig. 6A and B). Thus, deletion of HDAC9 in the pancreas enhances β-cell mass.
FIG. 7.

HDAC9 loss-of-function enhances β-cell mass. A: Immunohistological analyses of wild-type and Hdac9−/− pancreas at E18.5 and P7. β-Cells were detected with insulin (INS) staining (brown). B: Morphometric analysis of the β-cell surface by quantification of areas occupied by insulin-positive cells. β-Cell surfaces were normalized to wild-type (WT) values (100%). Data are shown as means ± SEM. At E18.5, we analyzed five WT and four Hdac9−/− pancreata. At P7, we analyzed four WT and three Hdac9−/− pancreata. *P < 0.05; ***P < 0.001. Scale bar, 100 μm. (A high-quality digital representation of this figure is available in the online issue.)
MC1568, a class IIa HDAC inhibitor, enhances β- and δ-cell development.
To further investigate how the class IIa HDACs control β- and δ-cell development, we assessed expression of MEF2 transcription factors, which associate with class IIa HDACs in cardiac muscle development, skeletal muscle differentiation, and T-cell apoptosis (7). We analyzed Mef2A, Mef2C, and Mef2D expression in the embryonic pancreas. Whereas Mef2C mRNA was detected at a low level (data not shown), Mef2A and Mef2D mRNAs, which are expressed at high levels in embryonic heart and skeletal muscle (32), were detected in E15.5 and E18.5 pancreata at levels similar to those observed in E18.5 heart and skeletal muscle (Fig. 8A and Supplementary Fig. 7A). Immunohistochemistry showed the expression of MEF2A in the pancreas at E18.5. MEF2A was expressed in endocrine cells and smooth muscle cells and not in acinar cells (Fig. 8B and Supplementary Fig. 7B–E).
FIG. 8.

The MEF2 transcription factors are expressed in the pancreas and the MC1568 inhibitor increases β- and δ-cell mass. A: qPCR analysis of Mef2A expression in E15.5 and E18.5 mouse pancreas, and E18.5 heart and muscle. B: Immunohistological analysis of MEF2A (red) in E18.5 mouse pancreas. β-Cells were detected with insulin (INS) staining (green). The arrow shows one cell coexpressing MEFA2 and insulin. C: qPCR analysis of Pax4 mRNA expression between 3 and 14 days in culture (D3 to D14), in E13.5 pancreatic explants that were treated or not with MC1568 during 14 days. D: qPCR analysis of insulin mRNA expression from D3 to D14 in cultured pancreatic explants that were treated or not with MC1568. E: Immunohistological analyses of pancreata after 7 days in culture, with and without MC1568 treatment. β-Cell development was evaluated with insulin staining (red). Absolute areas that were occupied by the insulin-positive cells were quantified. F: qPCR analysis of MafA mRNA expression from D3 to D14 in pancreatic explants treated or not with MC1568. G: qPCR analysis of Znt8 mRNA expression from D3 to D14 in pancreatic explants treated or not with MC1568. H: qPCR analysis of somatostatin mRNA expression from D3 to D14 in pancreatic explants treated or not with MC1568. I: Immunohistological analyses of pancreata after 7 days in culture, with and without MC1568 treatment. δ-Cell development was evaluated with somatostatin (SST) staining (green). Absolute areas that were occupied by the somatostatin-positive cells were quantified. In E and I, nuclei were stained with Hoechst stain (blue). qPCR data and immunohistological analyses are the means ± SEM of four and six independent experiments, respectively. *P < 0.05; **P < 0.005; ***P < 0.001. Scale bar, 50 μm. (A high-quality digital representation of this figure is available in the online issue.)
We next cultured E13.5 rat pancreata for up to 14 days under conditions that allowed endocrine and acinar cell development (14,28) and treated the explants with MC1568, a class II HDAC inhibitor that modulates the stability and activity of class IIa HDAC-MEF2 complexes, by inhibiting HDAC activity and blocking MEF2-mediated transactivation (15). Treatment of pancreas explants with MC1568 did not modify pancreatic shape or growth (Supplementary Fig. 8A). However, MC1568 enhanced expression of Pax4, which is involved in β/δ-cell fate (4) (a threefold increase at days 5 and 9; Fig. 8C). Induction of Pax4 expression was associated with a dramatic increase in both insulin (a twofold increase at day 7 and a 2.5-fold increase at day 14; Fig. 8D) and somatostatin mRNA (a fourfold increase at day 7 and a sixfold increase at day 14; Fig. 8H). Immunohistochemistry further supported such increases with a 2-fold increase in β-cell surface (Fig. 8E) and 2.3-fold increase in δ-cell surface (Fig. 8I) after 14 days of treatment. Furthermore, the expression of MafA, NeuroD1, Pdx1, Znt8, Ia1, and Nkx2.2 mRNA was significantly induced with MC1568 treatment (Fig. 8F and G and Supplementary Fig. 8C). By contrast, MC1568 treatment did not efficiently induce pancreatic polypeptide and glucagon expression (data not shown). Finally, MC1568 did not modify acinar cell development (Supplementary Fig. 8B). These results demonstrate that MC1568 treatment induces Pax4 expression and enhances the pool of β- and δ-cells, indicating a role for class IIa HDACs and MEF2 in the repression of the β/δ-cell differentiation lineage.
DISCUSSION
This work provides evidence that class IIa HDACs, which have specific expression patterns in the developing pancreas, control development of insulin-producing β-cells and somatostatin-producing endocrine δ-cells. Using loss- and gain-of-function approaches, our results highlight the epigenetic mechanisms underlying the regulation of endocrine cell development. We also show that a specific HDAC inhibitor MC1568 amplifies endocrine β- and δ-cells, revealing a potential new approach for β-cell differentiation-based therapies for diabetes.
In this study, we analyzed the pancreatic phenotype of Hdac4, Hdac5, and Hdac9 mutant mice. Hdac4−/− and Hdac5/9−/− mice were previously described and showed chondrocytes and cardiac defects, respectively. No physiological or metabolic studies were described concerning these mutants. Analyses taking in account the global metabolism of HDAC-deficient animals, including the role of muscle and liver, could be the topic of future study. Using loss- and gain-of-function approaches, we provide here evidence that class IIa HDACs are involved in β- and δ-cell development. Interestingly, two HDACs (HDAC4 and -5) are expressed in δ-cells, and both Hdac4−/− and Hdac5−/− pancreata showed an increased δ-cell mass. In the same manner, HDAC5 and -9 are both expressed in β-cells, and Hdac5−/− and Hdac9−/− pancreata showed an increased β-cell mass. Such a cooperative effect of class IIa HDACs was previously described. It is for example the case for HDAC4 and HDAC5 in skeletal muscle cells (12,29,33) and for HDAC5 and HDAC9 in the heart (9,10).
Pancreatic endocrine cell development depends on both cell proliferation and differentiation. The importance of cell proliferation in pancreatic endocrine cell development is well established, and both embryonic pancreatic progenitor and adult β-cell proliferate efficiently in rodents (34,35). Here, we demonstrate that pancreatic endocrine cell mass is increased in Hdac4, Hdac5, and Hdac9 mutant mice without any increase in cell proliferation at E18.5, suggesting an effect on cell differentiation. This result fits well with the roles of HDAC4, -5, and -9 in chondrocyte and cardiomyocyte differentiation (9,10,16). Many animal models were recently described with an increased pancreatic endocrine cell mass due to enhanced cell proliferation (36,37). To the best of our knowledge, class IIa HDAC-deficient mice represent the first example of animal models with an increased endocrine cell mass likely due to increased cell differentiation. Furthermore, we also analyzed individual β-cell size in Hdac5 and Hdac9 mutant mice, and we found no difference between mutants and wild-type mice (data not shown), indicating that the increased β-cell mass was not due to cell hypertrophy.
In previous work, using class I and pan HDAC inhibitors, we found that HDACs play a crucial role in the modulation of pancreatic cell fate. We demonstrated that class I HDAC inhibitors regulate a specific step in pancreatic endocrine cell differentiation, i.e., the development of NGN3-positive endocrine progenitor cells from PDX1-positive pancreatic progenitors (14). We also suggested a specific role of class II HDACs during the final steps of endocrine cell differentiation. However, at that time, the lack of a specific class II HDAC inhibitor did not allow further validation of this hypothesis. Here, we show that MC1568, a selective class IIa HDAC inhibitor, selectively amplifies endocrine β- and δ-cells, in correlation with the results obtained in Hdac mutant mice. Thus, our in vivo results were reproduced in vitro, implying that these findings represent direct effects on pancreatic cells.
Interestingly, MC1568 acts, at least in part, by stabilizing class IIa HDAC-MEF2 complexes, thus blocking MEF2 target genes (15). Class IIa HDACs bind MEF2 transcription factors and repress MEF2 targets (11,38). The four vertebrate Mef2 genes are highly expressed in skeletal muscle and brain, but are also highly expressed in neural crest, bone, lymphocytes, endothelium, and smooth muscle (39,40). Here, we show that Mef2A and Mef2D are expressed in the embryonic pancreas at levels in the same range as those observed in muscle and heart, and we detected MEF2A protein in endocrine cells. Thus, during pancreatic endocrine cell development, the function of class IIa HDACs could depend on MEF2A and MEF2D.
By treating pancreatic explants with MC1568, we determined the developmental step regulated by class IIa HDACs during endocrine differentiation. Whereas in our previous work, class I HDAC inhibitors induced Ngn3 expression after 5 days of culture (14), here, MC1568 did not induce Ngn3 expression (data not shown). Importantly, MC1568 acted downstream of NGN3 and enhanced the expression of PAX4, a downstream target of NGN3. During development, both β- and δ-cells originate from PAX4-expressing endocrine precursors (4). We demonstrate that MC1568 treatment activates Pax4 expression and gives rise to increased β- and δ-cell development. Future experiments will test whether a class IIa HDAC-MEF2 cooperation directly regulates Pax4 expression. In addition to MEF2, DACH2 and RUNX3 (41) are targets of class IIa HDACs and are expressed in pancreatic endocrine cells (42,43). Future studies will define whether they represent HDAC targets in pancreatic β-cells.
Class IIa HDACs shuttle between the nucleus and the cytoplasm (7). Here, we observed HDAC4 and HDAC9 immunoreactivity mainly in the cytoplasm and HDAC5 both in the cytoplasm and the nucleus of endocrine cells. A cytoplasmic subcellular localization of class IIa HDACs was previously described in retina for HDAC4 and in cortical neurons for HDAC9 (25,27), and cytoplasmic functions of class IIa HDACs are now emerging (26,27,44). As an example, HDAC4, which is predominantly cytoplasmic in neurons, regulates the survival of retinal neurons at least partly by regulating the hypoxia-inducible factor 1a (HIF1a) activity (27). Hif1a was recently shown to be a crucial regulator of β-cell differentiation (45). However, because Hif1a is expressed only during the early stages of development when class IIa HDACs are not yet detected, we exclude the possibility of a link between HIF1a and class IIa HDACs during endocrine differentiation. Further investigations will be necessary to study other putative cytoplasmic targets of class IIa HDACs in the differentiation of endocrine β- and δ-cells.
Together, our data define a novel site of expression of class IIa HDACs and MEF2. They highlight a specific role of these HDACs in the regulation of the pancreatic endocrine β- and δ-cells. From a therapeutic perspective, screenings are currently being performed to identify small molecules that favor β-cell differentiation from pancreatic progenitors (46). In this context, small molecules such as HDAC inhibitors are powerful tools to modulate cell differentiation programs. Thus, class IIa HDACs represent new targets to selectively enhance β- and δ-cell differentiation. Moreover, MC1568 could be used as a novel tool to generate β-cells from embryonic stem cells for cell therapies in diabetes.
ACKNOWLEDGMENTS
This work was supported by the Institut National de la Santé et de la Recherche Médicale (INSERM); funding to R.S. from the European Union 6th Framework Programs (512145); the Agence Nationale de la Recherche (ANR2008 Blanc); the bilateral program Bundesministerium fur Bildung und Forschung (BMBF) ANR, convention number 2009 GENO10502; and funding to C.H. from the Association pour la Recherche sur le Diabète (ARD) in the Programmme National de Recherche sur le Diabète (PNRD2007). O.L. was supported by fellowships from the Ministère de la Recherche et de la Technologie (MRT) and the Fondation pour la Recherche Médicale (FRM).
No potential conflicts of interest relevant to this article were reported.
O.L. performed the experiments, analyzed data, contributed to discussion, and wrote the manuscript. K.F. and F.X.M. performed the experiments. B.B. and A.M. contributed new reagents/analytic tools. R.B.-D. contributed new reagents/analytic tools and reviewed the manuscript. P.R. contributed new reagents/analytic tools and performed the experiments. E.N.O. contributed new reagents/analytic tools and reviewed the manuscript. C.H. designed the study, performed the experiments, analyzed data, contributed to discussion, and wrote the manuscript. R.S. designed the study, contributed to discussion, and wrote the manuscript.
The authors thank Claire Sauty and Blandine Bonnamy for lentiviruses constructs and productions (Institute of Brain and Spinal Cord Research Center, CNRS UMR 7225, INSERM UMRS 975, Pierre and Marie Curie University, Pitié Salpêtrière Hospital, Paris, France). The authors are grateful to Cheryl Nolen for the animal facility and John Shelton for the histology core facility (University of Texas Southwestern Medical Center, Dallas, TX).
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db11-0440/-/DC1.
C.H. is currently affiliated with CNRS UMR 7622, INSERM U969, Pierre and Marie Curie University, Paris, France.
REFERENCES
- 1.Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature 1994;371:606–609 [DOI] [PubMed] [Google Scholar]
- 2.Gradwohl G, Dierich A, LeMeur M, Guillemot F. Neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci U S A 2000;97:1607–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Collombat P, Hecksher-Sørensen J, Serup P, Mansouri A. Specifying pancreatic endocrine cell fates. Mech Dev 2006;123:501–512 [DOI] [PubMed] [Google Scholar]
- 4.Sosa-Pineda B, Chowdhury K, Torres M, Oliver G, Gruss P. The Pax4 gene is essential for differentiation of insulin-producing beta cells in the mammalian pancreas. Nature 1997;386:399–402 [DOI] [PubMed] [Google Scholar]
- 5.Collombat P, Mansouri A, Hecksher-Sorensen J, et al. Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev 2003;17:2591–2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaung HL. Growth dynamics of pancreatic islet cell populations during fetal and neonatal development of the rat. Dev Dyn 1994;200:163–175 [DOI] [PubMed] [Google Scholar]
- 7.Martin M, Kettmann R, Dequiedt F. Class IIa histone deacetylases: regulating the regulators. Oncogene 2007;26:5450–5467 [DOI] [PubMed] [Google Scholar]
- 8.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 2003;370:737–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 2002;110:479–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang S, McKinsey TA, Zhang CL, Richardson JA, Hill JA, Olson EN. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol Cell Biol 2004;24:8467–8476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu J, McKinsey TA, Zhang CL, Olson EN. Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol Cell 2000;6:233–244 [DOI] [PubMed] [Google Scholar]
- 12.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 2009;10:32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haumaitre C, Lenoir O, Scharfmann R. Directing cell differentiation with small-molecule histone deacetylase inhibitors: the example of promoting pancreatic endocrine cells. Cell Cycle 2009;8:536–544 [DOI] [PubMed] [Google Scholar]
- 14.Haumaitre C, Lenoir O, Scharfmann R. Histone deacetylase inhibitors modify pancreatic cell fate determination and amplify endocrine progenitors. Mol Cell Biol 2008;28:6373–6383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nebbioso A, Manzo F, Miceli M, et al. Selective class II HDAC inhibitors impair myogenesis by modulating the stability and activity of HDAC-MEF2 complexes. EMBO Rep 2009;10:776–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vega RB, Matsuda K, Oh J, et al. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell 2004;119:555–566 [DOI] [PubMed] [Google Scholar]
- 17.Miralles F, Battelino T, Czernichow P, Scharfmann R. TGF-beta plays a key role in morphogenesis of the pancreatic islets of Langerhans by controlling the activity of the matrix metalloproteinase MMP-2. J Cell Biol 1998;143:827–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zennou V, Petit C, Guetard D, Nerhbass U, Montagnier L, Charneau P. HIV-1 genome nuclear import is mediated by a central DNA flap. Cell 2000;101:173–185 [DOI] [PubMed] [Google Scholar]
- 19.Zougbédé S, Miller F, Ravassard P, et al. Metabolic acidosis induced by Plasmodium falciparum intraerythrocytic stages alters blood-brain barrier integrity. J Cereb Blood Flow Metab 2011;31:514–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castaing M, Guerci A, Mallet J, Czernichow P, Ravassard P, Scharfmann R. Efficient restricted gene expression in beta cells by lentivirus-mediated gene transfer into pancreatic stem/progenitor cells. Diabetologia 2005;48:709–719 [DOI] [PubMed] [Google Scholar]
- 21.Ravassard P, Bricout-Neveu E, Hazhouz Y, et al. A new strategy to generate functional insulin-producing cell lines by somatic gene transfer into pancreatic progenitors. PLoS ONE 2009;4:e4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duvillié B, Attali M, Aiello V, Quemeneur E, Scharfmann R. Label-retaining cells in the rat pancreas: location and differentiation potential in vitro. Diabetes 2003;52:2035–2042 [DOI] [PubMed] [Google Scholar]
- 23.Gesina E, Tronche F, Herrera P, et al. Dissecting the role of glucocorticoids on pancreas development. Diabetes 2004;53:2322–2329 [DOI] [PubMed] [Google Scholar]
- 24.Yang WM, Yao YL, Sun JM, Davie JR, Seto E. Isolation and characterization of cDNAs corresponding to an additional member of the human histone deacetylase gene family. J Biol Chem 1997;272:28001–28007 [DOI] [PubMed] [Google Scholar]
- 25.Sugo N, Oshiro H, Takemura M, et al. Nucleocytoplasmic translocation of HDAC9 regulates gene expression and dendritic growth in developing cortical neurons. Eur J Neurosci 2010;31:1521–1532 [DOI] [PubMed] [Google Scholar]
- 26.Gupta MP, Samant SA, Smith SH, Shroff SG. HDAC4 and PCAF bind to cardiac sarcomeres and play a role in regulating myofilament contractile activity. J Biol Chem 2008;283:10135–10146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen B, Cepko CL. HDAC4 regulates neuronal survival in normal and diseased retinas. Science 2009;323:256–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Attali M, Stetsyuk V, Basmaciogullari A, et al. Control of beta-cell differentiation by the pancreatic mesenchyme. Diabetes 2007;56:1248–1258 [DOI] [PubMed] [Google Scholar]
- 29.Potthoff MJ, Wu H, Arnold MA, et al. Histone deacetylase degradation and MEF2 activation promote the formation of slow-twitch myofibers. J Clin Invest 2007;117:2459–2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Méjat A, Ramond F, Bassel-Duby R, Khochbin S, Olson EN, Schaeffer L. Histone deacetylase 9 couples neuronal activity to muscle chromatin acetylation and gene expression. Nat Neurosci 2005;8:313–321 [DOI] [PubMed] [Google Scholar]
- 31.Muralidhar SA, Ramakrishnan V, Kalra IS, Li W, Pace BS. Histone deacetylase 9 activates gamma-globin gene expression in primary erythroid cells. J Biol Chem 2011;286:2343–2353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Potthoff MJ, Olson EN. MEF2: a central regulator of diverse developmental programs. Development 2007;134:4131–4140 [DOI] [PubMed] [Google Scholar]
- 33.Backs J, Backs T, Bezprozvannaya S, McKinsey TA, Olson EN. Histone deacetylase 5 acquires calcium/calmodulin-dependent kinase II responsiveness by oligomerization with histone deacetylase 4. Mol Cell Biol 2008;28:3437–3445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bhushan A, Itoh N, Kato S, et al. Fgf10 is essential for maintaining the proliferative capacity of epithelial progenitor cells during early pancreatic organogenesis. Development 2001;128:5109–5117 [DOI] [PubMed] [Google Scholar]
- 35.Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004;429:41–46 [DOI] [PubMed] [Google Scholar]
- 36.Rachdi L, Balcazar N, Osorio-Duque F, et al. Disruption of Tsc2 in pancreatic beta cells induces beta cell mass expansion and improved glucose tolerance in a TORC1-dependent manner. Proc Natl Acad Sci U S A 2008;105:9250–9255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Desgraz R, Bonal C, Herrera PL. β-Cell regeneration: the pancreatic intrinsic faculty. Trends Endocrinol Metab 2011;22:34–43 [DOI] [PubMed] [Google Scholar]
- 38.Miska EA, Karlsson C, Langley E, Nielsen SJ, Pines J, Kouzarides T. HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J 1999;18:5099–5107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edmondson DG, Lyons GE, Martin JF, Olson EN. Mef2 gene expression marks the cardiac and skeletal muscle lineages during mouse embryogenesis. Development 1994;120:1251–1263 [DOI] [PubMed] [Google Scholar]
- 40.Arnold MA, Kim Y, Czubryt MP, et al. MEF2C transcription factor controls chondrocyte hypertrophy and bone development. Dev Cell 2007;12:377–389 [DOI] [PubMed] [Google Scholar]
- 41.Cohen TJ, Waddell DS, Barrientos T, et al. The histone deacetylase HDAC4 connects neural activity to muscle transcriptional reprogramming. J Biol Chem 2007;282:33752–33759 [DOI] [PubMed] [Google Scholar]
- 42.Kalousova A, Mavropoulos A, Adams BA, et al. Dachshund homologues play a conserved role in islet cell development. Dev Biol 2010;348:143–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li J, Kleeff J, Guweidhi A, et al. RUNX3 expression in primary and metastatic pancreatic cancer. J Clin Pathol 2004;57:294–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Margariti A, Zampetaki A, Xiao Q, et al. Histone deacetylase 7 controls endothelial cell growth through modulation of beta-catenin. Circ Res 2010;106:1202–1211 [DOI] [PubMed] [Google Scholar]
- 45.Heinis M, Simon MT, Ilc K, et al. Oxygen tension regulates pancreatic beta-cell differentiation through hypoxia-inducible factor 1alpha. Diabetes 2010;59:662–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen S, Borowiak M, Fox JL, et al. A small molecule that directs differentiation of human ESCs into the pancreatic lineage. Nat Chem Biol 2009;5:258–265 [DOI] [PubMed] [Google Scholar]