

Abstract
OBJECTIVE
Insulin stimulates both nitric oxide (NO)-dependent vasodilation and endothelin-1 (ET-1)–dependent vasoconstriction. However, the cellular mechanisms that control the dual vascular effects of insulin remain unclear. This study aimed to investigate the roles of the multidomain adaptor protein APPL1 in modulating vascular actions of insulin in mice and in endothelial cells.
RESEARCH DESIGN AND METHODS
Both APPL1 knockout mice and APPL1 transgenic mice were generated to evaluate APPL1’s physiological roles in regulating vascular reactivity and insulin signaling in endothelial cells.
RESULTS
Insulin potently induced NO-dependent relaxations in mesenteric arteries of 8-week-old mice, whereas this effect of insulin was progressively impaired with ageing or upon development of obesity induced by high-fat diet. Transgenic expression of APPL1 prevented age- and obesity-induced impairment in insulin-induced vasodilation and reversed obesity-induced augmentation in insulin-evoked ET-1–dependent vasoconstriction. By contrast, genetic disruption of APPL1 shifted the effects of insulin from vasodilation to vasoconstriction. At the molecular level, insulin-elicited activation of protein kinase B (Akt) and endothelial NO synthase and production of NO were enhanced in APPL1 transgenic mice but were abrogated in APPL1 knockout mice. Conversely, insulin-induced extracellular signal–related kinase (ERK)1/2 phosphorylation and ET-1 expression was augmented in APPL1 knockout mice but was diminished in APPL1 transgenic mice. In endothelial cells, APPL1 potentiated insulin-stimulated Akt activation by competing with the Akt inhibitor Tribbles 3 (TRB3) and suppressed ERK1/2 signaling by altering the phosphorylation status of its upstream kinase Raf-1.
CONCLUSIONS
APPL1 plays a key role in coordinating the vasodilator and vasoconstrictor effects of insulin by modulating Akt-dependent NO production and ERK1/2-mediated ET-1 secretion in the endothelium.
Insulin is not only a principal regulator of glucose homeostasis but also a vasoactive hormone involved in modulation of vascular tone (1). In the vasculature, insulin exerts both vasodilator and vasoconstrictor effects by promoting the endothelial production of nitric oxide (NO) and the release of endothelin-1 (ET-1) (2). Insulin-stimulated NO production in endothelial cells is mediated by the phosphatidylinositol 3-kinase (PI 3-K)/protein kinase B (Akt) signaling cascade, which in turn phosphorylates and activates endothelial NO synthase (eNOS) (3). On the other hand, insulin-induced expression and secretion of the vasoconstrictor ET-1 is mediated by the extracellular signal–regulated kinase (ERK)1/2 mitogen-activated protein kinase (MAPK) signaling pathway in vascular endothelium (4). Activation of ERK1/2 increases both mRNA expression and secretion of ET-1 in endothelial cells (3). The balanced endothelial production of NO and ET-1 is critical in maintaining both metabolic and hemodynamic homeostasis under the healthy condition (1).
Vascular insulin resistance, manifested by impaired vasodilator effects and augmented vasoconstrictor actions of insulin, is a key phenomenon linking obesity, diabetes, and cardiovascular disease (5,6). In insulin-resistant states such as ageing and obesity, insulin-induced activation of PI 3-K/Akt signaling is selectively impaired, whereas the MAPK pathway is preserved or augmented (7). Endothelial dysfunction is not only a well-established antecedent of hypertension and atherosclerosis but also an important contributor to metabolic insulin resistance by reducing the capillary recruitment and blood flow in skeletal muscle (1). The existence of the vicious cycle between insulin resistance and endothelial dysfunction in the development of diabetes and cardiovascular disease has been documented in both animal studies (8) and clinical investigations (1,9). Therefore, therapeutic interventions that switch the vasoconstrictor action of insulin to its vasodilator effect may represent an effective strategy for treating both insulin resistance and endothelial dysfunction (5). However, the cellular pathways that control the balance between insulin-evoked NO production and ET-1 release remain poorly characterized.
APPL1, an adaptor protein containing an NH2-terminal Bin/Amphiphysin/Rvs domain, a central pleckstrin homology domain, and a COOH-terminal phosphotyrosine-binding domain was originally identified as an interacting partner of Akt in a yeast two-hybrid assay using Akt2 as bait (10). Several studies demonstrate an important role of APPL1 in mediating the metabolic actions of insulin, including stimulation of glucose uptake in muscle (11) and adipocytes (12) and inhibition of gluconeogenesis in hepatocytes (13). Furthermore, APPL1 is an essential signaling component of the insulin sensitizer adiponectin by its direct interaction with adiponectin receptors (14). In endothelial cells, APPL1 potentiates adiponectin-induced NO production by activation of AMP-activated protein kinase (AMPK) and eNOS (14) and is also required for adiponectin-mediated inhibition of cell apoptosis (15). Decreased APPL1 expression and impaired adiponectin-stimulated NO-dependent relaxations have been observed in mesenteric arteries of both db/db obese/diabetic mice (14) and Zucker diabetic fatty rats (16).
Although the aforementioned findings suggest a possible role of APPL1 in mediating NO production in endothelial cells, its physiological function in the vasculature has not been explored. Here we investigated the role of APPL1 in the vasoactive actions of insulin in both APPL1 transgenic (APPL1-Tg) and knockout (KO) mice and also determined the molecular basis whereby APPL1 modulates insulin signaling pathways in endothelial cells.
RESEARCH DESIGN AND METHODS
Generation and maintenance of APPL1-Tg and KO mice.
The cDNA encoding human APPL1 was cloned into pCAGGS vector, which consists of cytomegalovirus immediate early promoter and chicken β-actin promoter (17). The DNA fragment consisting of cytomegalovirus immediate early–β-actin promoter, APPL1 cDNA, and β-actin polyA was microinjected into F1 embryos (C57BL/6xCBA). APPL1-Tg mice were screened by PCR analysis of genomic DNA using β-actin promoter–specific oligonucleotides and human APPL1-specific oligonucleotides. The results reported here are comparable between mice from three founders with different levels of human APPL1 transgenic expression.
The APPL1 KO targeting vector was constructed from genomic DNA fragment derived from a C57BL/6 genomic bacterial artificial chromosome clone. The targeting vector, the left (4.7 kb) and right (4.2 kb) arms encompassing intron 17 and exon 19–21 of the APPL1 gene, respectively, were inserted into the ABRLFn-pBR32 vector. The linearized APPL1 KO targeting vector was electroporated into CJ7 (129SV/J) embryonic stem cells. The targeted clones were identified by PCR analysis. Correctly targeted embryonic stem cell clones were microinjected into blastocysts of C57 BL/6 J to generate chimeras that were then crossed onto a C57 BL/6 J genetic background. The offspring were screened by PCR analysis of genomic DNA using APPL1 wild type (WT)–specific primers and APPL1 KO–specific primers, respectively. APPL1 KO mice were generated by Shanghai Nanfang Research Center for Model Organisms.
Both APPL1-Tg and KO mice were crossed into a C57BL/6 background for at least six generations before use. All animal experimental protocols were approved by the animal ethics committee of the University of Hong Kong. The mice were housed in a room under controlled temperature (23 ± 1°C) with free access to water and standard chow diet (STD; 20% kcal protein, 10% kcal fat, and 70% kcal carbohydrates) or high-fat diet (HFD; Research Diet, 20% kcal protein, 45% kcal fat, and 35% kcal carbohydrates), respectively.
Isometric tension measurement.
Mesenteric arteries were isolated from mice as described (14) and cut into rings. In certain preparations, the endothelium was removed by gentle mechanical abrasion. After obtaining a sustained and comparable contraction induced by 10 μmol/L U46619, the functional integrity of endothelium was assessed by recording the relaxation to 10 μmol/L acetylcholine. Endothelium-dependent and -independent relaxations were determined with acetylcholine and sodium nitroprusside, respectively. Vascular responses to insulin in arteries were studied by exposing mesenteric arterial segments to six concentrations of insulin (0.001, 0.01, 0.1, 1, 10,100, and 1,000 nmol/L), and the resulting changes in isometric tension were recorded during 10 min after each concentration.
Cell culture, transfection, quantification of NO, and ET-1 release in endothelial cells.
Human umbilical vein endothelial cells (HUVECs) at passages 4–8 were cultured on gelatin-coated flasks and transfected with either a plasmid encoding HA-tagged Akt2 or small interfering RNA (siRNA) specific to human APPL1 or scramble control as previously described (13). At 40 h after transfection, cells were serum-starved for 6 h and treated with different concentrations of insulin for various periods as specified in each figure legend. NO release was determined by measurement of nitrite (NO2−) and nitrate (NO3−) levels using a Sievers NO analyzer (Boulder, CO) as previously described (14). ET-1 concentration in the conditioned medium was quantified by commercial ELISA kits from R&D Systems (Minneapolis, MN) (18).
Quantitative real-time PCR.
Total RNA were isolated from small mesentric arteries using TRIzol reagent according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA). For reverse transcription, 0.5 μg total RNA was converted into first-strand complementary DNA in 20 μL reactions using the ImProm-II Reverse Transcriptase kit (Promega, Madison, WI). Quantitative real-time PCR was performed as previously described (13), and the relative abundance of the genes was normalized against glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Specific primers used for mouse ET-1 and GAPDH are described in Supplementary Table 1.
Statistical analysis.
Relaxations are expressed as the decrease in tension in percentage of the contraction to U46619. The results were expressed as means ± SE. Statistical significance was determined by one-way ANOVA or Student t test. In all statistical comparisons, P values <0.05 were considered statistically significant differences.
RESULTS
Transgenic expression of APPL1 selectively potentiates insulin-induced endothelium-dependent relaxations in mesenteric arteries.
In both mice and rats with obesity, APPL1 expression in mesenteric arteries is reduced (14,16). To investigate the physiological roles of APPL1 in modulating vascular reactivity, we generated transgenic mice with overexpression of FLAG-tagged human APPL1 (APPL1-Tg) under control of the β-actin promoter (Fig. 1A). Transgenic expression of human APPL1 in mesenteric arteries was confirmed by PCR (Fig. 1B) and Western blot analysis (Fig. 1C). In APPL1-Tg mice, APPL1 protein level in mesenteric arteries was ∼2.5-fold higher than in that in WT littermates. A similar expression pattern was also observed in carotid arteries and aorta (data not shown).
FIG. 1.
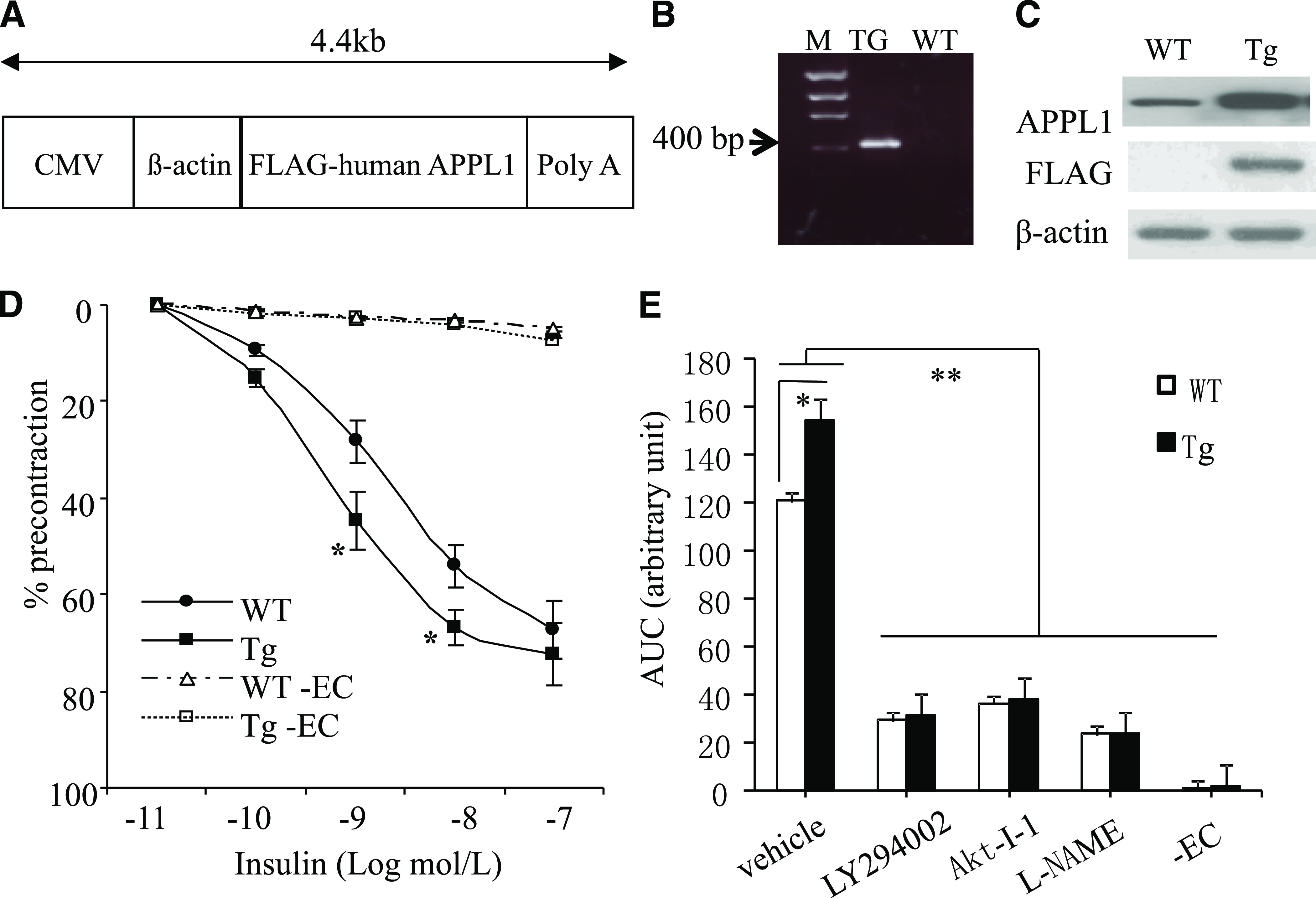
Effects of transgenic expression of APPL1 on insulin-induced relaxation in mouse arteries. A: Schematic diagram of the transgenic construct. Shown is cDNA encoding FLAG-tagged full-length human APPL1 under the control of CMV-β-actin promoter. CMV, cytomegalovirus. B: Confirmation of the APPL1 transgenic expression by PCR using genomic DNA as a template. M, DNA marker. C: Western blot analysis to detect APPL1 expression using anti-APPL1 or anti-FLAG antibody. D: Relaxations to increasing concentrations of insulin in mesenteric arterial rings from 8-week-old APPL1-Tg and WT during U46619-induced contractions. Data are expressed as percentage of the contraction to U46619. EC, endothelium. E: Effects of the PI 3-K inhibitor LY294002 (5 μmol/L), the Akt inhibitor Akt-I-1 (5 μmol/L), the NOS inhibitor l-NAME (100 μmol/L), and removal of EC on insulin-induced relaxations of mesenteric arteries from both APPL1-Tg and WT littermates fed STD. Data are shown as area under the curve (AUC). *P < 0.05, **P < 0.01 (n = 6–8).
In 8-week-old WT mice, insulin induced a dose-dependent relaxation of mesenteric arteries (Fig. 1D), and transgenic expression of APPL1 potentiated the sensitivity of insulin in inducing vasorelaxation. During contractions to the prostaglandin H2 analog U46619, the magnitude of relaxation to physiological concentrations of insulin was significantly greater in preparations from APPL1-Tg mice than from age-matched WT mice (1 nmol/L insulin: 44.6 ± 5.9% vs. 28.3 ± 3.7%, P < 0.05; 10 nmol/L insulin: 66.7 ± 5.8% vs. 54.1 ± 6.7%, P < 0.05). The EC50 value of insulin in APPL1-Tg mice was significantly lower than that in WT littermates (1.9 ± 0.4 vs. 8.1 ± 1.2 nmol/L, P < 0.01). However, the maximal relaxation response to the supraphysiological dose of insulin was comparable between these two groups of mice. The insulin-induced relaxation was abolished by the removal of the endothelium in rings of both APPL1-Tg and WT mice (Fig. 1E). Furthermore, the insulin-induced relaxation was attenuated significantly and to a comparable level in preparations of both types of mice by incubation with inhibitors of NOS (l-NG-nitro-l-arginine methyl ester [l-NAME; 100 μmol/L]), Akt (Akt-I-1 [5 μmol/L]), or PI 3-K (LY294002 [5 μmol/L]), suggesting that transgenic expression of APPL1 potentiates insulin-evoked relaxation by activating the PI 3-K/Akt/eNOS signaling cascade. On the other hand, endothelium-dependent relaxations to acetylcholine and bradykinin and endothelium-independent relaxation to sodium nitroprusside were not significantly different in preparations of APPL1-Tg and WT mice (Supplementary Fig. 1), suggesting that APPL1 selectively potentiates insulin-induced endothelium-dependent relaxation of mouse arteries.
APPL1 counteracts obesity-associated impairment in insulin-evoked relaxations in mesenteric arteries.
To further investigate the role of APPL1 in modulating vascular tone under pathological conditions, both APPL1-Tg mice and their WT littermates were fed HFD to induce obesity. Mice were killed after 8, 12, or 16 weeks of feeding with the diet. HFD induced a similar degree of body weight gain in APPL1-Tg mice and WT mice (data not shown). In WT mice, HFD resulted in a progressive impairment in insulin-evoked relaxation (Fig. 2). In 8- and 12-week-old WT mice, relaxations to insulin (100 nmol/L) were 71.6 ± 3.43% and 65.69 ± 2.77%, respectively, when fed STD but declined to 37.1 ± 13.8% and 23.7 ± 2.9% when fed HFD (Fig. 2A and B). In 16-week-old WT mice, insulin (100 nmol/L) induced relaxation under STD (52.2 ± 2.47%) but evoked contractions under HFD (−8.95 ± 3.07%) (Fig. 2C). The transgenic expression of APPL1 prevented the HFD-induced impairment in insulin-induced relaxation. In age-matched APPL1-Tg mice fed an HFD, the vasoconstrictor effect of insulin observed in 16-week-old WT mice was reversed to a relaxation (Fig. 2D).
FIG. 2.
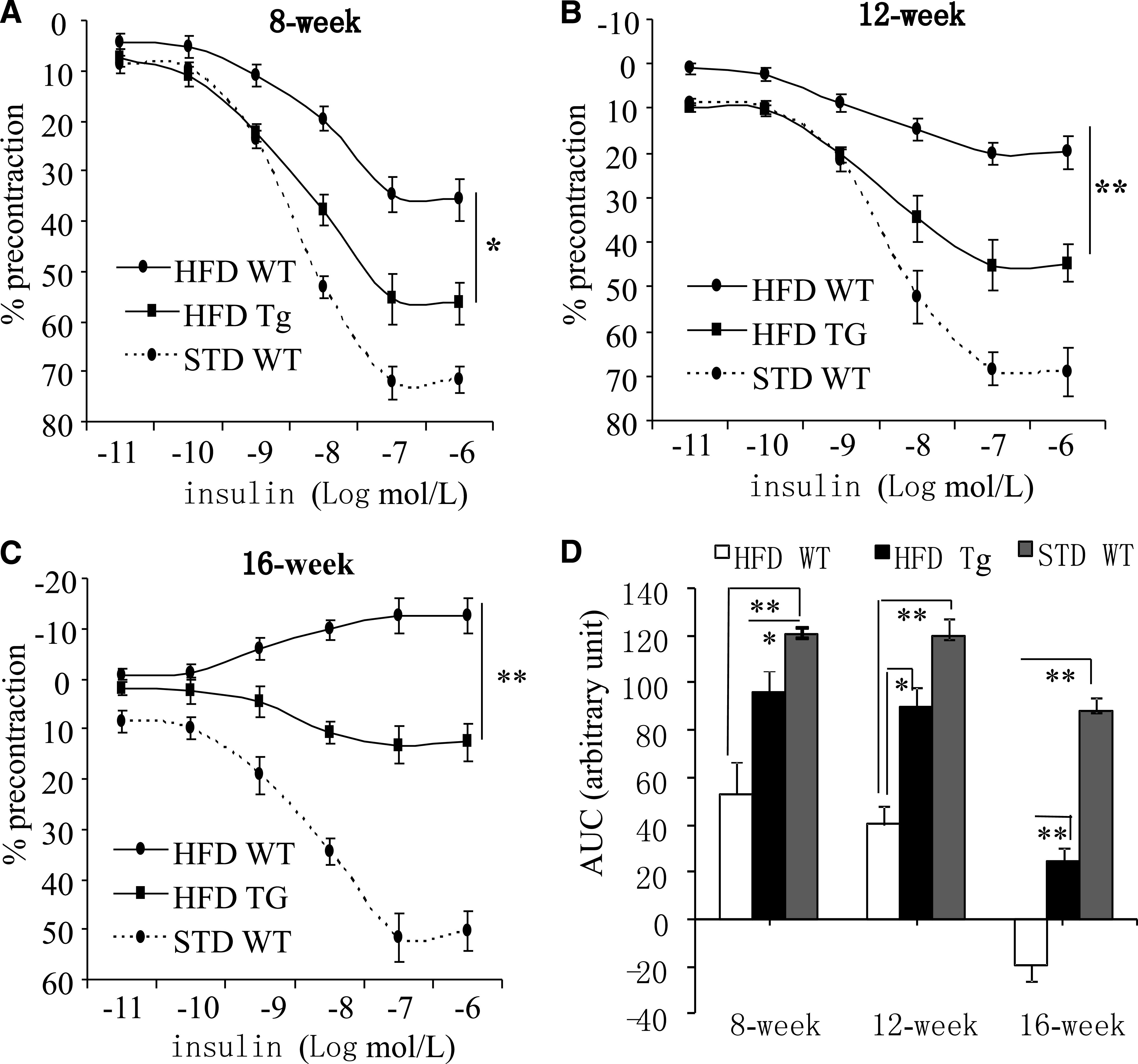
Transgenic expression of APPL1 counteracts dietary obesity–induced vascular insulin resistance. A–C: Concentration-dependent relaxations to insulin in mesenteric arteries isolated from APPL1-Tg mice and their WT littermates fed either STD or HFD at age of 8, 12, and 16 weeks. D: Data are shown as area under the curve (AUC). *P < 0.05; **P < 0.01 (n = 6–8).
Transgenic expression of APPL1 prevents insulin-induced ET-1–dependent vasoconstriction in HFD-fed obese mice.
In obesity, insulin promotes vasoconstriction by stimulating the release of ET-1 (19,20). We next investigated the impact of APPL1 overexpression on insulin-stimulated ET-1 expression and vasoconstriction in mice on HFD. Treatment of mesenteric arteries of 16-week-old WT mice on HFD with either the ERK1/2 inhibitor PD98059 (5 μmol/L) or the endothelin type a (ET-1A) receptor antagonist BQ123 (1 μmol/L) blocked insulin-elicited contractions and restored insulin-evoked relaxations (Fig. 3A), confirming that the insulin-induced vasoconstriction was mediated by ET-1 through ERK1/2 activation. In both APPL1-Tg mice and WT littermates on STD, l-NAME (100 μmol/L for 30 min) inhibited the relaxations to insulin (Fig. 3B). Treatment of mesenteric arteries with l-NAME potentiated insulin-elicited contractions in arteries of 16-week-old WT mice on HFD (Fig. 3B). By contrast, l-NAME blocked insulin-induced relaxations but did not unmask contractions to 100 nmol/L insulin in preparations of age-matched APPL1-Tg mice on HFD. These findings suggest that although the NO-mediated relaxation effects were inhibited by l-NAME, insulin-stimulated ET-1 production may not be abundant enough to induce vasoconstriction under STD.
FIG. 3.
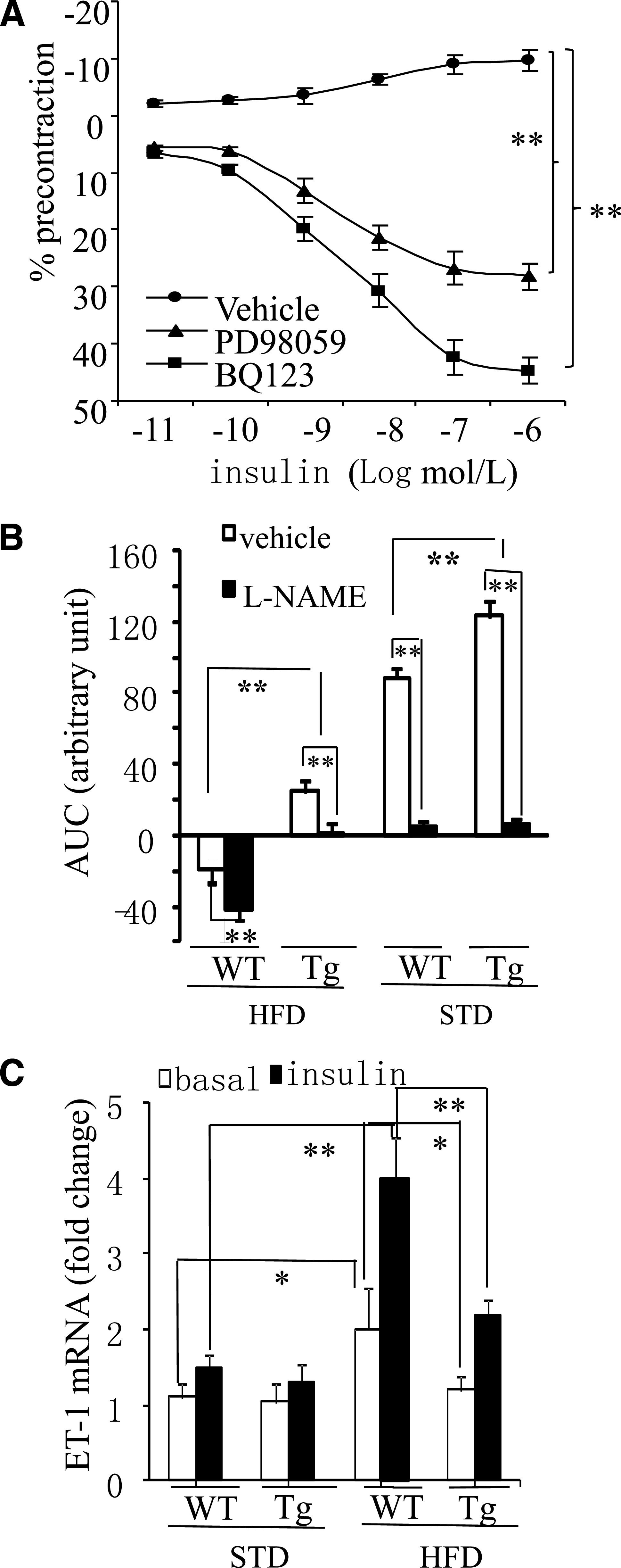
The insulin-induced ET-1–dependent contraction is suppressed in APPL1-Tg mice on HFD. A: Effect of insulin in mesenteric arteries of 16-week-old WT mice on HFD in the presence of the ERK1/2 inhibitor PD98059 (5 μmol/L) or the ET-1A receptor antagonist BQ123 (1 μmol/L). Data are expressed as percentage of the contraction to U46619. B: Effect of insulin in mesenteric arteries of 16-week-old APPL1-Tg and WT mice fed HFD or STD in the presence of l-NAME (100 μmol/L). Data are shown as area under the curve (AUC). C: The mRNA levels of ET-1 in mesenteric arteries from 16-week-old APPL1-Tg and WT mice fed STD or HFD were quantified by real-time quantitative-PCR and normalized against GAPDH. *P < 0.05, **P < 0.01 (n = 5–8).
Quantitative real-time PCR analysis demonstrated that the expression level of ET-1 in mesenteric arteries was comparable between APPL1-Tg mice and WT littermates on STD (Fig. 3C). Insulin stimulation caused a modest but not significant increase in ET-1 expression in both types of mice. When WT mice were fed HFD, both the basal and insulin-stimulated ET-1 expression levels in their mesenteric arteries were significantly elevated (by 1.5-fold and 2.7-fold, respectively) when compared with WT preparations on STD. By contrast, APPL1-Tg mice were resistant to HFD-induced elevation in ET-1 expression both under basal conditions and after insulin stimulation.
Transgenic expression of APPL1 potentiates insulin-stimulated Akt/eNOS signaling and suppresses ERK1/2 MAPK activation in mice with dietary obesity.
In endothelial cells, insulin-evoked production of NO and ET-1 is mediated by the Akt/eNOS and the ERK1/2 MAPK signaling cascades, respectively (3,4). To investigate the impact of APPL1 on these two signaling pathways, mesenteric arteries isolated from 16-week-old WT and APPL1-Tg mice on either STD or HFD were exposed to vehicle or 20 nmol/L insulin for 10 min and collected for Western blotting (Fig. 4). The basal levels of eNOS phosphorylation at Ser1177 and Akt at Thr308 were similar between WT mice and APPL1-Tg mice on either STD or HFD. Insulin caused an increase in phosphorylation of both eNOS at Ser1177 and Akt at Thr308 in arteries of WT mice, and this stimulatory effect of the hormone was enhanced significantly in preparations of APPL1-Tg mice (Fig. 4B and C). The insulin-induced phosphorylation of both eNOS and Akt was significantly impaired in arteries of WT mice on HFD. The HFD-induced impairment in insulin-stimulated phosphorylation of Akt and eNOS was prevented by the transgenic expression of APPL1.
FIG. 4.
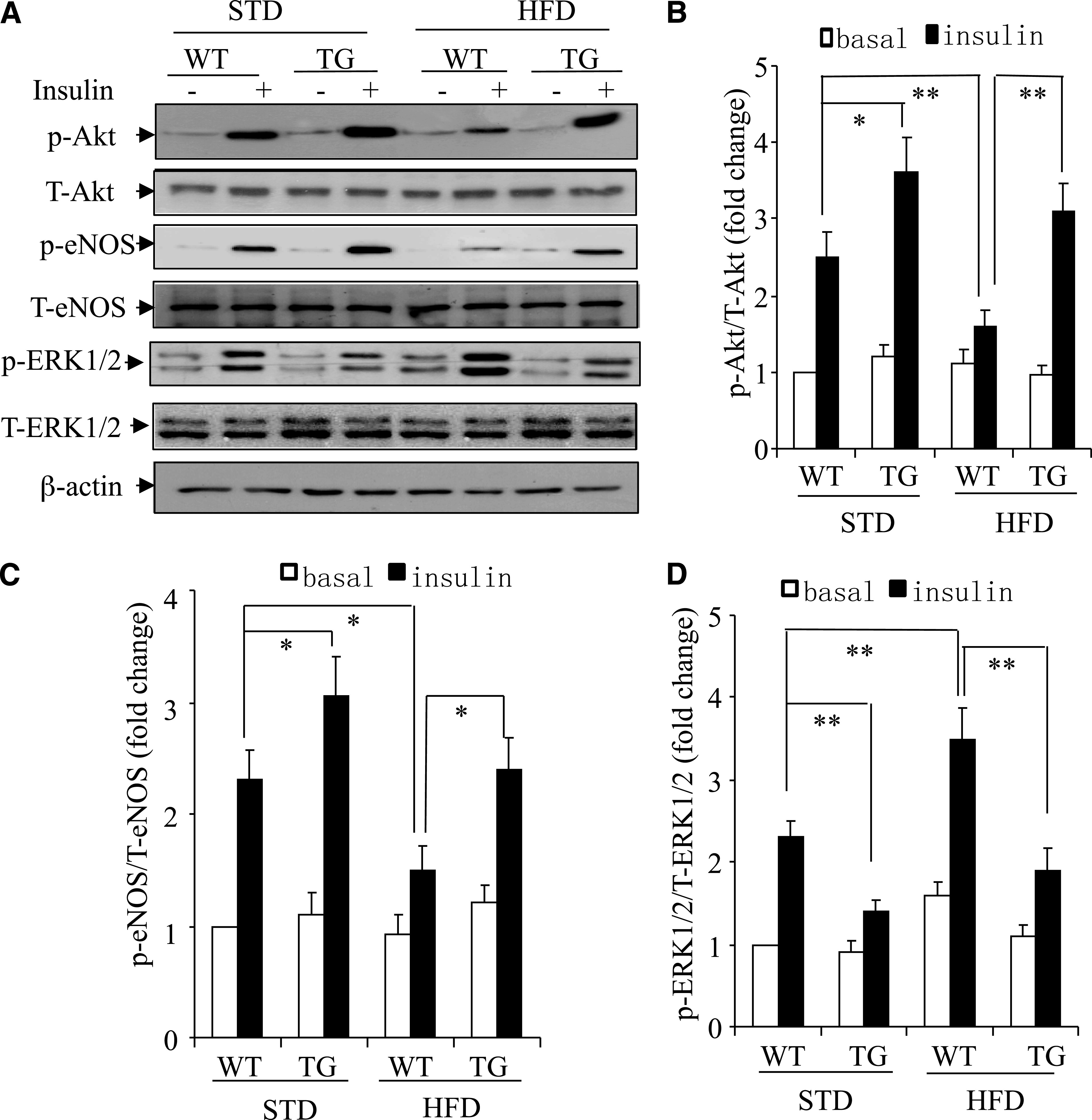
Opposite effects of APPL1 on insulin-evoked Akt/eNOS signaling and ERK1/2 activation in mesenteric arteries. Arteries isolated from 16-week-old WT and APPL1-Tg mice on STD or HFD were treated with vehicle or insulin (20 nmol/L). A: Total tissue lysates were subjected to immunoblotting using the antibodies against total or phospho-Akt (Thr308), total or phospho-eNOS (Ser1177), and total or phospho-ERK1/2 (Thr202/Tyr204), as specified. The ratio of p-Akt/t-Akt (B), p-eNOS/t-eNOS (C), and p-ERK/t-ERK (D) were quantified by densitometry and are expressed as fold changes relative to the basal levels observed in preparations of WT mice on STD. T, total; p, phosphorylated. *P < 0.05, **P < 0.01 (n = 5–7).
The basal levels of ERK1/2 phosphorylation were comparable in preparations of APPL1-Tg mice and WT littermates on STD. When WT mice were fed with HFD for 16 weeks, both basal and insulin-stimulated phosphorylation of ERK1/2 were significantly higher in their arteries than in those of mice on STD. By contrast, there was no significant difference in either basal or insulin-stimulated ERK1/2 phosphorylation between preparations of APPL1-Tg mice on either HFD or STD, suggesting that transgenic expression of APPL1 counteracts HFD-induced augmentation of ERK1/2 MAPK signaling pathway in mesenteric arteries.
Genetic disruption of the APPL1 gene aggravates endothelial dysfunction and hypertension in mice.
To investigate whether APPL1 is an indispensible regulator of insulin actions in the vasculature, we generated APPL1 KO mice by targeted disruption of the APPL1 gene at exon 17–18 (Supplementary Fig. 2A). PCR analysis was used to genotype mutant mice (Supplementary Fig. 2B). Western blotting confirmed the absence of APPL1 protein expression in APPL1 KO mice (Supplementary Fig. 2C). The relaxation of mesenteric arteries of APPL1 KO mice to insulin were impaired significantly compared with age-matched WT preparations, even when they were fed STD (Fig. 5A and B). The maximal relaxation to 100 nmol/L insulin in APPL1 KO and WT arteries was, respectively, 36.3 ± 5.8% and 71.6 ± 3.43% (P < 0.01) at the age of 8 weeks and 28.7 ± 7.7% and 56.8 ± 3.13% (P < 0.05) at the age of 16 weeks. In the presence of l-NAME, 100 nmol/L insulin caused a modest contraction in arteries of APPL1 KO mice even when they were fed STD (Fig. 5C). When fed HFD, the insulin-elicited increase in tension was significantly stronger in preparations of APPL1 KO than that in those of WT littermates (Fig. 5C). With both STD and HFD, the magnitude of insulin-induced increase in ET-1 expression in APPL1 KO was significantly higher than that in WT arteries (Fig. 5D). APPL1 KO mice exhibited significantly elevated systolic blood pressure when the mice aged beyond 16 weeks (data not shown). In 20-week-old APPL1 KO mice fed with HFD, systolic blood pressure was 10.5% higher than age-matched WT littermates. Western blotting analysis demonstrated that insulin-stimulated phosphorylation of eNOS at Ser1177 and Akt at Thr308 in mesenteric arteries of APPL1 KO mice was significantly blunted compared with WT controls (Supplementary Fig. 3). By contrast, insulin-stimulated ERK1/2 phosphorylation was elevated in arteries of APPL1 KO mice.
FIG. 5.
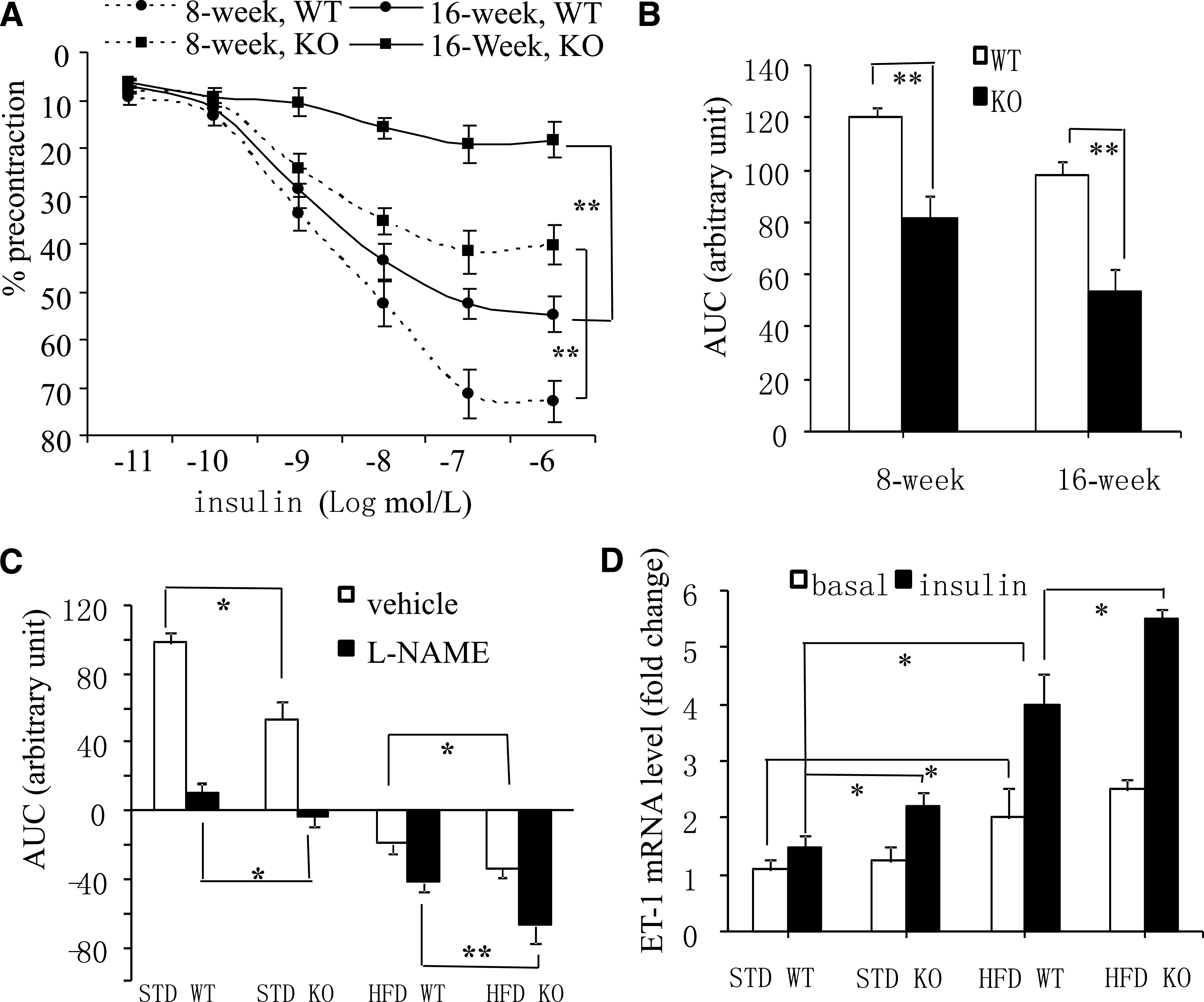
APPL1 KO mice display impaired relaxation and augmented ET-1–dependent contraction in response to insulin. Insulin-induced relaxations in mesenteric arteries of 8- and 16-week-old APPL1 KO and WT mice on STD. Data are expressed as percentage of the contraction to U46619 (A) or shown as area under the curve (AUC) (B). C: Effects of insulin in mesenteric arteries from 16-week-old APPL1 KO and WT mice fed HFD or STD in the presence of l-NAME (100 μmol/L). D: The mRNA levels of ET-1 in mesenteric arteries from 16-week-old APPL1 KO and WT mice fed STD or HFD were quantified using real-time PCR and normalized against GAPDH. *P < 0.05, **P < 0.01 (n = 5–7).
APPL1 exerts opposite effects on insulin-induced Akt/eNOS and ERK1/2 MAPK signaling cascades in cultured human endothelial cells.
Consistent with the above findings in APPL1 KO mice, insulin-induced phosphorylation of Akt at Thr308 and that of eNOS at Ser1177 was significantly decreased by siRNA-mediated knockdown of APPL1 in HUVECs (Fig. 6A–C). Insulin-induced elevation in endothelial NO production was also reduced by siRNA-mediated suppression of APPL1 expression (Fig. 6D). These changes were accompanied by an elevated interaction between Akt and its endogenous inhibitor Tribbles 3 (TRB3) (21), as determined by coimmunoprecipitation analysis (Fig. 6E).
FIG. 6.
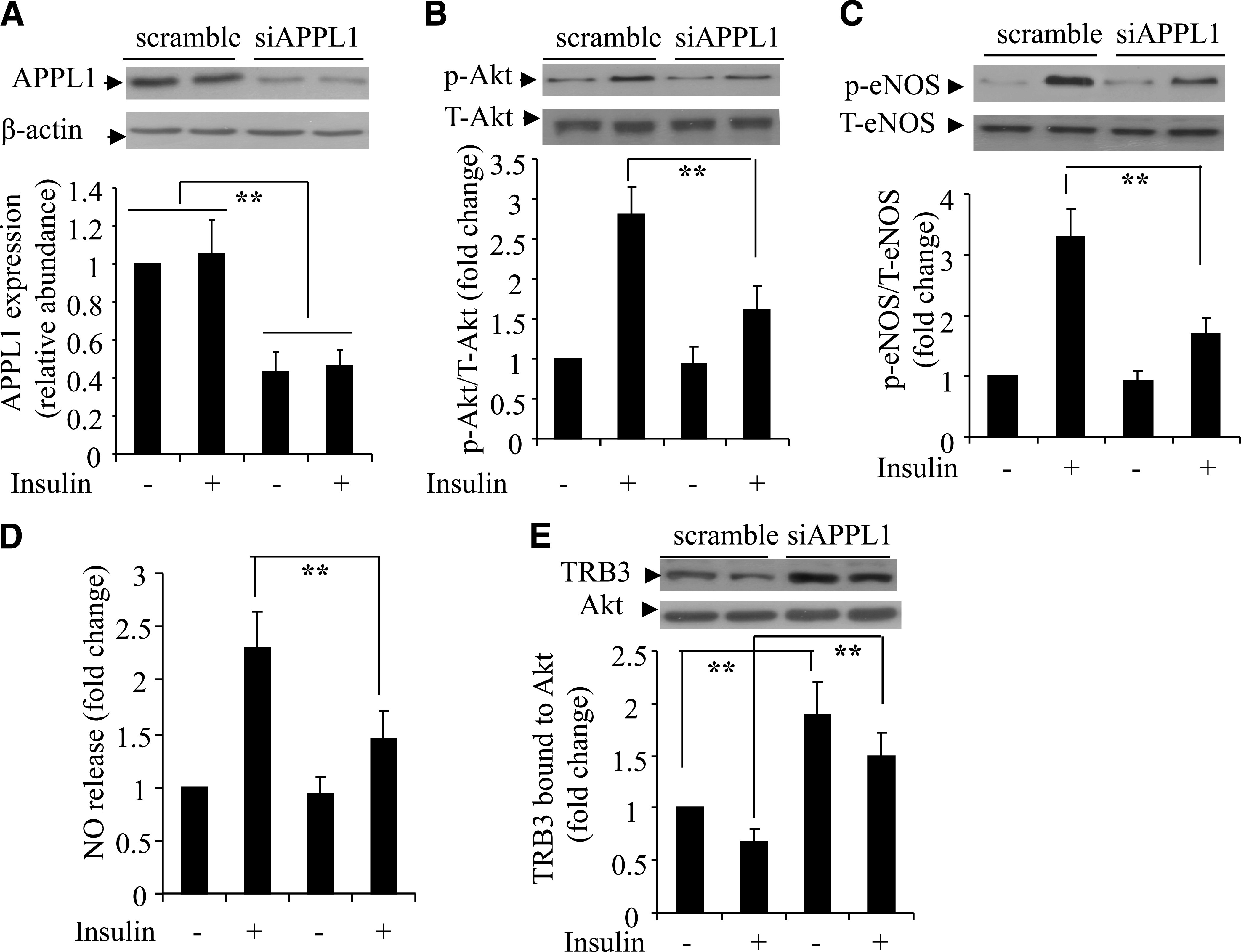
Effects of siRNA-mediated knockdown of APPL1 on insulin-induced Akt signaling and NO production in HUVECs. Cells were transfected with siRNA specific for APPL1 (siAPPL1) or scrambled control for 40 h, followed by serum starvation for 6 h and stimulation with insulin (50 nmol/L) for 10 min. The expression of APPL1 (A), phosphorylation of Akt at Thr308 (B), and phosphorylation of eNOS at Ser1177 (C) were determined by Western blotting. NO release in the conditioned medium was measured at 60 min after insulin treatment. The data were expressed as the fold over the control cells treated without insulin (D). E: Cells were cotransfected with siAPPL1 or scrambled control plus a plasmid encoding HA-tagged Akt, followed by insulin treatment as above. Cell lysates were subjected to immunoprecipitation with anti-HA antibodies and then probed with anti-Akt or anti-TRB3 antibody as indicated. T, total; p, phosphorylated. **P < 0.01 (n = 5–6).
In contrast to the changes in the Akt/eNOS signaling, siRNA-mediated suppression of APPL1 expression in HUVECs resulted in a significant elevation in insulin-induced phosphorylation of ERK1/2 MAPK at Thr202/Tyr204 and an increase in phosphorylation of the mitogen-activated protein kinase kinase (MEK1/2) at Ser217/Ser221 (Fig. 7A and B). Further analysis of the upstream signaling events of MEK1/2 showed that knockdown of APPL1 expression caused a significant elevation in insulin-induced phosphorylation of Raf-1 at its activation site Ser338 (Fig. 7C) but an attenuated phosphorylation of Raf-1 at its inhibitory site Ser259 (Fig. 7D). On the other hand, suppression of APPL1 expression had no effect on the insulin-induced increase in activated, guanosine-5'-triphosphate (GTP)-bound Ras, an upstream activator of Raf-1 (data not shown). In parallel with the changes in ERK1/2 MAPK signaling, insulin-induced secretion of ET-1 in HUVECs was significantly increased by knockdown of APPL1 expression (Fig. 7E). Taken together, these findings suggest that APPL1 modulates insulin-induced ERK1/2 MAPK signaling and ET-1 production by altering the phosphorylation of Raf-1.
FIG. 7.
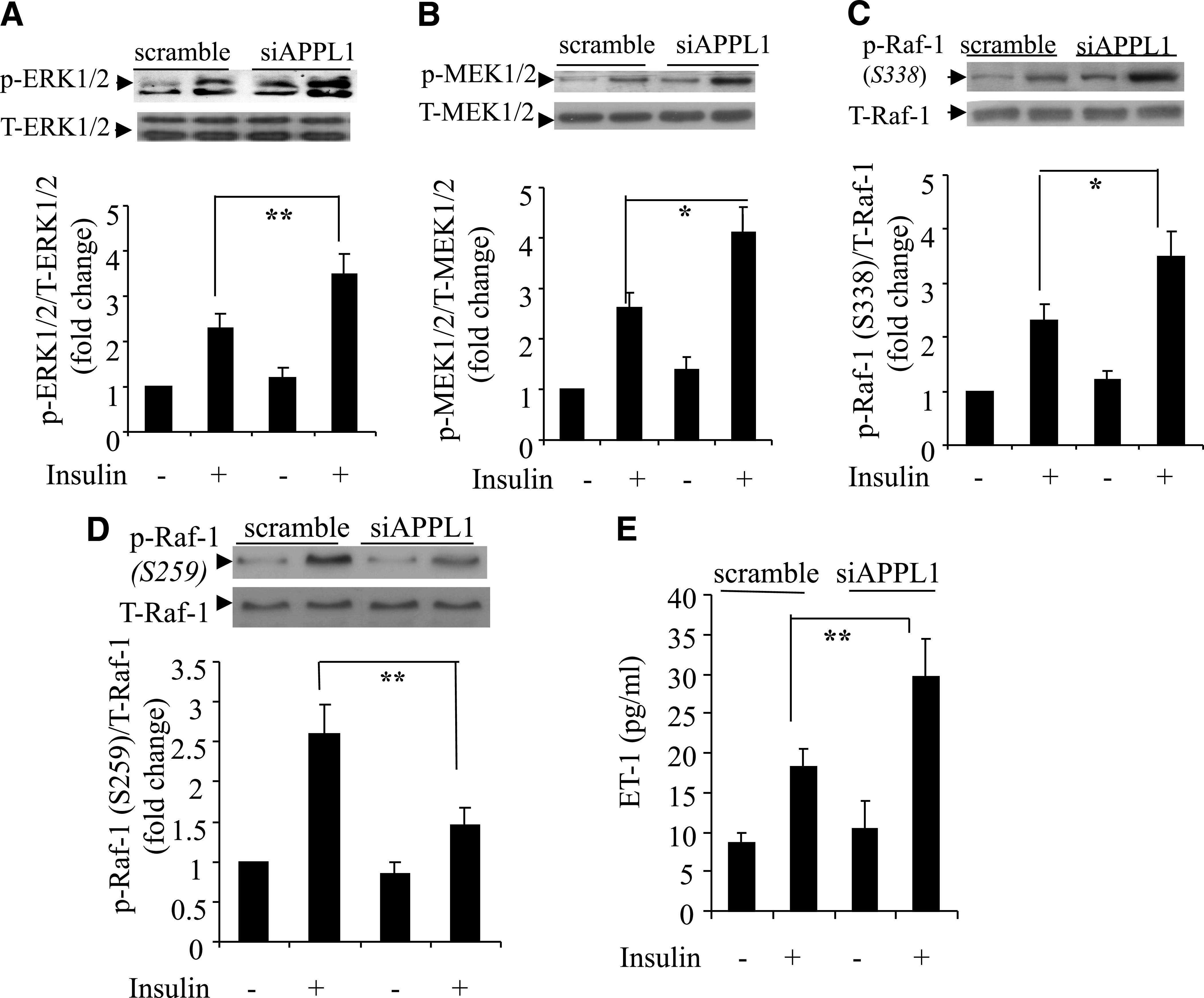
Effects of siRNA-mediated knockdown of APPL1 on insulin-stimulated ERK1/2 MAPK signaling cascades and ET-1 production in HUVECs. Cells were transfected with siRNA specific to APPL1 (siAPPL1) or scrambled control and then treated with insulin (50 nmol/L) for 10 min. Protein (40 μg) from total cell lysates was resolved by SDS-PAGE and probed for total ERK1/2 or phospho-ERK1/2 (A), total MEK1/2 and phospho-MEK1/2 (B), and total Raf-1 and phospho–Raf-1 at either Ser338 (C) or Ser259 (D). ET-1 concentration in the conditioned medium was measured 12 h after insulin treatment (E). T, total; p, phosphorylated. *P < 0.05, **P < 0.01 (n = 5–7).
DISCUSSION
APPL1 in the control of vasodilator and vasoconstrictor effects of insulin.
Vascular actions of insulin play an important role in maintaining both hemodynamic and metabolic homeostasis under healthy conditions. In insulin-resistant states, the selective impairment in the PI 3-K/Akt/eNOS pathway and the augmented ERK1/2 signaling cascade in vascular endothelium lead to decreased NO availability and enhanced ET-1 production, thereby tilting the balance between the vasodilator and vasoconstrictor actions of insulin toward endothelial dysfunction and hypertension (6). However, the cellular mechanisms that regulate the balance of the two major branches of insulin-induced signaling pathways remain poorly characterized. The current study provides both in vivo and ex vivo evidence demonstrating that APPL1 is a key intracellular adaptor protein that coordinates the two major insulin-evoked signaling cascades leading to the production of NO and ET-1 in endothelial cells. Transgenic expression of APPL1 is sufficient to reverse both obesity- and ageing-induced imbalances between insulin-induced Akt-dependent NO production and ERK1/2-dependent ET-1 release. On the other hand, APPL1 deficiency impairs insulin-induced vasodilatation and augments the vasoconstrictor effect of the hormone. These findings raise the possibility that reduced APPL1 expression, as observed in mesenteric arteries of obese mice and rats (14,16), is causally associated with vascular insulin resistance and endothelial dysfunction.
The role of insulin as a vasodilator has been well documented in both animals (1) and humans (22). However, the net vascular outcome of insulin administration depends on a balance between its vasodilator and vasoconstrictor effects. In fact, a number of earlier studies failed to demonstrate acute stimulation of NO-dependent relaxations in isolated arteries by insulin (23). In this connection, the current study demonstrates a potent effect of insulin in causing NO-dependent relaxations in mesenteric arteries of young C57 mice, whereas this effect is progressively blunted with ageing and obesity. Furthermore, a vasoconstrictor effect of higher concentrations of the hormone becomes predominant when obesity progresses to a more severe form. The switch from the vasodilator to vasoconstrictor effects of insulin in obese mice is accompanied by blunted activation of the Akt/eNOS signaling cascade, augmented ERK1/2 signaling, and elevated ET-1 production. These findings further support the notion that the imbalance between insulin-evoked production of NO and ET-1 is an important contributor to insulin resistance and endothelial dysfunction induced by obesity and may explain some of the discrepancies in the literature concerning the direct vascular effects of insulin.
Potentiation of insulin-induced Akt/eNOS signaling by APPL1.
As an intracellular adaptor protein with multiple domains involved in cellular signaling, APPL1 has been found to interact with a number of cell surface receptors—TrkA (24,25), adiponectin (14,16), and follicle-stimulating hormone (26)—and the intracellular signaling molecules small GTPase Rab5 (27), GIPC (25), inositol 5-phosphatase (28), and Akt (10). The interaction of APPL1 with adiponectin receptors is essential in mediating the insulin-sensitizing actions of the adipokine in skeletal muscle (12) and endothelial cells (14). Furthermore, APPL1 potentiates insulin-induced Akt activation in its several metabolic targets, including adipocytes (29), muscle cells (12), and hepatocytes (13). In hepatocytes, APPL1 potentiates insulin-induced Akt activation by competing with the intracellular pseudokinase TRB3 for binding to Akt (13). In unstimulated cells, Akt preferentially interacts with TRB3, which serves as an endogenous inhibitor of Akt by trapping it in the cytosol (21). Upon insulin stimulation, APPL1 replaces TRB3 for binding to Akt, thereby promoting Akt translocation to the plasma membrane and endosome for further activation. In the liver tissue of db/db mice with type 2 diabetes, the interaction between APPL1 and Akt is reduced, whereas the association of Akt with TRB3 is augmented, and these changes may represent a key mechanism accounting for hepatic insulin resistance in mice (13). Noticeably, TRB3 suppresses insulin-induced activation of Akt and phosphorylation of eNOS at Ser1177 and thereby attenuates NO production in HUVECs (30). In the current study, we demonstrated that siRNA-mediated knockdown of APPL1 expression causes an increase in Akt-TRB3 interaction and a concurrent decrease in Akt/eNOS/NO signaling. Furthermore, we found that APPL1 expression in mesenteric arteries is decreased in obesity, whereas TRB3 expression is increased. Taken in conjunction, these findings suggest that potentiation of insulin-induced Akt/eNOS/NO signaling and vasodilatation by APPL1 is attributed partly to its ability to compete with the endogenous Akt inhibitor TRB3 in the endothelium.
In addition to quenching the inhibitory effects of TRB3 on Akt activation, APPL1 may also play a role in modulating insulin-induced activation of PI 3-K. APPL1 possesses phosphoinositide-binding properties (31,32) and also interacts with both the regulatory (p85) and the catalytic (p110) subunit of PI 3-K (10). Therefore, APPL1 may act as a dynamic scaffold protein to recruit PI 3-K and its substrate phosphoinositide in a specialized complex to facilitate the signal transmission. Likewise, a recent study demonstrates that APPL1 is a key mediator in coupling the follicle-stimulating hormone receptor to inositol 1,4,5-trisphosphate production in human granulosa cells (33).
Suppression of insulin-induced ET-1 production by APPL1.
Although APPL1 interacts with several components of the PI 3-K/Akt signaling cascade, our coimmunoprecipitation and proteomic analysis found that it does not bind to any molecule known to be involved in the ERK1/2 MAPK signaling pathway, suggesting that the suppressive effect of APPL1 on insulin-induced MAPK in endothelial cells may be attributed to an indirect mechanism, secondary to Akt activation. Indeed, selective inhibition of PI 3-K in human endothelial cells has been shown to enhance insulin-induced ERK1/2 MAPK activation (34). Akt has been reported to inactivate Raf-1 through phosphorylation at Ser259 (35). The Raf-1 kinase lies at the heart of the ERK1/2 MAPK signaling cascade, and its activity is regulated by phosphorylation at multiple sites (36). Phosphorylation of Raf-1 at Ser259 negatively regulates the Raf-1–MEK1/2–ERK1/2 signaling by suppressing the phosphorylation of Raf-1 at its activation site Ser338 (36). In the current study, we found that siRNA-mediated knockdown of APPL1 expression suppressed phosphorylation of Raf-1 at its inhibitory site Ser259 but augmented insulin-induced Raf-1 phosphorylation at its activation site Ser338. Furthermore, we also observed a similar change in the pattern of Raf-1 phosphorylation in endothelial cells pretreated with the PI 3-K inhibitor LY294002. Taken together, these findings imply that the suppression of insulin-induced ERK1/2 MAPK signaling by APPL1 in endothelial cells is mediated by the direct cross talk between Akt and Raf-1, which in turn switches the phosphorylation of Raf-1 from its active form to the inhibitory status in vascular endothelium.
In summary, both in vivo and in vitro evidence from the current study support a key role of APPL1 in modulating the vascular actions of insulin by controlling the balance between insulin-induced Akt-dependent NO production and ERK1/2 MAPK–dependent ET-1 expression in endothelial cells (Fig. 8). Because an imbalance between the vasodilator and vasoconstrictor effects of insulin is an important contributor to vascular insulin resistance, endothelial dysfunction, and hypertension, APPL1 may represent an attractive therapeutic target for the treatment of both metabolic and cardiovascular diseases.
FIG. 8.

A schematic representation of the proposed molecular pathways whereby APPL1 enhances insulin-evoked Akt-dependent NO production and blocks insulin-induced ERK1/2-dependent ET-1 expression in endothelial cells. IRS, insulin receptor substrate; MEK1/2, mitogen-activated protein kinase kinase 1/2. (A high-quality color representation of this figure is available in the online issue.)
ACKNOWLEDGMENTS
A.X. has received a grant from the general research fund of the Research Grants Council of Hong Kong (HKU 779707M). This study has received support from the collaborative research fund of the Research Grants Council of Hong Kong (HKU 2/07C and HKU4/CRF/10) and the General Program of the National Natural Science Foundation of China (NSF 30771024 and 30811120429).
No potential conflicts of interest relevant to this article were reported.
Y.W. and K.K.Y.C. researched and analyzed data. K.S.L.L. supervised the study and edited the manuscript. D.W. generated the APPL1 KO mice. Y.W., Y.H., and P.M.V. supervised the study and edited the manuscript. G.S. researched data and edited the manuscript. Y.L. designed and supervised the study. A.X. designed and supervised the study and wrote the manuscript.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db11-0666/-/DC1.
REFERENCES
- 1.Muniyappa R, Montagnani M, Koh KK, Quon MJ. Cardiovascular actions of insulin. Endocr Rev 2007;28:463–491 [DOI] [PubMed] [Google Scholar]
- 2.Cardillo C, Nambi SS, Kilcoyne CM, et al. Insulin stimulates both endothelin and nitric oxide activity in the human forearm. Circulation 1999;100:820–825 [DOI] [PubMed] [Google Scholar]
- 3.Zeng G, Nystrom FH, Ravichandran LV, et al. Roles for insulin receptor, PI3-kinase, and Akt in insulin-signaling pathways related to production of nitric oxide in human vascular endothelial cells. Circulation 2000;101:1539–1545 [DOI] [PubMed] [Google Scholar]
- 4.Eringa EC, Stehouwer CD, van Nieuw Amerongen GP, Ouwehand L, Westerhof N, Sipkema P. Vasoconstrictor effects of insulin in skeletal muscle arterioles are mediated by ERK1/2 activation in endothelium. Am J Physiol Heart Circ Physiol 2004;287:H2043–H2048 [DOI] [PubMed] [Google Scholar]
- 5.Ko SH, Cao W, Liu Z. Hypertension management and microvascular insulin resistance in diabetes. Curr Hypertens Rep 2010;12:243–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang PL. eNOS, metabolic syndrome and cardiovascular disease. Trends Endocrinol Metab 2009;20:295–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bourgoin F, Bachelard H, Badeau M, et al. Endothelial and vascular dysfunctions and insulin resistance in rats fed a high-fat, high-sucrose diet. Am J Physiol Heart Circ Physiol 2008;295:H1044–H1055 [DOI] [PubMed] [Google Scholar]
- 8.Kubota T, Kubota N, Kumagai H, et al. Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell Metab 2011;13:294–307 [DOI] [PubMed] [Google Scholar]
- 9.Pistrosch F, Passauer J, Fischer S, Fuecker K, Hanefeld M, Gross P. In type 2 diabetes, rosiglitazone therapy for insulin resistance ameliorates endothelial dysfunction independent of glucose control. Diabetes Care 2004;27:484–490 [DOI] [PubMed] [Google Scholar]
- 10.Mitsuuchi Y, Johnson SW, Sonoda G, Tanno S, Golemis EA, Testa JR. Identification of a chromosome 3p14.3-21.1 gene, APPL, encoding an adaptor molecule that interacts with the oncoprotein-serine/threonine kinase AKT2. Oncogene 1999;18:4891–4898 [DOI] [PubMed] [Google Scholar]
- 11.Wang C, Xin X, Xiang R, et al. Yin-Yang regulation of adiponectin signaling by APPL isoforms in muscle cells. J Biol Chem 2009;284:31608–31615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mao X, Kikani CK, Riojas RA, et al. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat Cell Biol 2006;8:516–523 [DOI] [PubMed] [Google Scholar]
- 13.Cheng KK, Iglesias MA, Lam KS, et al. APPL1 potentiates insulin-mediated inhibition of hepatic glucose production and alleviates diabetes via Akt activation in mice. Cell Metab 2009;9:417–427 [DOI] [PubMed] [Google Scholar]
- 14.Cheng KK, Lam KS, Wang Y, et al. Adiponectin-induced endothelial nitric oxide synthase activation and nitric oxide production are mediated by APPL1 in endothelial cells. Diabetes 2007;56:1387–1394 [DOI] [PubMed] [Google Scholar]
- 15.Chandrasekar B, Boylston WH, Venkatachalam K, Webster NJ, Prabhu SD, Valente AJ. Adiponectin blocks interleukin-18-mediated endothelial cell death via APPL1-dependent AMP-activated protein kinase (AMPK) activation and IKK/NF-kappaB/PTEN suppression. J Biol Chem 2008;283:24889–24898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmid PM, Resch M, Steege A, et al. Globular and full-length adiponectin induce NO-dependent vasodilation in resistance arteries of Zucker lean but not Zucker diabetic fatty rats. Am J Hypertens 2011;24:270–277 [DOI] [PubMed] [Google Scholar]
- 17.Kagawa S, Soeda Y, Ishihara H, et al. Impact of transgenic overexpression of SH2-containing inositol 5′-phosphatase 2 on glucose metabolism and insulin signaling in mice. Endocrinology 2008;149:642–650 [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Huang Y, Lam KS, et al. Berberine prevents hyperglycemia-induced endothelial injury and enhances vasodilatation via adenosine monophosphate-activated protein kinase and endothelial nitric oxide synthase. Cardiovasc Res 2009;82:484–492 [DOI] [PubMed] [Google Scholar]
- 19.Andrawis N, Jones DS, Abernethy DR. Aging is associated with endothelial dysfunction in the human forearm vasculature. J Am Geriatr Soc 2000;48:193–198 [PubMed] [Google Scholar]
- 20.Cardillo C, Campia U, Iantorno M, Panza JA. Enhanced vascular activity of endogenous endothelin-1 in obese hypertensive patients. Hypertension 2004;43:36–40 [DOI] [PubMed] [Google Scholar]
- 21.Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science 2003;300:1574–1577 [DOI] [PubMed] [Google Scholar]
- 22.Steinberg HO, Brechtel G, Johnson A, Fineberg N, Baron AD. Insulin-mediated skeletal muscle vasodilation is nitric oxide dependent. A novel action of insulin to increase nitric oxide release. J Clin Invest 1994;94:1172–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Randriamboavonjy V, Schrader J, Busse R, Fleming I. Insulin induces the release of vasodilator compounds from platelets by a nitric oxide-G kinase-VAMP-3-dependent pathway. J Exp Med 2004;199:347–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin DC, Quevedo C, Brewer NE, et al. APPL1 associates with TrkA and GIPC1 and is required for nerve growth factor-mediated signal transduction. Mol Cell Biol 2006;26:8928–8941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Varsano T, Dong MQ, Niesman I, et al. GIPC is recruited by APPL to peripheral TrkA endosomes and regulates TrkA trafficking and signaling. Mol Cell Biol 2006;26:8942–8952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nechamen CA, Thomas RM, Cohen BD, et al. Human follicle-stimulating hormone (FSH) receptor interacts with the adaptor protein APPL1 in HEK 293 cells: potential involvement of the PI3K pathway in FSH signaling. Biol Reprod 2004;71:629–636 [DOI] [PubMed] [Google Scholar]
- 27.Miaczynska M, Christoforidis S, Giner A, et al. APPL proteins link Rab5 to nuclear signal transduction via an endosomal compartment. Cell 2004;116:445–456 [DOI] [PubMed] [Google Scholar]
- 28.Erdmann KS, Mao Y, McCrea HJ, et al. A role of the Lowe syndrome protein OCRL in early steps of the endocytic pathway. Dev Cell 2007;13:377–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saito T, Jones CC, Huang S, Czech MP, Pilch PF. The interaction of Akt with APPL1 is required for insulin-stimulated Glut4 translocation. J Biol Chem 2007;282:32280–32287 [DOI] [PubMed] [Google Scholar]
- 30.Andreozzi F, Formoso G, Prudente S, et al. TRIB3 R84 variant is associated with impaired insulin-mediated nitric oxide production in human endothelial cells. Arterioscler Thromb Vasc Biol 2008;28:1355–1360 [DOI] [PubMed] [Google Scholar]
- 31.Chial HJ, Wu R, Ustach CV, McPhail LC, Mobley WC, Chen YQ. Membrane targeting by APPL1 and APPL2: dynamic scaffolds that oligomerize and bind phosphoinositides. Traffic 2008;9:215–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J, Mao X, Dong LQ, Liu F, Tong L. Crystal structures of the BAR-PH and PTB domains of human APPL1. Structure 2007;15:525–533 [DOI] [PubMed] [Google Scholar]
- 33.Thomas RM, Nechamen CA, Mazurkiewicz JE, Ulloa-Aguirre A, Dias JA. The adapter protein APPL1 links FSH receptor to inositol 1,4,5-trisphosphate production and is implicated in intracellular Ca(2+) mobilization. Endocrinology 2011;152:1691–1701 [DOI] [PMC free article] [PubMed]
- 34.Montagnani M, Golovchenko I, Kim I, et al. Inhibition of phosphatidylinositol 3-kinase enhances mitogenic actions of insulin in endothelial cells. J Biol Chem 2002;277:1794–1799 [DOI] [PubMed] [Google Scholar]
- 35.Zimmermann S, Moelling K. Phosphorylation and regulation of Raf by Akt (protein kinase B). Science 1999;286:1741–1744 [DOI] [PubMed] [Google Scholar]
- 36.Dhillon AS, Meikle S, Yazici Z, Eulitz M, Kolch W. Regulation of Raf-1 activation and signalling by dephosphorylation. EMBO J 2002;21:64–71 [DOI] [PMC free article] [PubMed] [Google Scholar]