

Abstract
OBJECTIVE
To investigate deep and comprehensive analysis of gut microbial communities and biological parameters after prebiotic administration in obese and diabetic mice.
RESEARCH DESIGN AND METHODS
Genetic (ob/ob) or diet-induced obese and diabetic mice were chronically fed with prebiotic-enriched diet or with a control diet. Extensive gut microbiota analyses, including quantitative PCR, pyrosequencing of the 16S rRNA, and phylogenetic microarrays, were performed in ob/ob mice. The impact of gut microbiota modulation on leptin sensitivity was investigated in diet-induced leptin-resistant mice. Metabolic parameters, gene expression, glucose homeostasis, and enteroendocrine-related L-cell function were documented in both models.
RESULTS
In ob/ob mice, prebiotic feeding decreased Firmicutes and increased Bacteroidetes phyla, but also changed 102 distinct taxa, 16 of which displayed a >10-fold change in abundance. In addition, prebiotics improved glucose tolerance, increased L-cell number and associated parameters (intestinal proglucagon mRNA expression and plasma glucagon-like peptide-1 levels), and reduced fat-mass development, oxidative stress, and low-grade inflammation. In high fat–fed mice, prebiotic treatment improved leptin sensitivity as well as metabolic parameters.
CONCLUSIONS
We conclude that specific gut microbiota modulation improves glucose homeostasis, leptin sensitivity, and target enteroendocrine cell activity in obese and diabetic mice. By profiling the gut microbiota, we identified a catalog of putative bacterial targets that may affect host metabolism in obesity and diabetes.
Obesity and related metabolic disorders are closely associated with a low-grade inflammatory state (1). Growing evidence also demonstrates that the gut microbiota plays a critical role in the development of obesity, type 2 diabetes, and insulin resistance (2–9). Given that the total number of bacteria in the gut is estimated at ∼1014, it has been proposed that the genome size of this exteriorized organ largely exceeds the human genome size (10,11). However, the composition of the gut microbiota and the exact role of microorganisms present in the gut remain poorly defined. Nonetheless, advances in metagenomic methods for characterizing microbial diversity have helped to evaluate the functional contribution of this large collection of microbes to host metabolism (12). For instance, recent evidence suggests that changes in gut microbiota composition may play a critical role in the development of obesity-associated inflammation (7,8,13,14). Accordingly, obesity-associated, low-grade inflammation may be related to the gut microbiota by mechanisms involving bacterially derived lipopolysaccharide (LPS) (6–8,14–16).
In light of these recent findings, understanding the role of microbial communities and identifying molecular targets related to metabolism regulation are of the utmost importance. Addressing these issues is challenging because of the lack of well-characterized models. Nevertheless, germ-free animals have led to striking and unequivocal findings regarding the role of gut microbiota in host energy metabolism (3,17,18). In addition to these highly specific models, approaches that are more generously applicable, including prebiotic-induced modulation of the gut microbiota, have been developed and widely used. Prebiotics are nondigestible carbohydrates that beneficially affect host health by selectively stimulating the growth and/or activity of a limited number of bacteria (e.g., bifidobacteria and lactobacilli) (19). We have previously shown that prebiotics improve gut barrier function and alleviate inflammation and insulin resistance associated with obesity by increasing the release of gut hormones, such as glucagon-like peptide 1 and 2 (GLP-1 and GLP-2), and by modulating the endocannabinoid system (8,15,20). Although the beneficial effects of prebiotics have been linked to a concomitant effect on Bifidobacteriaceae, no clear causal relationship has been established between this family and their beneficial metabolic effects (15,21). Thus, to obtain a more deterministic analysis of the gut microbiota, we combined multiple molecular methods, including quantitative PCR (qPCR), barcoded pyrosequencing, and phylogenetic microarrays of 16S rRNA, to generate comprehensive microbial community profiles of obese mice with or without prebiotics in their diet. Furthermore, we identified novel mechanisms by which prebiotics change obesity-associated metabolic disorders in both genetic and diet-induced leptin-resistant mice.
RESEARCH DESIGN AND METHODS
Mice
Ob/ob experiments.
Six-week-old ob/ob (n = 10/group) mice (C57BL/6 background; Jackson Laboratory, Bar Harbor, ME) were housed in a controlled environment (12-h daylight cycle; lights off at 6:00 p.m.) in groups of two mice per cage, with free access to food and water. The mice were fed a control diet (Ob-CT) (A04, Villemoisson-sur-Orge, France) or a control diet supplemented with prebiotics, such as oligofructose (Ob-Pre) (Orafti, Tienen, Belgium) for 5 weeks as previously described (8,15). A second set of mice was provided with the same dietary treatments to investigate tight-junction proteins and body composition.
High-fat diet experiments.
A set of 10-week-old C57BL/6J mice (40 mice; n = 10/group) (Charles River, Brussels, Belgium) were housed in groups of five mice per cage, with free access to food and water. All of the mice were fed a high-fat (HF) diet (60% fat and 20% carbohydrates [kcal/100 g], D12492; Research Diets, Inc., New Brunswick, NJ) or an HF diet supplemented with oligofructose (0.3 g/mouse/day) added in tap water (HF-Pre). Treatment continued for 8 weeks. Food and water intake were recorded twice a week. Body composition was assessed by using a 7.5-MHz time-domain nuclear magnetic resonance (LF50 minispec; Bruker, Rheinstetten, Germany).
All mouse experiments were approved by and performed in accordance with the guidelines of the local ethics committee. Housing conditions were specified by the Belgian Law of 6 April 2010, regarding the protection of laboratory animals (Agreement LA1230314).
Leptin sensitivity measurement.
After 5 weeks of HF or HF-Pre treatment (n = 10/group), mice were individually housed 5 days prior to drug treatment. Mice were divided into two groups that received intraperitoneal injections for 4 days. The first 2 days, all of the mice received twice-daily intraperitoneal saline and the last 2 days intraperitoneal recombinant leptin (750 μg/kg/day) (Bachem, Bubendorf, Switzerland). Body weights and food intake were measured daily. After 1 week of recovery, mice were fasted for 6 h, treated with either saline (n = 4/group) or leptin (n = 6/group) (1 mg/kg), and killed 6 h later.
Tissue sampling.
Mice were anesthetized by intraperitoneal injection of 100 mg/kg ketamine and 10 mg/kg xylazine or by isoflurane gas (Forene; Abbott Laboratories, Queenborough, U.K.) after a 6-h fasting period. Blood samples and tissues were harvested for further analysis. Mice were killed by cervical dislocation. Epididymal, subcutaneous, and visceral adipose deposits and muscles (Vastus lateralis) were precisely dissected and weighed. The intestinal segments (jejunum and colon) and adipose tissues were immediately immersed in liquid nitrogen and stored at −80°C for further analysis.
Immunofluorescent analysis of occludin, zonula occludens-1, and L cells.
Jejunum and colon segments were immediately removed, washed with PBS, mounted in embedding medium (Tissue-Tek; Sakura, Zoeterwoude, the Netherlands), and stored (−80°C) until use. The expression of occludin and zonula occludens-1 (ZO-1) tight-junction proteins was assessed as previously described (8). The L-cell number was determined with rabbit anti–GLP-1 (1:200; Abcam, Cambridge, U.K.). Four to eight fields were used to quantify the L-cell number for each intestinal segment. Using the same material, the mucosal area was manually delineated by the investigator and measured by an image analyzer (Motic Image Plus 2.0ML; Xiamen, China). The results are expressed as the L-cell number per mucosal area (number/millimeter squared). Both L-cell and tight-junction determinations were analyzed in double-blind fashion by two different investigators.
Oral glucose tolerance test.
Oral glucose tolerance tests were performed after 4 weeks (ob/ob study) or 6 weeks (HF study). Food was removed 2 h after the onset of the daylight cycle, and mice were treated after a 6-h fasting period as previously described (6,7).
Biochemical analyses.
Muscle lipid content and the adipose tissue oxidative stress level were evaluated as previously described (6,8). Portal plasma LPS concentration was measured using Endosafe-MCS (Charles River Laboratories, Lyon, France) as previously described (22). Portal GLP-1 and glucose-dependent insulinotropic peptide (GIP) were determined in duplicate using a Bio-Plex Pro Assays kit (Bio-Rad, Nazareth, Belgium) and measured using Luminex (Bio-Rad Bioplex; Bio-Rad) according to the manufacturer’s instructions.
In vivo intestinal permeability.
Intestinal permeability was measured as described previously (8).
DNA isolation from mouse cecal samples.
The cecal content of mice collected post mortem was stored at −80°C. Metagenomic DNA was extracted from the cecal content using a QIAamp-DNA stool minikit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions.
qPCR: primers and conditions.
qPCR for total bacteria, Firmicutes, Bacteroidetes, Bifidobacterium spp., Lactobacillus spp., Roseburia spp., Eubacterium rectale/Clostridium coccoides group, and Bacteroides-Prevotella spp. was also performed as previously described (21).
Sequencing: PCR primers and conditions.
For each sample, we amplified the V1–3 region of the 16S rRNA gene corresponding to the Escherichia coli 16S rRNA gene positions 28–514, excluding primer sequences. The PCRs included 1 μL of 50× diluted, purified DNA as previously described (23). The composite PCR primers included: 1) the 454 Life Science 19-base adaptors A or B; 2) an eight-base, sample-specific barcode sequence (NNNNNNNN; designated 672–691 in Hamady et al. [24]; Supplementary Table 1); 3) the sequence of the broad-range 16S forward or reverse primer; and 4) a dinucleotide sequence introduced between the 16S primer and the barcode sequence designed to prevent pairing of different barcodes with rDNA targets. The products were generated and sequenced on a Genome Sequencer FLX system (Roche, Basel, Switzerland), as previously described (23). A total of 83,522 reads, which had a quality score <20, corresponded to cecal samples of 20 ob/ob mice presented in this study. Sequences containing uncalled bases, incorrect primer sequences, or runs of ≥10 identical nucleotides were removed. Reads with the 16S rDNA forward oligonucleotide sequence CCGCGRCTGCTGGCGC, containing G instead of A at the penultimate position of the 3′ end, were relatively frequent (60.7%). They are likely due to a primer synthesis or sequencing artifact (25) and were not removed from the dataset provided that other quality criteria were met. After trimming primer sequences, reads <200 nucleotides or >290 nucleotides and those that incompletely covered the Escherichia coli 16S rRNA gene positions 288–514 (determined using the Ribosomal Database Project [RDP] pyrosequencing tool Aligner [26]) were discarded, leaving 68,163 sequences. Informatic analyses were described as detailed previously (23).
Each distinct sequence was assigned to representative phylotypes at 100 or 97% identity (100–97% ID phylotype) using CD-HIT (27). Distances between phylotypes were calculated using MUSCLE (28) (with parameters maxiters 2 and diags). Hierarchical clustering and Principal Coordinates Analyses (PCoA) were carried out using UniFrac (29). The taxonomic composition was assigned using the RDP Classifier (30) with a 50% confidence cutoff. The sequences (68,163 reads) are publicly available at the MG-RAST repository (31) under ID 4449917.3.
Mouse Intestinal Tract Chip: PCR primers and conditions.
The Mouse Intestinal Tract Chip (MITChip) procedure was performed as previously described (23,32).
RNA preparation and real-time qPCR analysis.
Total RNA was prepared from tissues using TriPure reagent (Roche), and primer sequences for the targeted mouse genes were previously described (7,8,21). RPL19 was chosen as the housekeeping gene.
Statistical analyses.
Data are expressed as the mean ± SEM. Differences between two groups were assessed using two-tailed Student t test. Data were analyzed using GraphPad Prism 5.00 (GraphPad Software, San Diego, CA) and JMP 8.0.1 (SAS Institute, Inc., Cary, NC). The results were considered statistically significant at P < 0.05.
RESULTS
16S rRNA analysis of gut bacterial populations in prebiotic-treated obese mice: qPCR analyses.
We found that the prebiotic treatment significantly increased the abundance of Bifidobacterium spp. and the E. rectale/C. coccoides group (Supplementary Fig. 1A and B). In addition, the abundance of Firmicutes and Roseburia spp. decreased after the treatment (Supplementary Fig. 1C and D), whereas the abundance of Bacteroidetes, Lactobacillus spp., and the Bacteroides-Prevotella group and the total bacteria number were not affected by the treatment (Supplementary Fig. 1E–H).
16S rRNA analysis of gut bacterial populations in prebiotic-treated obese mice: pyrosequencing analysis.
We observed a significant phylum-wide shift between Bacteroidetes and Firmicutes, of which the abundance increased and decreased, respectively, after the prebiotic treatment, compared with the control (Fig. 1A and D). The abundance of Actinobacteria and Proteobacteria tended to increase in the prebiotic group (P = 0.07 and P = 0.051, respectively) (Fig. 1D).
FIG. 1.
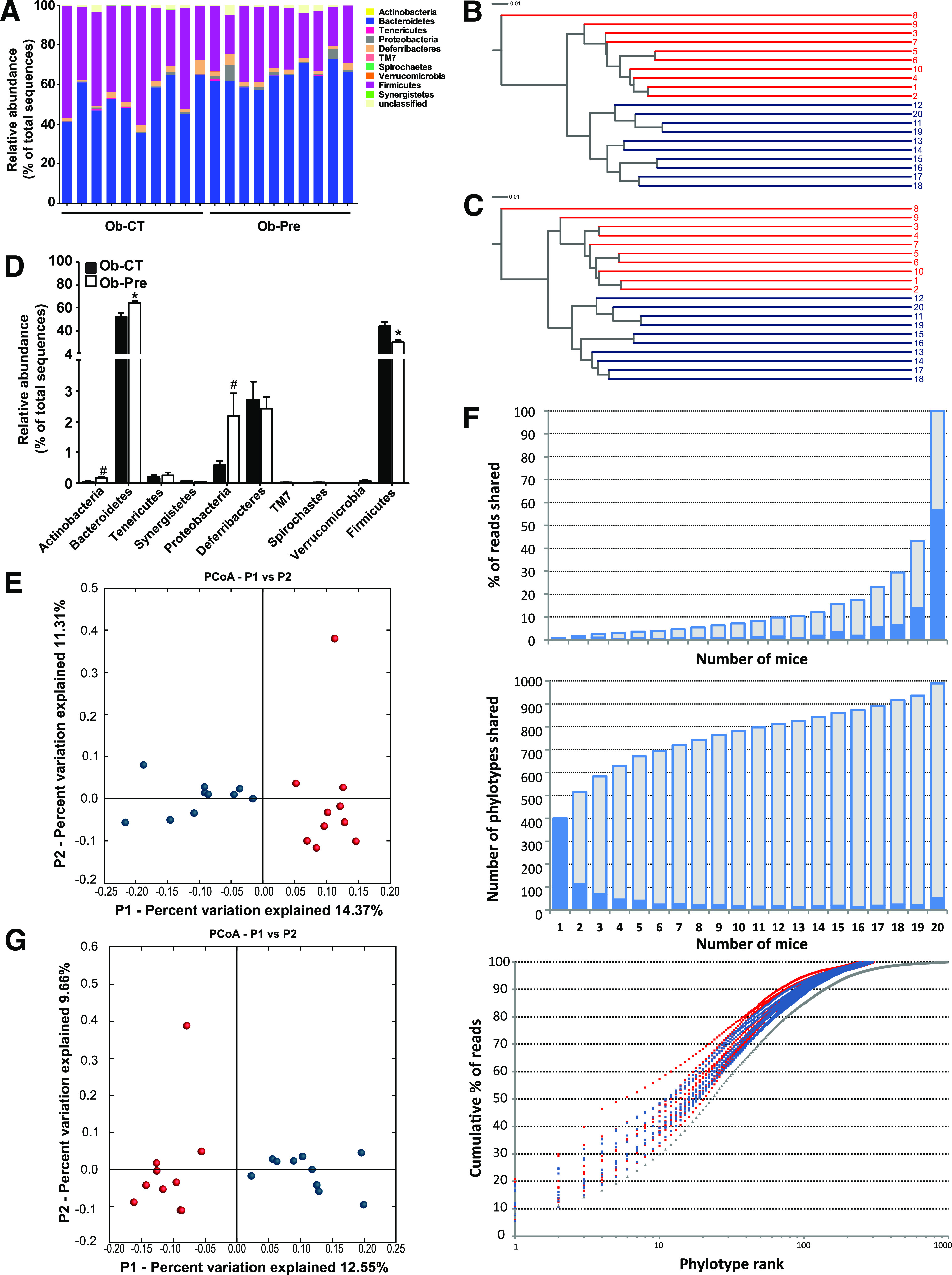
Analysis of the gut bacterial community by 16S rRNA pyrosequencing from obese mice on standard chow and prebiotic diets. A: Percentage of each community contributed by the indicated phyla. B: Clustering of mice cecal microbial communities in the two tested groups based on the unweighted UniFrac analysis and 97% ID phylotypes or (C) 100% ID phylotypes. Red corresponds to the standard chow diet (Ob-CT), and blue corresponds to the prebiotic diet (Ob-Pre). Branch length represents distance between environments in UniFrac units, indicated by the scale bar. D: Relative abundance of different phyla expressed as the percentage of total sequence reads. Mean ± SEM. n = 10 mice/group. *P < 0.05; #P < 0.1, determined by a two-tailed Student t test. PCoA based on the unweighted (presence/absence) UniFrac analysis and (E) 97% ID phylotypes or (G) 100% ID phylotypes. Each circle representing a single sample is colored according to the dietary conditions; red corresponds to Ob-CT and blue corresponds to Ob-Pre. F: Number of 97% ID phylotypes shared among a given number of mice (middle panel) and their corresponding abundance expressed as the percentage of total reads (top panel) indicated by blue bars. Cumulative data are indicated in gray. The bottom panel shows the relative abundance of 97% ID phylotypes, in which the x-axis indicates individual phylotypes ranked according to their relative abundance from high to low, and the y-axis indicates the cumulative abundance (the percentage of total reads). Gray triangles correspond to a pooled data set from 20 mice.
Here we identified 11 genera whose abundance was significantly affected by prebiotics (Table 1). Interestingly, the Bifidobacterium and Syntrophococcus genera were identified exclusively in the prebiotic group and the control group, respectively. A phylogenetic tree including phylotypes significantly affected by prebiotic intake shows (Supplementary Fig. 2) that many lineages are composed of members following the same trend in the change of their relative abundance (decrease or increase). Certain closely related 97% ID phylotypes belonging to the family Porphyromonadaceae appear to be confined to one of the two mice groups (Supplementary Fig. 2).
TABLE 1.
Phylogenetic analysis of the taxa enriched or depleted in prebiotic-fed mice using pyrosequencing
| Abundance (percent of total sequences) |
||||
|---|---|---|---|---|
| Rank | RDP classification | Ob-CT | Ob-Pre | Change (%)* |
| Subclass | Actinobacteria; Actinobacteria; Actinobacteridae | 0.006 ± 0.004 | 0.12 ± 0.05 | 1,971 |
| Order | Actinobacteria; Actinobacteria; Actinobacteridae; Bifidobacteriales | 0 ± 0 | 0.12 ± 0.05 | Ob-Pre |
| Family | Actinobacteria; Actinobacteria; Actinobacteridae; Bifidobacteriales; Bifidobacteriaceae | 0 ± 0 | 0.12 ± 0.05 | Ob-Pre |
| Genus | Actinobacteria; Actinobacteria; Actinobacteridae; Bifidobacteriales; Bifidobacteriaceae; Bifidobacterium | 0 ± 0 | 0.12 ± 0.05 | Ob-Pre |
| Phylum | Bacteroidetes | 52 ± 3 | 64 ± 2 | 24 |
| Class | Bacteroidetes; Bacteroidia | 52 ± 3 | 64 ± 2 | 23 |
| Order | Bacteroidetes; Bacteroidia; Bacteroidales | 52 ± 3 | 64 ± 2 | 23 |
| Family | Bacteroidetes; Bacteroidia; Bacteroidales; Prevotellaceae | 2.6 ± 0.5 | 4.1 ± 0.5 | 55 |
| Genus | Bacteroidetes; Bacteroidia; Bacteroidales; Prevotellaceae; Prevotella | 1.4 ± 0.2 | 2.4 ± 0.2 | 73 |
| Genus | Bacteroidetes; Bacteroidia; Bacteroidales; Porphyromonadaceae; Tannerella | 1.4 ± 0.2 | 1.9 ± 0.1 | 33 |
| Genus | Bacteroidetes; Bacteroidia; Bacteroidales; Porphyromonadaceae; Barnesiella | 8.5 ± 0.8 | 10.7 ± 0.6 | 26 |
| Phylum | Firmicutes | 44 ± 4 | 29 ± 2 | −33 |
| Class | Firmicutes; Clostridia; Clostridia | 42 ± 4 | 28 ± 2 | −34 |
| Order | Firmicutes; Clostridia; Clostridiales | 42 ± 4 | 27 ± 2 | −35 |
| Genus | Firmicutes; Clostridia; Clostridiales; Ruminococcaceae; Anaerofilum | 0.04 ± 0.01 | 0.003 ± 0.003 | −93 |
| Genus | Firmicutes; Clostridia; Clostridiales; Ruminococcaceae; Anaerotruncus | 1.0 ± 0.1 | 0.26 ± 0.04 | −74 |
| Genus | Firmicutes; Clostridia; Clostridiales; Ruminococcaceae; Subdoligranulum | 0.015 ± 0.007 | 0.07 ± 0.02 | 376 |
| Family | Lachnospiraceae | 33 ± 3 | 18 ± 2 | −44 |
| Genus | Firmicutes; Clostridia; Clostridiales; Lachnospiraceae; Syntrophococcus | 0.012 ± 0.005 | 0 ± 0 | Ob-CT |
| Genus | Firmicutes; Clostridia; Clostridiales; Lachnospiraceae; Marvinbryantia | 2.1 ± 0.6 | 0.8 ± 0.2 | −63 |
| Genus | Firmicutes; Clostridia; Clostridiales; Lachnospiraceae; Anaerostipes | 0.023 ± 0.008 | 0.003 ± 0.003 | −88 |
| Class | Proteobacteria; Betaproteobacteria | 0.18 ± 0.08 | 0.6 ± 0.1 | 259 |
| Order | Proteobacteria; Betaproteobacteria; Burkholderiales | 0.17 ± 0.08 | 0.6 ± 0.1 | 271 |
| Family | Proteobacteria; Betaproteobacteria; Burkholderiales; Alcaligenaceae | 0.17 ± 0.08 | 0.6 ± 0.1 | 269 |
| Genus | Proteobacteria; Betaproteobacteria; Burkholderiales; Alcaligenaceae; Parasutterella | 0.17 ± 0.08 | 0.6 ± 0.1 | 269 |
Data are mean ± SEM.
*P < 0.05, as determined by a two-tailed Student t test with equal variance. Ob-CT, found only in the control group; Ob-Pre, found only in prebiotic-fed mice.
We also found that a small percentage of species-level phylotypes (53 of 990, 5.4%) shared by all 20 investigated mice contributed to more than half (56.7%) of all reads in the pooled dataset (Fig. 1F and Supplementary Table 2). Likewise, 35 of 6,885 distinct sequences (0.5%) contributed to 36.7% of the total number of all sequence reads (Supplementary Table 3).
Supplementary Tables 4 and 5, respectively, show the significant differences in 69 phylotypes defined at 97% identity (97% ID phylotypes) or 102 distinct sequences (100% ID phylotypes) significantly enriched or depleted in the prebiotic-treated mice. Among these sequences, eight displayed a >10-fold increase, and another eight demonstrated a >10-fold decrease in average frequency in the Ob-Pre group. Importantly, both 97% ID and 100% ID phylotypes belonging to the genus Butyricimonas were observed in all mice in the Ob-Pre group but were absent in all control mice. Similarly, Barnesiella were 63-and 55-fold higher in the Ob-Pre group compared with the Ob-CT group, respectively. Furthermore, hierarchical clustering based on UniFrac analysis (29) clearly showed that the cecal communities of the prebiotic-treated mice were more similar to each other than to the communities of the 10 control mice (Fig. 1B and C). Moreover, PCoA of UniFrac-based pairwise comparisons of community structures revealed two clusters corresponding to the two dietary conditions (Fig. 1E and G). Finally, the average fraction of shared species-level phylotypes for paired samples was higher within the same group, compared with that between the two groups (62.3 + 0.3 vs. 58.9 + 0.3; P < 1012).
16S rRNA analysis of gut bacterial populations in prebiotic-treated obese mice: phylogenetic microarray analysis.
We also performed gut microbiota analysis using a high-throughput phylogenetic microarray, called MITChip (23,32), and previously compared with 454 pyrosequencing (23,33). The profiles of the cecal microbiota were obtained based on the intensity of 3,580 distinct oligonucleotide probes. The profiles visualized the presence or absence of all targeted operational taxonomic units. Hierarchical clustering analyses of the MITChip phylogenetic fingerprints showed separate clusters between the two treatment groups (Fig. 2A). A Monte Carlo permutation procedure indicated that the overall microbiota detected by MITChip of the control mice was significantly different from that of the prebiotic-treated mice (P = 0.002). Similar to the results obtained by pyrosequencing analysis, we observed a lower relative and absolute abundance of Firmicutes and a higher abundance of Bacteroidetes in the Ob-Pre group compared with the Ob-CT group (Fig. 2B and C). In contrast, the abundance of Actinobacteria was not significantly affected. In addition, a significant decrease in the abundance of Proteobacteria and the class Deltaproteobacteria was observed after the prebiotic treatment (Fig. 2C). Interestingly, the abundance of Verrucomicrobia dramatically increased in the Ob-Pre mice (Fig. 2C). Although the absolute abundance was still low, this increase was, on average, >80-fold higher than in the control mice (Table 2). Importantly, the specific species responsible for the increased abundance of Verrucomicrobia was identified as Akkermansia muciniphila (Table 2). In accordance with the pyrosequencing analysis, the different hierarchical clustering analyses and PCoA showed separate clusters corresponding to the dietary treatment (data not shown). In addition to specific changes observed in the pyrosequencing analyses, we found several previously unidentified modifications at level 2 (Table 2).
FIG. 2.
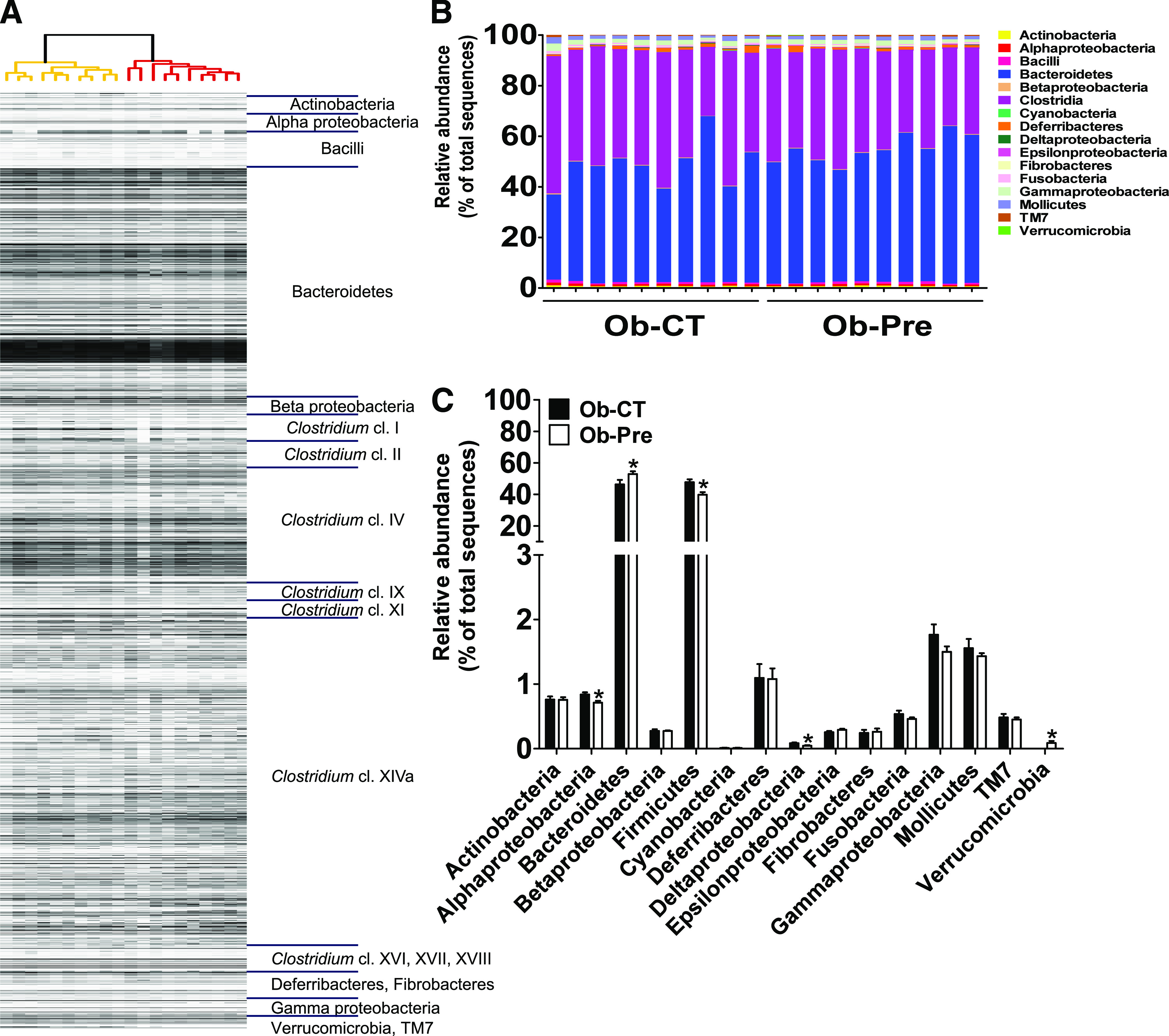
Phylogenetic microarray analysis of gut bacterial community from the Ob-CT and the Ob-Pre mice. A: Clustering of the MITChip phylogenetic fingerprints of the gut microbiota from the cecal content of the Ob-CT and the Ob-Pre mice (n = 10/group). The highest phylogenetic level of specificity of probes (level 1) is depicted on the right. B: Percentage of each community contributed by the indicated taxa. C: Relative abundance of different taxa expressed as the percentage of total sequence reads. Mean ± SEM. n = 10 mice/group. *P < 0.05, determined by a two-tailed Student t test.
TABLE 2.
Phylogenetic analysis of the gut microbiota upon prebiotic administration using MITChip
| Abundance (percent of total sequences) |
||||
|---|---|---|---|---|
| Phylum | Bacterial group | Ob-CT | Ob-Pre | Change (%)* |
| Mollicutes | Acholeplasma | 0.073 ± 0.008 | 0.049 ± 0.009 | −33.6 |
| Verrucomicrobia | A. muciniphila | 0.001 ± 0.0 | 0.089 ± 0.031 | 8,003.7 |
| Firmicutes | Allobaculum | 0.221 ± 0.045 | 0.103 ± 0.009 | −53.3 |
| Firmicutes | Anaerotruncus | 1.666 ± 0.199 | 0.984 ± 0.038 | −40.9 |
| Firmicutes | Anaerovorax | 0.048 ± 0.007 | 0.027 ± 0.002 | −44.8 |
| Proteobacteria | Bilophila | 0.019 ± 0.002 | 0.011 ± 0.002 | −42.2 |
| Firmicutes | Catenibacterium | 0.440 ± 0.056 | 0.316 ± 0.018 | −28.2 |
| Firmicutes | C. lactatifermentans | 0.175 ± 0.011 | 0.117 ± 0.009 | −32.9 |
| Firmicutes | Clostridium perfringens | 0.393 ± 0.021 | 0.507 ± 0.024 | 28.9 |
| Actinobacteria | Collinsella | 0.009 ± 0.001 | 0.007 ± 0.0003 | −18.9 |
| Proteobacteria | Desulfovibrio | 0.066 ± 0.009 | 0.033 ± 0.009 | −49.8 |
| Firmicutes | Eubacterium plexicaudatum | 3.030 ± 0.307 | 2.308 ± 0.149 | −23.8 |
| Firmicutes | Lachnospira pectinoschiza | 0.788 ± 0.057 | 0.589 ± 0.067 | −25.2 |
| Firmicutes | Lactococcus | 0.002 ± 0.0005 | 0.001 ± 0.0001 | −46.4 |
| Firmicutes | Peptococcus niger | 0.227 ± 0.022 | 0.453 ± 0.059 | 99.8 |
| Bacteroidetes | Rikenella | 0.735 ± 0.062 | 0.978 ± 0.037 | 33 |
| Firmicutes | S. intermedius | 0.005 ± 0.001 | 0.003 ± 0.0003 | −39.1 |
| Firmicutes | Turicibacter | 0.001 ± 0.0002 | 0.001 ± 0.0001 | −31.1 |
| Bacteroidetes | Uncultured Bacteroidetes | 0.003 ± 0.003 | 0.005 ± 0.0003 | 63.6 |
| Firmicutes | Unclassified Clostridium cluster I | 0.308 ± 0.112 | 1.030 ± 0.278 | 234.6 |
| Firmicutes | Unclassified Clostridium cluster II | 0.951 ± 0.076 | 0.679 ± 0.037 | −28.5 |
| Firmicutes | Unclassified Clostridium cluster XIVa | 14.331 ± 1.161 | 11.354 ± 0.581 | −20.8 |
| Firmicutes | Uncultured Clostridiales | 0.458 ± 0.053 | 0.304 ± 0.021 | −33.6 |
Bacterial groups that were changed significantly are listed. Data are mean ± SEM.
*P < 0.05, as determined by a two-tailed Student t test.
Prebiotics improve glucose and lipid metabolism in obese mice.
The changes in the gut microbiota composition were associated with significantly lower fasting glycemia and markedly improved glucose tolerance (Fig. 3A). However, it should be noted that body weight was not significantly affected by the treatment (body weight [g]: Ob-CT 46.79 ± 1.28, Ob-Pre 43.06 ± 1.58; P = 0.1), whereas fat mass (Fig. 3B) and cumulative food intake (g) (Ob-CT 466.8 ± 13.8, Ob-Pre 319.6 ± 20.6; P = 0.00034) were significantly lower than Ob-CT. In contrast, muscle mass significantly increased (Fig. 3C). Overall, these data indicate a decreased fat to muscle mass ratio in the Ob-Pre group. Interestingly, plasma triglycerides (Fig. 3D) and muscle lipid (total, triglycerides, and phospholipids) content were dramatically reduced in the prebiotic-treated mice (Fig. 3E and F) (nanogram of phospholipids per microgram of tissue: Ob-CT 29.05 ± 2.55, Ob-Pre 20.05 ± 2.49; P = 0.02). In addition, we found that prebiotic treatment significantly increased muscle lipoprotein lipase mRNA expression (about 70%) (Fig. 3G). This increase may be one of the mechanisms leading to the reduced plasma and muscle lipid content observed in Ob-Pre mice. Further supporting the link between oxidative stress and metabolic disturbances, we found that the prebiotic treatment reduced the adipose tissue lipid peroxide content by ∼50% (Fig. 3H). Moreover, multivariate analyses suggested that metabolic footprints (e.g., the content of plasma triglycerides and fat deposit lipid peroxides) can be used as potential biomarkers of glucose tolerance (Supplementary Fig. 3).
FIG. 3.

Changes in the gut microbiota improve glucose tolerance and reduce plasma triglyceride content, tissue weight, oxidative stress, and muscle lipid accumulation. A: Plasma glucose profile following 1 g/kg glucose oral challenge in freely moving mice. Inset shows the mean area under the curve (AUC) measured between 0 and 120 min after glucose load in the Ob-CT (■) and the Ob-Pre (○) mice. Mean ± SEM. n = 10 mice/group. *P < 0.05, determined by a two-tailed Student t test. B: White adipose tissue weight expressed as the percentage of total body weight of the Ob-CT and Ob-Pre mice. Mean ± SEM. n = 8 mice/group. *P < 0.05, determined by a two-tailed Student t test. C: Muscle weight (Vastus lateralis) expressed as the percentage of total body weight. D: Plasma triglyceride content. E: Muscle lipid content. F: Muscle triglycerides. G: Muscle lipoprotein lipase (LPL) mRNA expression. H: Adipose tissue lipid peroxidation markers (TBARS). I: Plasma LPS levels in the same set of mice. Mean ± SEM. n = 10 mice/group. *P < 0.05, determined by a two-tailed Student t test.
Prebiotics reduce plasma LPS and improve gut barrier function.
We have previously found that prebiotic feeding improves gut barrier function (8). Here, we found that Ob-Pre mice exhibited twofold lower plasma LPS levels (Fig. 3I) and fluorescein isothiocyanate (FITC)–dextran levels (Supplementary Fig. 4A) than Ob-CT mice. In accordance with these and our previous findings, we found that prebiotic treatment improved jejunum ZO-1 and occludin distribution (Supplementary Fig. 4B and C). In addition to these findings, we found that the prebiotic treatment significantly reduced the expression of oxidative stress (NADPHoxidase) and inflammatory (IL-1) mRNA markers in the colon (Table 3).
TABLE 3.
Changes in the gut microbiota upon prebiotic administration impacts on cecal, colon weight, and inflammatory markers
| Ob-CT | Ob-Pre | |
|---|---|---|
| Full cecum (g/100 g body wt) | 1.1 ± 0.13 | 1.52 ± 0.09* |
| Empty cecum (g/100 g body wt) | 0.27 ± 0.03 | 0.34 ± 0.02* |
| Colon weight (g/100 g body wt) | 0.46 ± 0.02 | 0.57 ± 0.02* |
| Colon length (cm) | 8.14 ± 0.24 | 9.63 ± 0.16* |
| Colon NADPHox mRNA levels | 1.0 ± 0.09 | 0.75 ± 0.05* |
| Colon IL-1 mRNA levels | 1.0 ± 0.14 | 0.62 ± 0.09* |
Data are mean ± SEM.
*P < 0.05, as determined by a two-tailed Student t test. NADPHox, NADPHoxidase.
Importantly, multiple correlation analyses revealed that plasma FITC–dextran levels, glucose intolerance, plasma triglycerides, and muscle lipid content were positively or negatively correlated with the abundance of several genera (Supplementary Table 6).
Prebiotic-induced changes in gut microbiota are associated with increased enteroendocrine L-cell number in obese mice.
Several lines of evidence suggest that prebiotic treatment promotes the production of GLP-1 and GLP-2 by enteroendocrine L cells (8,16,20,34,35). However, the exact contribution of the gut microbiota modulation associated with prebiotic treatment to L-cell number in obese mice is unclear. Strikingly, the prebiotic-treated mice exhibited a twofold increase in the L-cell number in the colon (Fig. 4C) and a similar increase in the proglucagon mRNA level (Fig. 4B). L-cell number and proglucagon mRNA level similarly increased in the jejunum after the prebiotic-induced gut microbiota modulation (Supplementary Fig. 5A–C). In accordance with these findings, we found that prebiotic feeding increases portal plasma GLP-1 levels (Fig. 4A), whereas GIP tends to decrease in Ob-pre mice (Ob-CT 280.4 ± 42.9, Ob-Pre 204.7 ± 15.8; P = 0.1). Given that the prebiotic treatment significantly increased colon weight and length (Table 3), it is likely that this effect is attributed to a greater pool of L cells within the intestine.
FIG. 4.
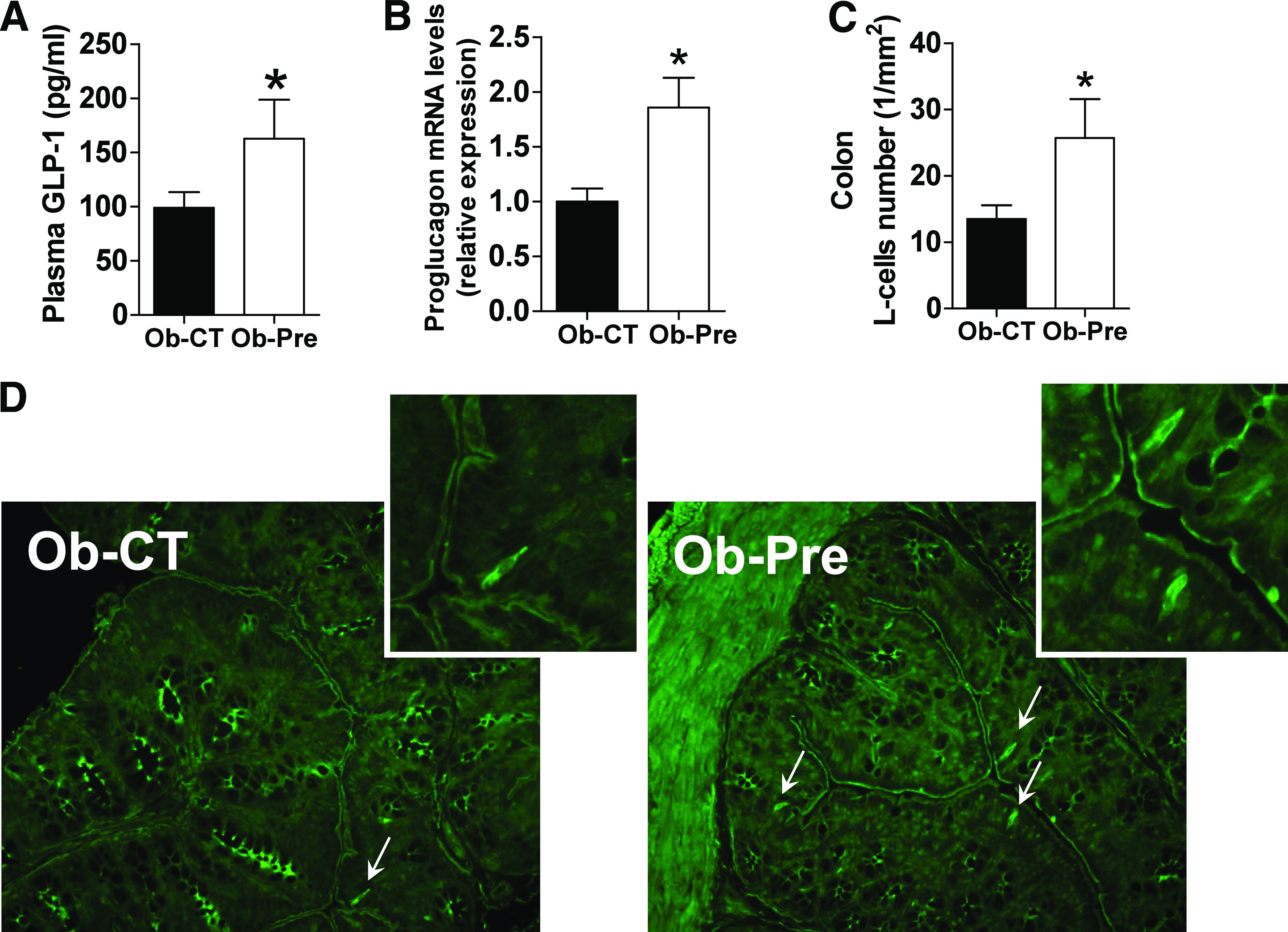
Prebiotic-induced changes in gut microbiota are associated with increased enteroendocrine L-cell number in obese mice. A: Portal plasma GLP-1 levels. B: Proglucagon mRNA expression measured in the colon. Mean ± SEM. n = 10 mice/group. *P < 0.05, determined by a two-tailed Student t test. C: L-cell number expressed per mm2 of colon. D: Representative immunofluorescence staining of L cells using a GLP-1 antibody. Mean ± SEM. n = 4–6 mice/group. *P < 0.05, determined by a two-tailed Student t test. (A high-quality digital representation of this figure is available in the online issue.)
Next, we performed pairwise correlation analyses to obtain a broader view of the intestinal responses regarding the L-cell number due to microbiome-wide variation observed by means of pyrosequencing and phyloarray. By combining the two approaches, we found that the abundance of 25 taxa was correlated with the L-cell number (Supplementary Fig. 6A and B).
Prebiotic-induced changes in gut microbiota are associated with improved leptin sensitivity and glucose homeostasis in diet-induced obese and diabetic mice.
We further investigated a dietary obesity model to identify the impact of prebiotic feeding when leptin signaling becomes compromised. Here we found that prebiotic feeding markedly improved glucose tolerance, reduced body weight and fat mass, and increased muscle mass (Fig. 5A–D). Mean food intake (kcal/mice/day) (HF 20.9 ± 0.6, HF-Pre 19.6 ± 0.3) was not significantly affected.
FIG. 5.
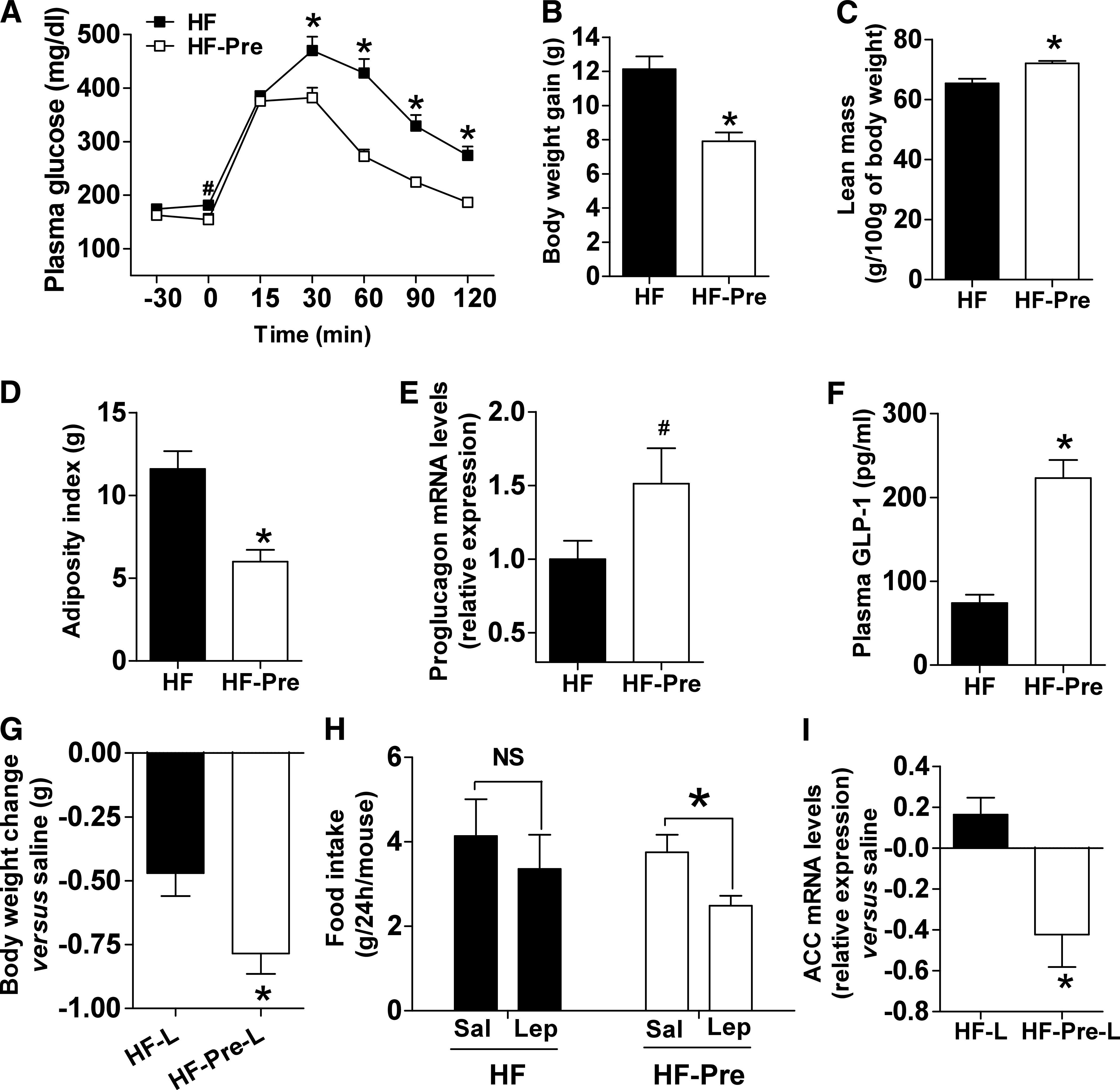
Prebiotic-induced changes in gut microbiota are associated with improved leptin sensitivity and glucose homeostasis in diet-induced obese and diabetic mice. A: Plasma glucose profile after 2 g/kg glucose oral challenge in freely moving mice in the HF (■) and the HF-Pre (□) mice. B: Body weight gain. C: Lean body mass measured by nuclear magnetic resonance. D: Adiposity index, corresponding to the sum of the subcutaneous, the visceral, and the epididymal adipose depot weights. E: Colon proglucagon mRNA expression. F: Portal plasma GLP-1 level content. Mean ± SEM. n = 8 (HF) and 9 (HF-Pre). *P < 0.05; #P = 0.08, determined by a two-tailed Student t test. G: Body weight changes 2 days after twice-daily intraperitoneal leptin (0.375 mg/kg) in the HF (HF-L) and the HF-Pre mice (HF-Pre-L). Data from each group were normalized to their own paired saline control. Mean ± SEM. n = 10 mice/group. *P < 0.05, determined by a two-tailed Student t test. H: Food intake 24 h after two doses of intraperitoneal leptin (Lep) (0.375 mg/kg). Data from each group are compared with their own paired saline control (Sal) vs. leptin. Mean ± SEM. n = 10 mice/group. *P < 0.05, determined by a paired Student t test. I: Adipose tissue acetyl-CoA carboxylase (ACC) mRNA expression 6 h after intraperitoneal leptin (1 mg/kg) or saline in the HF (HF-L) and the HF-Pre mice (HF-Pre-L). Data from each group were normalized to their own saline control. Mean ± SEM. Saline, n = 4 mice/group; leptin, n = 6 mice/group. *P < 0.05, determined by a two-tailed Student t test.
Similar to the ob/ob mice study, we found that prebiotic feeding significantly increased portal plasma GLP-1 levels (threefold), whereas colon proglucagon mRNA was increased by ∼50% (Fig. 5E and F). In contrast, portal plasma GIP levels were not affected by the treatment (HF 79.6 ± 10.9, HF-Pre 63.1 ± 6.8 pg/mL; P = 0.19).
Given that the vast majority of obesity is associated with leptin resistance, and that leptin, primarily involved in food intake and energy homeostasis, is also linked to the regulation of glucose homeostasis and numerous gastrointestinal functions (36), we may not exclude that the impact of prebiotics in this model interferes with leptin sensitivity.
To this aim, we compared the impact of leptin administration versus saline in mice fed with HF or HF and prebiotics. We found that leptin treatment induced a stronger decrease in body weight in HF-Pre than HF mice versus saline treatment (Fig. 5G). This last effect was associated with a significant decrease in food intake after leptin treatment in HF-Pre mice (Fig. 5H). In addition, in prebiotic-treated mice, leptin administration reduced adipose tissue acetyl-CoA carboxylase mRNA expression versus saline-treated mice. In contrast, HF-fed mice showed no expression changes (Fig. 5I). Thus, this analysis revealed that prebiotic treatment improved the anorexigenic, weight-, and lipogenesis-reducing effect of leptin compared with control obese mice.
DISCUSSION
Our findings provide new evidence for an important modification of the gut microbiota upon prebiotic treatment and indicate its contribution to host metabolism. The first methodological approach (qPCR) confirmed our previous findings that prebiotic treatment significantly increases the number of Bifidobacterium spp. (8). Furthermore, using 16S rDNA pyrosequencing, we observed an increase of five genera belonging to three phyla and a decrease in the abundance of six genera belonging to the Clostridiales order of the phylum Firmicutes. Similarly, a phylogenetic array approach revealed a lower abundance of 10 genera or species of Firmicutes. Estimates of relative taxa abundance by pyrosequencing and phylogenetic microarrays depend, among other factors, on taxonomic assignment methods, sequencing depth, taxonomic coverage of the microarray probes, and choice of 16S PCR primers (32,33,37). For instance, the difference observed in the abundance of the phylum Verrucomicrobia can be attributed to the choice of primers that can poorly detect this phylum. Nevertheless, it is worth mentioning that other recent studies also strongly support the interest of studies comparing culture-dependent microbiological techniques and next-generation sequencing technologies as performed in the current study (38). Although our data relied on different technologies and rDNA identification methods, clustering of bacterial communities showed similar patterns, reinforcing the idea that prebiotics induce profound changes in the gut bacteria composition.
These findings challenge the concept that prebiotics affect only a minor part of the gut microbial community. Initial observations in obese leptin-deficient mice (ob/ob) have shown decreased Bacteroidetes, whereas the proportion of Firmicutes was increased compared with lean mice (2). Similar shifts in the two dominant phyla were observed in the gut microbiota of obese humans (4,39). Importantly, several other studies have already characterized the gut microbiota composition of ob/ob and diet-induced obese mice by using similar metagenomic approaches (2,12,18,36,40,41). Interestingly, all of these studies are relatively concordant regarding the modulation of the gut microbiota in both ob/ob and HF-fed mice. There was an increase in Firmicutes and a decrease in Bacteriodetes in these obese mice models. Similar results linking gut microbiota to fat mass and body weight have been described in diet-induced obese mice. For instance, ingestion of an HF diet resulted in a bloom in Firmicutes and a decrease in Bacteroidetes. In addition, Hildebrandt et al. (40) showed that when switching lean mice to an HF diet, there was the expected decrease in Bacteriodetes and an increase in Firmicutes and Proteobacteria. Murphy et al. (41) found an increase in the proportions of Firmicutes in both HF-fed and ob/ob mice and a reduction in Bacteroidetes. More recently, Ravussin et al. (36) found that mice eating an HF diet have greater abundances of Firmicutes excluding Allobaculum operational taxonomic units. The current study demonstrates that prebiotic intake in mice impacts the relative abundance of the two dominating gut phyla, Bacteroidetes and Firmicutes, in a manner resembling the shift observed when comparing obese to lean humans or mice.
Given that prebiotic treatment can reduce obesity and associated metabolic disorders, the discovery of bacteria or bacterial group(s) that is able to shape host metabolism provides an attractive mechanistic explanation. Interestingly, both 16S rRNA analyses identified significant correlations between the genus Anaerotruncus and several metabolic parameters, such as glucose intolerance, gut permeability, plasma triglyceride content, and muscle lipid content. Similarly, Clostridium lactifermentans was positively correlated with all of these parameters, except plasma triglyceride content. Desulfovibrionaceae (i.e., Bilophila and Desulfovibrio, both gram-negative bacteria) were less prevalent in the prebiotic-treated mice. Interestingly, two recent studies demonstrate that diet-induced obesity and diabetes are associated with a bloom of this family (40,42). Some members of Desulfovibrionaceae, shown to be involved in gut barrier disruption (43), are able to reduce sulfate to H2S. In agreement with these reports, we found a very strong correlation between gut permeability and the abundance of Streptococcus intermedius. It is important to note that this species produces a specific cytolysin (intermedilysin) (44) that leads to altered tight-junction architecture (45,46). Therefore, it is tempting to speculate that the lower abundance of cytolysin-producing bacteria may participate in the control of gut barrier function through these mechanisms.
Among the factors recently identified to play a key role in the control of gut barrier function and glucose homeostasis, we demonstrated that the abundance of two peptides produced by enteroendocrine L cells (GLP-1 and GLP-2) specifically increases upon prebiotic treatment (47). Although the mechanisms seem to be related to enhanced proglucagon expression, in the current study, we found both increased L-cell number and portal plasma GLP-1 levels. There were wide variations of gut microbial communities between the control mice and the prebiotic-treated mice. The extent to which these changes correlated with metabolic parameters and the L-cell number suggests the presence of specific targets. For instance, the bloom in A. muciniphila was strongly and positively correlated with the L-cell number (r = 0.72; P = 0.01). The presence of this bacterium is not only associated with healthy mucosa, compared with that of patients with Crohn disease or ulcerative colitis (48), but also inversely correlated to body weight (49), increasing after the surgical weight loss procedure Roux-en-Y gastric bypass (50).
We previously found lower fasting glycemic levels and improved glucose tolerance observed upon prebiotic-induced gut microbiota modulation (20). However, in the current study, these changes were also associated with reduced plasma triglyceride levels, muscle lipid infiltration, adipose tissue mass, and oxidative stress and higher leptin sensitivity. This finding suggests that the improved metabolic phenotype observed in the prebiotic-treated mice is dependent on multiple mechanisms.
Here, we unraveled novel mechanisms linking gut microbiota changes and metabolism in genetic obese mice and found that prebiotics improved leptin sensitivity in diet-induced leptin-resistant mice. Further work is required to understand the functional links between the metabolic/catabolic activities of gut bacteria and their impact on host metabolism. For instance, it would be of interest to establish a causal relationship, instead of correlations as shown here, by using transfer of bacterial communities. An alternative experiment would be to analyze intestinal (fecal) microbiota in a time-series study in view of identifying the specific impact of prebiotics and the gut microbes on the onset of obesity and type 2 diabetes.
Taken together, the microbiota analyses revealed an unexpectedly wide shift in the gut microbiota profiles, which represent an important proportion of the total number of the sequence reads. First, this finding indicates that, in addition to the previously largely considered family Bifidobacteriaceae (8,16,19), prebiotic treatment profoundly modifies several other gut bacterial taxa from the phylum level down to the 100% ID phylotype level. Second, based on our findings, we proposed additional mechanisms and relationships between specific gut bacteria and metabolic alterations characterizing the obesity phenotype (e.g., leptin sensitivity). Third, a combination of two complementary 16S rRNA–based approaches with the use of prebiotics represents a promising approach to identify novel bacterial targets that may affect host metabolism in a given pathological context, such as obesity or type 2 diabetes.
ACKNOWLEDGMENTS
P.D.C. is a research associate from the Fonds de la Recherche Scientifique (Belgium). J.S. and P.F. were supported by grants from the Swiss National Science Foundation (31003A-124717/1 and 3100A0-116075, respectively). N.M.D. and P.D.C. are recipients of Fonds Spéciaux de Recherches (Université Catholique de Louvain) and Fonds de la Recherche Scientifique Médicale (Belgium) grants, respectively. P.D.C. is a recipient of grants from the Société Francophone du Diabète (France).
No potential conflicts of interest relevant to this article were reported.
A.E., V.L., and M.D. researched data and analyzed and wrote the manuscript. M.G. prepared the pool of barcoded PCR amplicons. G.M.M. and A.M.N. helped with tissue sampling. S.P. and A.V.H. performed gut microbiota qPCR analyses. P.F. and W.M.d.V. researched data and analyzed and wrote the manuscript. N.M.D. participated in discussion and wrote the manuscript. J.S. researched data and analyzed and wrote the manuscript. P.D.C. conceptualized, supervised, performed, analyzed, and interpreted all studies, wrote the manuscript, and is the guarantor of the study.
The authors thank Y. Guiot and R.M. Goebbels (Université Catholique de Louvain) for histological assistance; and Damien Naslain, Marie Van Roye, Florence Sohet, Laure Bindels, Céline Druart, Lucie Geurts, and Melania Osto (Université Catholique de Louvain) for helpful discussion and technical support.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db11-0227/-/DC1.
REFERENCES
- 1.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 2010;72:219–246 [DOI] [PubMed] [Google Scholar]
- 2.Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA 2005;102:11070–11075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bäckhed F, Ding H, Wang T, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA 2004;101:15718–15723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature 2006;444:1022–1023 [DOI] [PubMed] [Google Scholar]
- 5.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006;444:1027–1031 [DOI] [PubMed] [Google Scholar]
- 6.Cani PD, Amar J, Iglesias MA, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007;56:1761–1772 [DOI] [PubMed] [Google Scholar]
- 7.Cani PD, Bibiloni R, Knauf C, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008;57:1470–1481 [DOI] [PubMed] [Google Scholar]
- 8.Cani PD, Possemiers S, Van de Wiele T, et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 2009;58:1091–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vijay-Kumar M, Aitken JD, Carvalho FA, et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 2010;328:228–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 2006;124:837–848 [DOI] [PubMed] [Google Scholar]
- 11.Qin J, Li R, Raes J, et al. ; MetaHIT Consortium. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010;464:59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med 2009;1:6ra14 [DOI] [PMC free article] [PubMed]
- 13.Brun P, Castagliuolo I, Di Leo V, et al. Increased intestinal permeability in obese mice: new evidence in the pathogenesis of nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol 2007;292:G518–G525 [DOI] [PubMed] [Google Scholar]
- 14.de La Serre CB, Ellis CL, Lee J, Hartman AL, Rutledge JC, Raybould HE. Propensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. Am J Physiol Gastrointest Liver Physiol 2010;299:G440–G448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muccioli GG, Naslain D, Bäckhed F, et al. The endocannabinoid system links gut microbiota to adipogenesis. Mol Syst Biol 2010;6:392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cani PD, Neyrinck AM, Fava F, et al. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia 2007;50:2374–2383 [DOI] [PubMed] [Google Scholar]
- 17.Bäckhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci USA 2007;104:979–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turnbaugh PJ, Bäckhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 2008;3:213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roberfroid M, Gibson GR, Hoyles L, et al. Prebiotic effects: metabolic and health benefits. Br J Nutr 2010;104(Suppl. 2):S1–S63 [DOI] [PubMed] [Google Scholar]
- 20.Cani PD, Knauf C, Iglesias MA, Drucker DJ, Delzenne NM, Burcelin R. Improvement of glucose tolerance and hepatic insulin sensitivity by oligofructose requires a functional glucagon-like peptide 1 receptor. Diabetes 2006;55:1484–1490 [DOI] [PubMed] [Google Scholar]
- 21.Dewulf EM, Cani PD, Neyrinck AM, et al. Inulin-type fructans with prebiotic properties counteract GPR43 overexpression and PPARγ-related adipogenesis in the white adipose tissue of high-fat diet-fed mice. J Nutr Biochem 2011;22:712–722 [DOI] [PubMed] [Google Scholar]
- 22.Alhouayek M, Lambert DM, Delzenne NM, Cani PD, Muccioli GG. Increasing endogenous 2-arachidonoylglycerol levels counteracts colitis and related systemic inflammation. FASEB J 2011;25:2711–2721 [DOI] [PubMed] [Google Scholar]
- 23.Geurts L, Lazarevic V, Derrien M, et al. Altered gut microbiota and endocannabinoid system tone in obese and diabetic leptin-resistant mice: impact on apelin regulation in adipose tissue. Front Microbiol 2011;2:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R. Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat Methods 2008;5:235–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lazarevic V, Whiteson K, Hernandez D, François P, Schrenzel J. Study of inter- and intra-individual variations in the salivary microbiota. BMC Genomics 2010;11:523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cole JR, Wang Q, Cardenas E, et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 2009;37(Database issue):D141–D145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang Y, Niu B, Gao Y, Fu L, Li W. CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 2010;26:680–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 2004;32:1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lozupone C, Hamady M, Knight R. UniFrac—an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics 2006;7:371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 2007;73:5261–5267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meyer F, Paarmann D, D’Souza M, et al. The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 2008;9:386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rajilić-Stojanović M, Heilig HG, Molenaar D, et al. Development and application of the human intestinal tract chip, a phylogenetic microarray: analysis of universally conserved phylotypes in the abundant microbiota of young and elderly adults. Environ Microbiol 2009;11:1736–1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Claesson MJ, O’Sullivan O, Wang Q, et al. Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS ONE 2009;4:e6669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cani PD, Dewever C, Delzenne NM. Inulin-type fructans modulate gastrointestinal peptides involved in appetite regulation (glucagon-like peptide-1 and ghrelin) in rats. Br J Nutr 2004;92:521–526 [DOI] [PubMed] [Google Scholar]
- 35.Cani PD, Hoste S, Guiot Y, Delzenne NM. Dietary non-digestible carbohydrates promote L-cell differentiation in the proximal colon of rats. Br J Nutr 2007;98:32–37 [DOI] [PubMed] [Google Scholar]
- 36.Ravussin Y, Koren O, Spor A, et al. Responses of gut microbiota to diet composition and weight loss in lean and obese mice. Obesity (Silver Spring). 19 May 2011 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van den Bogert B, de Vos WM, Zoetendal EG, Kleerebezem M. Microarray analysis and barcoded pyrosequencing provide consistent microbial profiles depending on the source of human intestinal samples. Appl Environ Microbiol 2011;77:2071–2080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Sullivan O, Coakley M, Lakshminarayanan B, et al. ; ELDERMET consortium (http://eldermet.ucc.ie). Correlation of rRNA gene amplicon pyrosequencing and bacterial culture for microbial compositional analysis of faecal samples from elderly Irish subjects. J Appl Microbiol 2011;111:467–473 [DOI] [PubMed] [Google Scholar]
- 39.Turnbaugh PJ, Hamady M, Yatsunenko T, et al. A core gut microbiome in obese and lean twins. Nature 2009;457:480–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hildebrandt MA, Hoffmann C, Sherrill-Mix SA, et al. High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology 2009;137:1716–1724, e1–e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murphy EF, Cotter PD, Healy S, et al. Composition and energy harvesting capacity of the gut microbiota: relationship to diet, obesity and time in mouse models. Gut 2010;59:1635–1642 [DOI] [PubMed] [Google Scholar]
- 42.Zhang C, Zhang M, Wang S, et al. Interactions between gut microbiota, host genetics and diet relevant to development of metabolic syndromes in mice. ISME J 2010;4:232–241 [DOI] [PubMed] [Google Scholar]
- 43.Loubinoux J, Bronowicki JP, Pereira IA, Mougenel JL, Faou AE. Sulfate-reducing bacteria in human feces and their association with inflammatory bowel diseases. FEMS Microbiol Ecol 2002;40:107–112 [DOI] [PubMed] [Google Scholar]
- 44.Nagamune H, Ohnishi C, Katsuura A, et al. Intermedilysin, a novel cytotoxin specific for human cells secreted by Streptococcus intermedius UNS46 isolated from a human liver abscess. Infect Immun 1996;64:3093–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Welch RA. RTX toxin structure and function: a story of numerous anomalies and few analogies in toxin biology. Curr Top Microbiol Immunol 2001;257:85–111 [DOI] [PubMed] [Google Scholar]
- 46.Ma TY, Tran D, Hoa N, Nguyen D, Merryfield M, Tarnawski A. Mechanism of extracellular calcium regulation of intestinal epithelial tight junction permeability: role of cytoskeletal involvement. Microsc Res Tech 2000;51:156–168 [DOI] [PubMed] [Google Scholar]
- 47.Delzenne NM, Cani PD. Interaction between obesity and the gut microbiota: relevance in nutrition. Annu Rev Nutr 2011;31:15–31 [DOI] [PubMed] [Google Scholar]
- 48.Png CW, Lindén SK, Gilshenan KS, et al. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am J Gastroenterol 2010;105:2420–2428 [DOI] [PubMed] [Google Scholar]
- 49.Santacruz A, Collado MC, García-Valdés L, et al. Gut microbiota composition is associated with body weight, weight gain and biochemical parameters in pregnant women. Br J Nutr 2010;104:83–92 [DOI] [PubMed] [Google Scholar]
- 50.Zhang H, DiBaise JK, Zuccolo A, et al. Human gut microbiota in obesity and after gastric bypass. Proc Natl Acad Sci USA 2009;106:2365–2370 [DOI] [PMC free article] [PubMed] [Google Scholar]