

History, to the Roman Cicero, is “the witness of time, the life of memory, the mistress of life.” But history can be a cruel mistress, illustrating all we have not achieved. A glance back challenges us to reconsider what we have learned about type 1 diabetes and to what effect. More specifically, what happened after 1974, the annus mirabilis, when The Lancet reported the genetic basis of this disease (through HLA genes) (1) and a principle disease-associated biomarker (islet cell autoantibodies) (2). Certainly, we can predict the disease broadly based on these two observations, allied to progressive loss of insulin secretion, but without any tangible clinical benenfit. For the purpose of prediction is prevention, and that, thus far, we cannot do. As a result, attention has focused on nongenetic, specifically environmental, factors that lead to type 1 diabetes and might be modified. Two features of such environmental effects have had an impact on our understanding of the disease pathogenesis, as illustrated in this month’s Diabetes (3). First, early environmental events appear to cause type 1 diabetes (4). Second, the age at diagnosis of this disease impacts the clinical phenotype (5).
Twin, migration, population, and birth cohort studies emphasize the importance of environmental factors operating in early childhood when disease-predictive autoantibodies appear (6). Even disproportionate maternal and birth-related events can influence the disease risk (Table 1). Other putative factors include temperate climate, increased hygiene, increasing wealth, overcrowding in childhood, virus infections, early diet including exposure to cow’s milk, reduced rates or duration of breast feeding, and vitamin D (4,5). Paradoxically, the most powerful evidence characterizing such factors comes from genetic studies because genetic associations unequivocally point toward predisposition and cannot be an epiphenomenon. For example, two genes—DHCR7 and CYP2R1—that are each directly linked to vitamin D levels, are among three key genes associated with 25(OH) vitamin D metabolism and diabetes susceptibility (7).
TABLE 1.
List of potential birth-related risk factors for type 1 diabetes
| Born to diabetic fathers rather than diabetic mothers |
| Having a diabetic mother aged <8 years at diagnosis compared with a mother diagnosed later |
| Increasing maternal age at delivery |
| First born |
| ABO incompatibility with the mother |
| Season of delivery |
| More maternal enterovirus infections |
| Early cessation of breast feeding |
Virus infections have also been implicated, and enterovirus infections presage the appearance of autoantibodies, potentially contributing to rapid disease progression (8). Cellular factors that mediate defense against viruses include products of many interferon-stimulated genes, which encompass inhibitory activities (9). Of these genes, one cluster containing the macrophage interferon regulatory factor 7–driven inflammatory network is enriched for diabetes-associated genes and includes rare genetic variants of IFIH1, a cytoplasmic helicase involved in protection from viruses (10). Autoantibody-positive children, with an IFIH1 disease-protective genotype, progress less rapidly to diabetes than those with different genotypes (11).
Much of our current knowledge is based on this relatively modest armamentarium of genetic analyses plus autoantibodies. Therefore, any additional biomarker would be enormously valuable. Metabolomics, the detection and quantifiction of small molecules in a biological sample, has the advantage of being unbiased and so hypothesis-free as with genome-wide associated studies. It is also less complex than genomic or proteomic analyses, given the numbers of human endogenous metabolites (several thousand) compared with gene variants (multiple polymorphisms of ∼3 × 104 human genes) and proteins (∼5 × 105 –106, including splicing variants and posttranslational modifications). A remarkable study of 56 children who progressed to type 1 diabetes found that phosphatidylcholine was reduced at birth, independent of HLA risk, with increased levels of proinflammatory lysophosphatidylcholine several months before seroconversion to autoantibody positivity, but not thereafter (12). Thus, early lipid dysregulation with increased oxidative stress may influence disease pathogenensis. Now the same metabolomic method has been applied to the Munich birth cohort, which demonstrates, after seroconversion, increased odd-chain triglycerides and polyunsaturated fatty acid–containing phospholipids in autoantibody-positive compared with autoantibody-negative children, who are at high and low risk, respectively, for diabetes (3). But children who developed autoantibodies by age 2 years also had persistent twofold lower concentrations of methionine compared with children who either developed autoantibodies later or were autoantibody negative. By implication, pathways utilizing methionine could be relevant to time to appearance of autoantibodies and, by inference, clinical disease. Methionine, like choline, is an epigenetic regulator, the former because it is a methyl-donor, important in transmethylation and one-carbon moiety pathways. Methionine is, therefore, involved in DNA methylation, an epigenetic effect putatively involved in autoimmunity (13). Moreover, an imprinted gene, a quintessential epigenetic effect, has been implicated in diabetes genetic susceptibility (14). However, the low plasma methionine in the young high disease-risk subset is not likely to cause diabetes, since it is only found in very young autoantibody-positive children, more likely reflecting a marker for rapid disease progression.
These observations bring us back to the heterogeneity of autoimmune diabetes according to age at diagnosis, which is severe and insulin dependent in childhood but often mild and noninsulin requiring in adulthood (15). That heterogeneity highlights the differential character of the disease process beyond clinical differences. Thus, children at diagnosis compared with adults have higher HLA genetic risk, more antigen-specific autoantibodies, and greater insulin deficiency (15). Genetic risk aside, such heterogeneity could result from similar environmental events operating at disparate times, different factors operating at the same time, or both. Evidence supports the latter (Fig. 1). Age at diagnosis is strongly correlated with age of appearance of the first autoantibody; when autoantibodies appear before age 5 years, they tend to be multiple, antigen-specific, isotype restricted, high titer, and highly predictive of disease (4). Autoantibodies appearing after age 8 years are often directed against single antigens, at low titer, with reduced predictive value (4). If, as seems likely, autoantibodies reflect disease-associated environmental events, then the timing of that critical exposure impacts clinical outcome. Such distinctions are now shown to encompass metabolic changes, including low plasma methionine, with the caveat that metabolomics, like autoantibodies, may be an epiphenomenon. As when immunogenetic changes were initially identified, future developments will be methodological and descriptive—the former through improved assays and sample collection, the latter by exploiting cohort studies, other diseases, and animal models to define the robustness, specificity, and causation of the results (16).
FIG. 1.
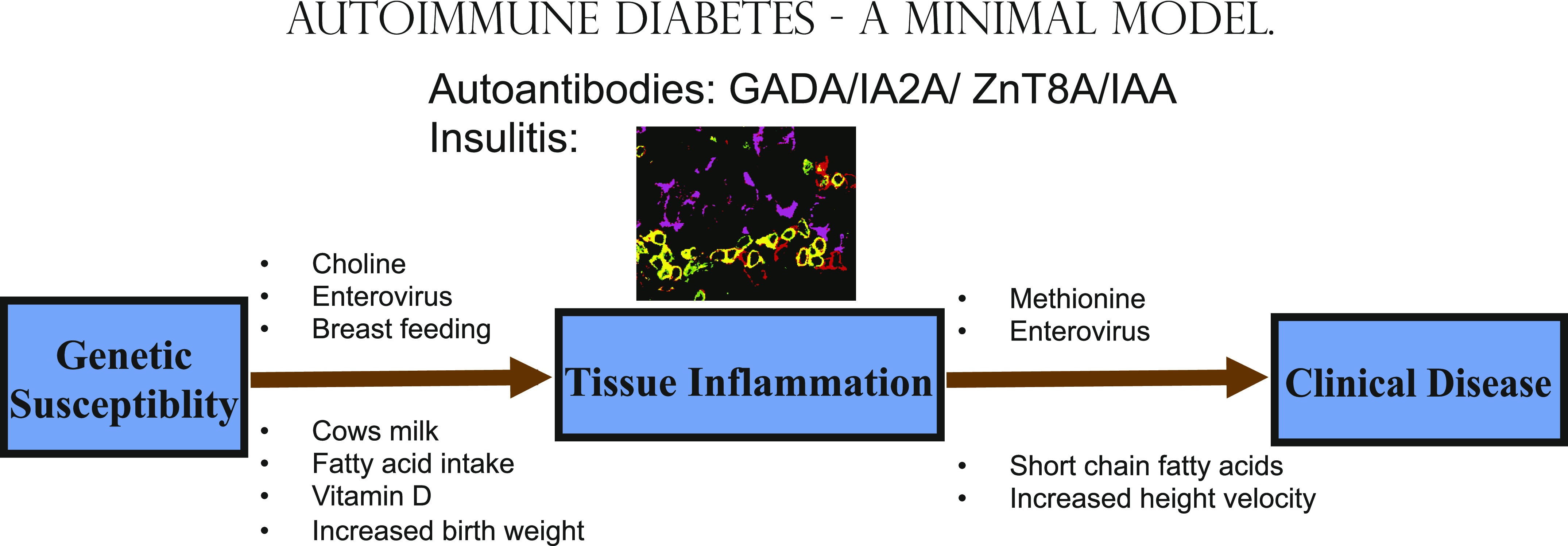
Minimal model of autoimmune diabetes. Tissue inflammation (around insulin secreting cells in purple with CD3 cells in green and CD3/CD45 cells in yellow) is reflected in the production of serum autoantibodies. Factors are detailed that may predispose in genetically susceptible subjects to 1) autoantibodies and 2) progression to clinical disease. Autoantibodies to glutamic acid decarboxylase (GADA); insulinoma-associated antigen 2 (IA-2A); zinc transporter 8 (ZnT8A); insulin (IAA). (A high-quality digital representation of this figure is available in the online issue.)
Autoimmune diabetes has a broad clinical spectrum according to age at diagnosis. That spectrum is now shown to result from differential immunogenetic and nongenetic effects, including metabolic effects, in the prediabetic period, and these effects influence the likelihood and rate of disease progression (4,5,17–20) (Fig. 1). We have been the witness of time, and time in science offers hope. For with the most modest of tools and with time, we have transformed our understanding of this disease. The addition of the metabolome as a further biomarker promises a rich harvest.
ACKNOWLEDGMENTS
No potential conflicts of interest relevant to this article were reported.
Images in this article were provided by Network for Pancreatic Organ Donors with Diabetes (nPOD) online pathology site. nPOD is a collaborative type 1 diabetes research project sponsored by the Juvenile Diabetes Research Foundation International. Organ Procurement Organizations partnering with nPOD to provide research resources are listed at www.jdrfnpod.org/our-partners.php. <http://www.jdrfnpod.org/our-partners.php>.
Footnotes
See accompanying original article, p. 2740.
REFERENCES
- 1.Nerup J, Platz P, Andersen OO, et al. HL-A antigens and diabetes mellitus. Lancet 1974;2:864–866 [DOI] [PubMed] [Google Scholar]
- 2.Bottazzo GF, Florin-Christensen A, Doniach D. Islet-cell antibodies in diabetes mellitus with autoimmune polyendocrine deficiencies. Lancet 1974;2:1279–1283 [DOI] [PubMed] [Google Scholar]
- 3.Pflueger M, Seppänen-Laakso T, Suortti T, et al. Age- and islet autoimmunity–associated differences in amino acid and lipid metabolites in children at risk for type 1 diabetes. Diabetes 2011;60:2740–2747 [DOI] [PMC free article] [PubMed]
- 4.Ziegler AG, Nepom GT. Prediction and pathogenesis in type 1 diabetes. Immunity 2010;32:468–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leslie RD, Delli Castelli M. Age-dependent influences on the origins of autoimmune diabetes: evidence and implications. Diabetes 2004;53:3033–3040 [DOI] [PubMed] [Google Scholar]
- 6.Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 2010;464:1293–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cooper JD, Smyth DJ, Walker NM, et al. Inherited variation in vitamin D genes is associated with predisposition to autoimmune disease type 1 diabetes. Diabetes 2011;60:1624–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stene LC, Oikarinen S, Hyöty H, et al. Enterovirus infection and progression from islet autoimmunity to type 1 diabetes: the Diabetes and Autoimmunity Study in the Young (DAISY). Diabetes 2010;59:3174–3180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schoggins JW, Wilson SJ, Panis M, et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011;472:481–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heinig M, Petretto E, Wallace C, et al. ; Cardiogenics Consortium. A trans-acting locus regulates an anti-viral expression network and type 1 diabetes risk. Nature 2010;467:460–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Winkler C, Lauber C, Adler K, et al. An interferon-induced helicase (IFIH1) gene polymorphism associates with different rates of progression from autoimmunity to type 1 diabetes. Diabetes 2011;60:685–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oresic M, Simell S, Sysi-Aho M, et al. Dysregulation of lipid and amino acid metabolism precedes islet autoimmunity in children who later progress to type 1 diabetes. J Exp Med 2008;205:2975–2984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet 2011;12:529–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wallace C, Smyth DJ, Maisuria-Armer M, Walker NM, Todd JA, Clayton DG. The imprinted DLK1-MEG3 gene region on chromosome 14q32.2 alters susceptibility to type 1 diabetes. Nat Genet 2010;42:68–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leslie RD. Predicting adult-onset autoimmune diabetes: clarity from complexity. Diabetes 2010;59:330–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang TJ, Larson MG, Vasan RS, et al. Metabolite profiles and the risk of developing diabetes. Nat Med 2011;17:448–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lamb MM, Yin X, Zerbe GO, et al. Height growth velocity, islet autoimmunity and type 1 diabetes development: the Diabetes Autoimmunity Study in the Young. Diabetologia 2009;52:2064–2071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Norris JM, Yin X, Lamb MM, et al. Omega-3 polyunsaturated fatty acid intake and islet autoimmunity in children at increased risk for type 1 diabetes. JAMA 2007;298:1420–1428 [DOI] [PubMed] [Google Scholar]
- 19.Knip M, Virtanen SM, Seppä K, et al. ; Finnish TRIGR Study Group. Dietary intervention in infancy and later signs of beta-cell autoimmunity. N Engl J Med 2010;363:1900–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ziegler AG, Schmid S, Huber D, Hummel M, Bonifacio E. Early infant feeding and risk of developing type 1 diabetes-associated autoantibodies. JAMA 2003;290:1721–1728 [DOI] [PubMed] [Google Scholar]