

Abstract
OBJECTIVE
Insulin receptor substrate-2 (IRS-2) plays an essential role in pancreatic islet β-cells by promoting growth and survival. IRS-2 turnover is rapid in primary β-cells, but its expression is highly regulated at the transcriptional level, especially by glucose. The aim was to investigate the molecular mechanism on how glucose regulates IRS-2 gene expression in β-cells.
RESEARCH DESIGN AND METHODS
Rat islets were exposed to inhibitors or subjected to adenoviral vector–mediated gene manipulations and then to glucose-induced IRS-2 expression analyzed by real-time PCR and immunoblotting. Transcription factor nuclear factor of activated T cells (NFAT) interaction with IRS-2 promoter was analyzed by chromatin immunoprecipitation assay and glucose-induced NFAT translocation by immunohistochemistry.
RESULTS
Glucose-induced IRS-2 expression occurred in pancreatic islet β-cells in vivo but not in liver. Modulating rat islet β-cell Ca2+ influx with nifedipine or depolarization demonstrated that glucose-induced IRS-2 gene expression was dependent on a rise in intracellular calcium concentration derived from extracellular sources. Calcineurin inhibitors (FK506, cyclosporin A, and a peptide calcineurin inhibitor [CAIN]) abolished glucose-induced IRS-2 mRNA and protein levels, whereas expression of a constitutively active calcineurin increased them. Specific inhibition of NFAT with the peptide inhibitor VIVIT prevented a glucose-induced IRS-2 transcription. NFATc1 translocation to the nucleus in response to glucose and association of NFATc1 to conserved NFAT binding sites in the IRS-2 promoter were demonstrated.
CONCLUSIONS
The mechanism behind glucose-induced transcriptional control of IRS-2 gene expression specific to the islet β-cell is mediated by the Ca2+/calcineurin/NFAT pathway. This insight into the IRS-2 regulation could provide novel therapeutic means in type 2 diabetes to maintain an adequate functional mass.
The pancreatic β-cell plays a pivotal role in control of metabolic glucose homeostasis by secreting insulin in response to elevated glucose concentrations. During insulin-resistant states, such as in obesity, increased β-cell mass and function are critical to compensate for decreased insulin sensitivity. Failure of the functional β-cell mass to compensate for insulin resistance results in the onset of type 2 diabetes (1). As such, there is interest to better understand molecular mechanisms that regulate specific β-cell growth, regeneration, and/or survival, which could reveal novel means to maintain a critical functional β-cell mass and avoid the onset of type 2 diabetes (2,3).
It is established that insulin receptor substrate-2 (IRS-2) is required for maintaining an optimal population of functional β-cells and necessary for the β-cell mass to increase in compensation for peripheral insulin resistance (4–6). IRS-2 is a member of the IRS family of proteins, which are adaptor molecules that are tyrosine phosphorylated by plasma membrane associated tyrosine kinases (including insulin and insulin growth factor I receptors) (7). This tyrosine phosphorylation leads to downstream activation of the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (also known as Akt) pathway and the Ras/Raf-1/mitogen-activated protein kinase pathway (7,8). IRS-2 appears to have a uniquely important role in β-cells, playing a part in the control of β-cell growth and, especially, β-cell survival (5,7). This is illustrated when comparing the phenotypes of IRS-1 versus IRS-2-null mice (4,5). In IRS-1-null mice, there is marked insulin resistance (since without IRS-1, insulin signaling is impaired), but the β-cell mass compensates so that the animals are only mildly glucose-intolerant. However, in IRS-2-null mice, where there is a similar degree of insulin resistance, the β-cell mass fails to compensate. Indeed, there is a marked loss of β-cells and decreased insulin production that leads to a severe diabetes state at 10 weeks of age (4). In contrast, when IRS-2 expression is specifically increased in β-cells, IRS-2 downstream signaling is enhanced, which in turn promotes β-cell survival and maintains an optimal population of functional β-cells to avoid diabetes in vivo (9). As such, there has been interest in whether a means of increasing IRS-2 expression in β-cells may have therapeutic potential to treat type 2 diabetes. However, the molecular mechanisms that control IRS-2 expression in β-cells are relatively ill defined.
The turnover of IRS-2 in β-cells, at both mRNA and protein levels, appears to be relatively rapid in having a half-life of only ∼2 h (10). However, to counteract this, IRS-2 expression is dynamically regulated in primary pancreatic islet β-cells by physiologically relevant stimuli, especially glucose (10,11). Certain peptide hormones, such as glucagon-like peptide 1 (GLP-1), have also been shown to specifically upregulate IRS-2 expression (12) but are glucose dependent (13). As such, glucose is the major regulator of IRS-2 expression in β-cells. Intriguingly, glucose has been suggested to play a prominent role in β-cell compensation for insulin resistance (14,15). But the molecular mechanism as to how glucose might control β-cell growth and survival remains illusive and is only partly understood (16). Considering the critical role that IRS-2 plays in influencing β-cell mass, glucose regulation of IRS-2 expression in β-cells could be a major factor in maintaining an optimal functional mass of β-cells to meet the metabolic demand. Here, we show that specific glucose-induced increase in IRS-2 expression in primary islet β-cells is mediated by a Ca2+-dependent activation of Ca2+/calmodulin-dependent protein phosphatase-2B (or calcineurin) and dephosphorylation activation of the transcription factor nuclear factor of activated T cells (NFAT) to drive IRS-2 gene transcription. Considering the role that IRS-2 plays in β-cell mass compensation (7), our findings complement recent observations that calcineurin inhibitors compromise both β-cell proliferation (17) and survival (18).
RESEARCH DESIGN AND METHODS
Materials.
Nifedipine, FK506, and cyclosporin A (CsA) were purchased from Sigma-Aldrich (St. Louis, MO). Anti–IRS-2 antibody was provided by Dr. M. White (Children’s Hospital and Harvard Medical School, Boston, MA) or purchased from Upstate Biotechnology (Lake Placid, NY). Antibody to the p85 subunit of PI3K was also purchased from Upstate Biotechnology. Antibody to NFATc1 was purchased from BD Biosciences PharMingen (San Jose, CA) and Santa Cruz Biotechnology. Adenovirus construct expressing luciferase was generated as described previously (19). Adenoviruses expressing CAIN peptide (AdV-CAIN) or a constitutively active form of calcineurin (AdV-CNca) were a gift from Dr. J.D. Molkentin (Cincinnati Children's Hospital Medical Center, Cincinnati, OH). Adenovirus expressing peptide VIVIT (AdV-VIVIT) was a gift from Dr. C.M. Norris (Lexington, KY). Adenovirus expressing NFATc1 (AdV-NFATc1) was purchased from Seven Hills Bioreagents (Cincinnati, OH).
Animals and pancreatic islet isolation.
Male Wistar rats and C57Bl6/6J mice were obtained from Charles River Laboratories (Wilmington, MA). All animal care, use, and experiment protocols were approved by the Institutional Animal and Use Committee of the University of Chicago. Rodent pancreatic islets were isolated by collagenase digestion and Histopaque density gradient centrifugation as described (20). For the in vivo studies, where efficient tissue harvesting was an issue, the technique was modified. Preparation and excision of the pancreas was conducted first; the liver was then flushed through with buffer in situ, and the lobes were excised. Islets were isolated from the pancreas by collagenase digestion (20) but with a shorter density gradient centrifugation (≤ 10 min) to save time. Islets were then handpicked from the density gradient interface as described (20). From the time of death, it took ~12–15 min to harvest the liver and around 25 min to isolate sufficient pancreatic islets (∼100) for subsequent immunoblot analysis of IRS-2 expression. As such, for liver there is ∼15-min delay and for islets an ∼25–30-min delay, after the time points on the glucose/saline tolerance test. For in vitro studies, isolated islets were incubated in RPMI 1640 medium containing 3 mmol/L glucose and 0.1% fatty acid–free bovine serum albumin and at 37°C, in 5% CO2, for 16 h before the experiment. For adenovirus infection, dispersed islet cells were obtained using trypsin and DNase to obtain islet monolayer.
Intraperitoneal glucose tolerance test.
The C57Blk6/6J mice were fasted for 16 h before intraperitoneal glucose injection at a concentration of 2 mg glucose/g body wt. Blood glucose was measured at indicated time points using an AccuChek Compact Plus glucometer (Roche).
Cell culture.
The pancreatic β-cell line INS-1 was maintained as previously defined (21).
Real-time fluorescence-based reverse transcription.
Total RNA was isolated using an RNeasy Plus Mini Kit from Qiagen (Valencia, CA) and quantified using Spectrophotometer Nanodrop 2000 (Thermo Scientific). RNA (40 ng) was submitted to the Power SYBR Green RNA-to-Ct 1 step kit (Applied Biosystems) using a ABI Prism 7700 Sequence Detector System according to the manufacturer’s instructions. Oligonucleotide primers used are: IRS-2, reverse, TTTCCTGAGAGAGACGTTTTCCA, forward, CCCCAGTGTCCCCATCCT; and β-actin, reverse, GGGGTGTTGAAGGTCTCAAA, and forward, TGTCACCAACTGGGACGATA. Results are shown relative to β-actin (control) mRNA.
Nuclear extracts.
Nuclear extracts were prepared as described by Schreiber et al. (22). In brief, islets were lysed in a hypotonic buffer containing proteases inhibitors and 0.6% (v/v) Nonidet P-40 (NP-40). The lysates were centrifuged, and the pellets were rocked at 4°C in a high salt buffer. After centrifugation, supernatants containing nuclear proteins were collected.
Immunoblot analysis.
Immunoblots were performed on cell lysates from isolated rat islets or INS-1 cells as described previously (10).
Chromatin immunoprecipitation assay.
Chromatin immunoprecipitation (ChIP) assay was performed using the ChIP-IT Express kit (Active Motif, Carlsbad, CA). In brief, INS-1 cells were grown and then infected with various adenovirus constructs as indicated. Chromatin was fixed in 1% (v/v) formaldehyde for 10 min before cell lysis. Nuclear pellets were extracted and then sheared by sonication using the Bioruptor UCD-200 from Diagenode Inc. (Sparta, NJ). DNA concentration from each condition was measured, and 75 μg of DNA were immunoprecipitated overnight at 4°C with an antibody to NFATc1 or Mouse IgG as a control, together with protein G-coated magnetic beads. Magnetic beads were washed three times, and DNA-protein complexes were then eluted and reversed cross-link. Both DNA isolated from ChIP and input corresponding to DNA aliquot not submitted to immunoprecipitation were analyzed by PCR using five different primer pairs (Supplementary Table 1).
Immunofluorescent analysis.
INS-1 pancreatic β-cells were cultured on glass coverslips, and after incubation at 3 mmol/L glucose or 15 mmol/L glucose ± FK506 (10 µmol/L) were washed with ice-cold PBS, fixed in 4% (v/v) paraformaldehyde/PBS for 15 min at 4°C, washed three times in PBS, and then permeabilized with 0.3% (v/v) Triton X-100/PBS for 30 min at room temperature. Cells were then incubated with 10% donkey serum/PBS for 1 h at room temperature, before incubation with antibodies to NFATc1, or to insulin (Millipore, Billerica, MA) for 16 h at 4°C. Cells were then washed three times in PBS before incubation with anti-mouse or anti–guinea pig antibodies for 1 h at room temperature. For nuclear staining, cells were mounted with a reagent containing DAPI (Invitrogen, Carlsbad, CA). Slides were analyzed using confocal microscopy.
Statistical analysis.
Data are presented as means ± SE. Statistical analysis between groups was performed using Student t test, where P ≤ 0.05 was considered statistically significant.
RESULTS
Specific glucose-induced regulation of IRS-2 expression in pancreatic islets but not in hepatocytes in vivo.
It has been shown previously that glucose can specifically increase IRS-2 expression in islet β-cells in vitro, predominately at the transcriptional level (10). Here, normal mice were subjected to an intraperitoneal glucose tolerance test using a dose of 2 mg glucose/g body wt or saline as a control where excursions in blood glucose were indicated (Fig. 1A). In islets isolated at 2 h, and more so at 4 h, after the introduction of glucose, IRS-2 protein levels were specifically increased compared with saline-injected controls (Fig. 1B), complementary to previous in vitro observations (10,11). However, in parallel experiments, no increase in IRS-2 expression was found in the liver (Fig. 1C), suggesting that glucose regulation of IRS-2 expression may be unique to pancreatic β-cells.
FIG. 1.

Specific glucose-induced regulation of IRS-2 expression in pancreatic islets, but not in hepatocytes in vivo. Normal C57Blk6/6J mice (aged 12 weeks) were fasted overnight and then subjected to an intraperitoneal glucose tolerance test (2 mg/g body wt) or using saline as a control as described (42). Pancreatic islets were isolated, and a liver biopsy was conducted at the 2- and 4-h time point and then subjected to immunoblot (IB) analysis for IRS-2 protein expression relative to PI3K(p85) as a loading control. A: Excursion in circulating glucose in the mice after a glucose (●) or saline (○) injection. A mean ± SE is shown (n ≥ 3). B and C: Example IB analyses of IRS-2 and PI3K(p85) in islets (B) and liver (C) from saline (S)- or glucose (G)-treated mice at 2 or 4 h are shown from two separate experiments. A quantification of a series of experiments is also depicted, where gray bars are S- and black bars are G-treated animals. Data are a mean ± SE (n = 3), where * indicates statistically significant difference (P ≤ 0.05) from the equivalent saline control.
Glucose regulation of IRS-2 expression in β-cells is dependent on increased cytosolic [Ca2+]i flux.
It has been indicated previously that control of glucose-induced IRS-2 expression in β-cells is likely Ca2+ dependent (10). It was investigated whether this Ca2+ dependency was based on an influx of extracellular Ca2+. Specifically, blocking L-type voltage-dependent Ca2+ channels (L-type VDCCs) in isolated rat islets with the inhibitor nifedipine had little effect on basal IRS-2 expression at 3 mmol/L glucose, but prevented a 15 mmol/L glucose-induced specific increase in the expression of IRS-2 mRNA (P ≤ 0.05) (Fig. 2A) and protein (P ≤ 0.05) (Fig. 2B). The L-type VDCCs in β-cells can be opened via a depolarization of the plasma membrane (23). Depolarization of primary islet β-cells in vitro, by addition of 30 mmol/L KCl, resulted in a significant increase in IRS-2 mRNA (Fig. 2C) and protein (Fig. 2D) at basal 3 mmol/L glucose and further increased IRS-2 mRNA and protein expression levels at a stimulatory 15 mmol/L glucose relative to control islets. Collectively, these data indicate that a glucose-induced increase in β-cell cytosolic intracellular calcium concentration ([Ca2+]i) flux via depolarization and subsequent opening of L-type VDCCs is required for glucose-induced increases in IRS-2 expression in β-cells.
FIG. 2.
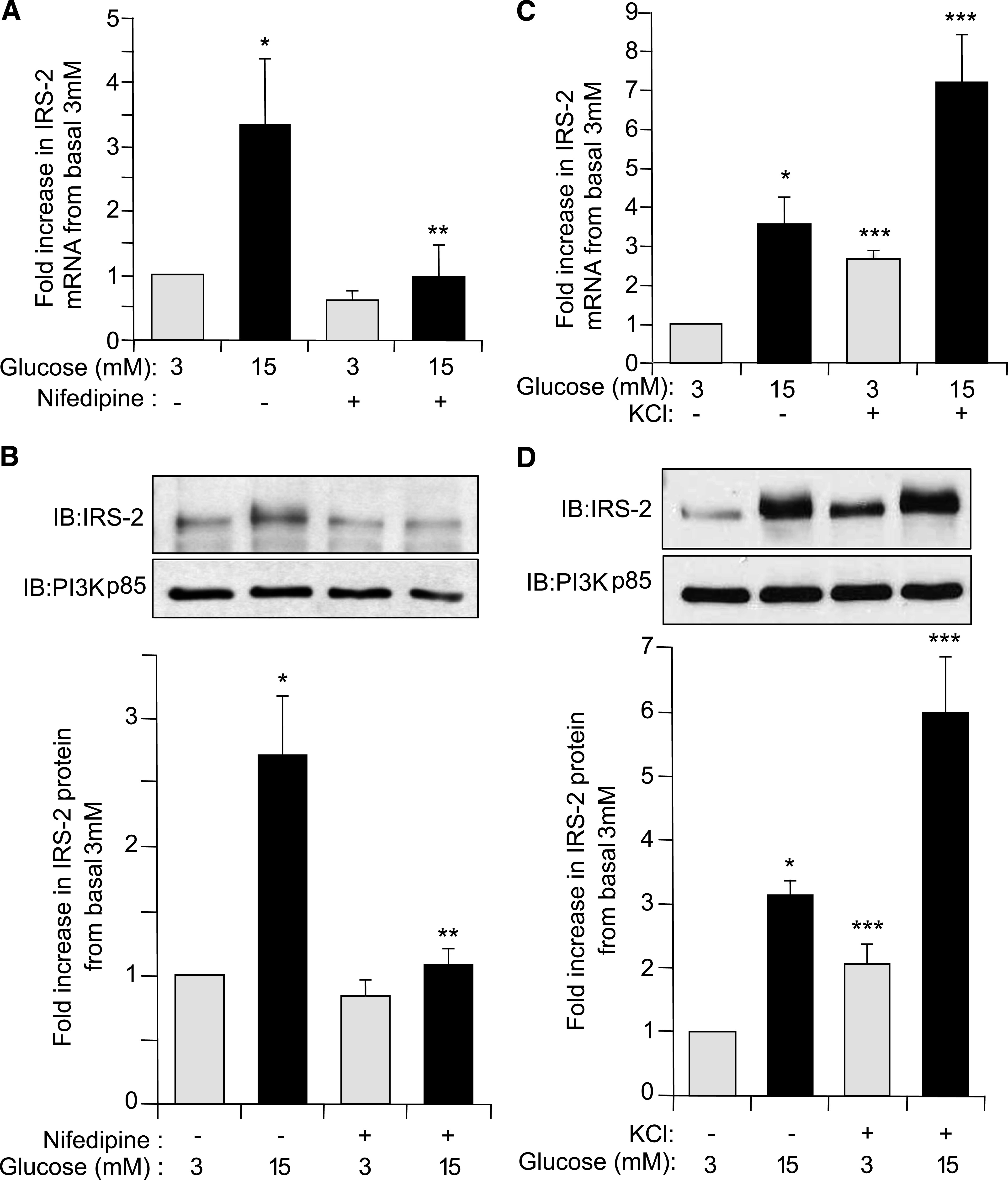
Glucose regulation of IRS-2 expression in β-cells is dependent on increased cytosolic [Ca2+]i flux. Isolated rat pancreatic islets were cultured overnight at normal 5.6 mmol/L glucose and then incubated at either basal 3 mmol/L (gray bars) or stimulatory 15 mmol/L (black bars) glucose concentration for 6 h in presence or absence of either the voltage-sensitive L-type Ca2+-channel inhibitor nifedipine (50 µmol/L) (A and B) or KCl to depolarize β-cells (30 mmol/L) (C and D). A and C: IRS-2 and β-actin (internal reference control) mRNA expression levels were measured by real-time fluorescence-based quantitative RT-PCR. The data are shown as a mean ± SE above basal 3 mmol/L glucose control, where * indicates statistically significant difference (P ≤ 0.05) at 15 mmol/L glucose in the presence of nifedipine or KCl versus control (n ≥ 4). B and D: IRS-2 and PI3K(p85) (control) protein expression levels were analyzed by immunoblotting. Example immunoblots (IBs) are shown. Quantitative measurements are also shown as a mean ± SE, where * indicates statistically significant increase at 15 mmol/L glucose compared with basal 3 mmol/L glucose control, ** indicates statistically significant inhibition in the presence of nifedipine at 15 mmol/L glucose, and *** indicates statistically significant potentiation by KCl relative to the equivalent glucose concentration in the absence of KCl (P ≤ 0.05; n ≥ 4).
Glucose regulation of IRS-2 expression in β-cells is mediated by calcineurin.
A potential downstream target that responds to a rise in cytosolic [Ca2+]i in β-cells is the Ca2+/calmodulin-dependent phosphoprotein phosphatase-2B (or calcineurin) (24,25). To assess whether calcineurin was involved in specific glucose regulation of IRS-2 expression, we used two immunosuppressive drugs, FK506 (also named tacrolimus) and CsA that specifically inhibit calcineurin activity (26). In the presence of FK506, glucose-induced IRS-2 mRNA and protein levels were significantly inhibited by ∼90% in rat islets (P ≤ 0.05) (Fig. 3A and B). Likewise, CsA blocked glucose-induced IRS-2 protein levels compared with control rat islets (P ≤ 0.05) (Fig. 2C). It is noteworthy that neither FK506 nor CsA significantly affected islet IRS-2 mRNA or protein expression levels at basal 3 mmol/L glucose (Fig. 3A–C), indicating that calcineurin only mediated glucose-induced regulation of IRS-2 expression in islet β-cells. An alternative, more specific, inhibition of calcineurin was examined using recombinant adenovirus (AdV-CAIN), which expresses an endogenous peptide inhibitor of calcineurin activity (CAIN) (25,27). In AdV-CAIN–infected rat islets, glucose-induced IRS-2 mRNA and protein expression were significantly inhibited ≥90% (P ≤ 0.05) compared with control islets infected with a recombinant adenovirus expressing Firefly luciferase (AdV-FLuc) (Fig. 3D and E). There was no apparent effect on IRS-2 mRNA or protein expression by inhibiting calcineurin with AdV-CAIN at basal 3 mmol/L glucose (Fig. 3D and E). Islets were also infected with a recombinant adenovirus expressing a constitutively activated variant of calcineurin (AdV-CNCa). In AdV-CNca–infected islets there was a significant increase in IRS-2 mRNA and protein expression at basal 3 mmol/L glucose that was further augmented at a stimulatory 15 mmol/L glucose concentration compared with AdV-FLuc–infected control islets (Fig. 3F and G). Collectively, these data show that calcineurin is required to control glucose-induced IRS-2 expression in rat islets.
FIG. 3.
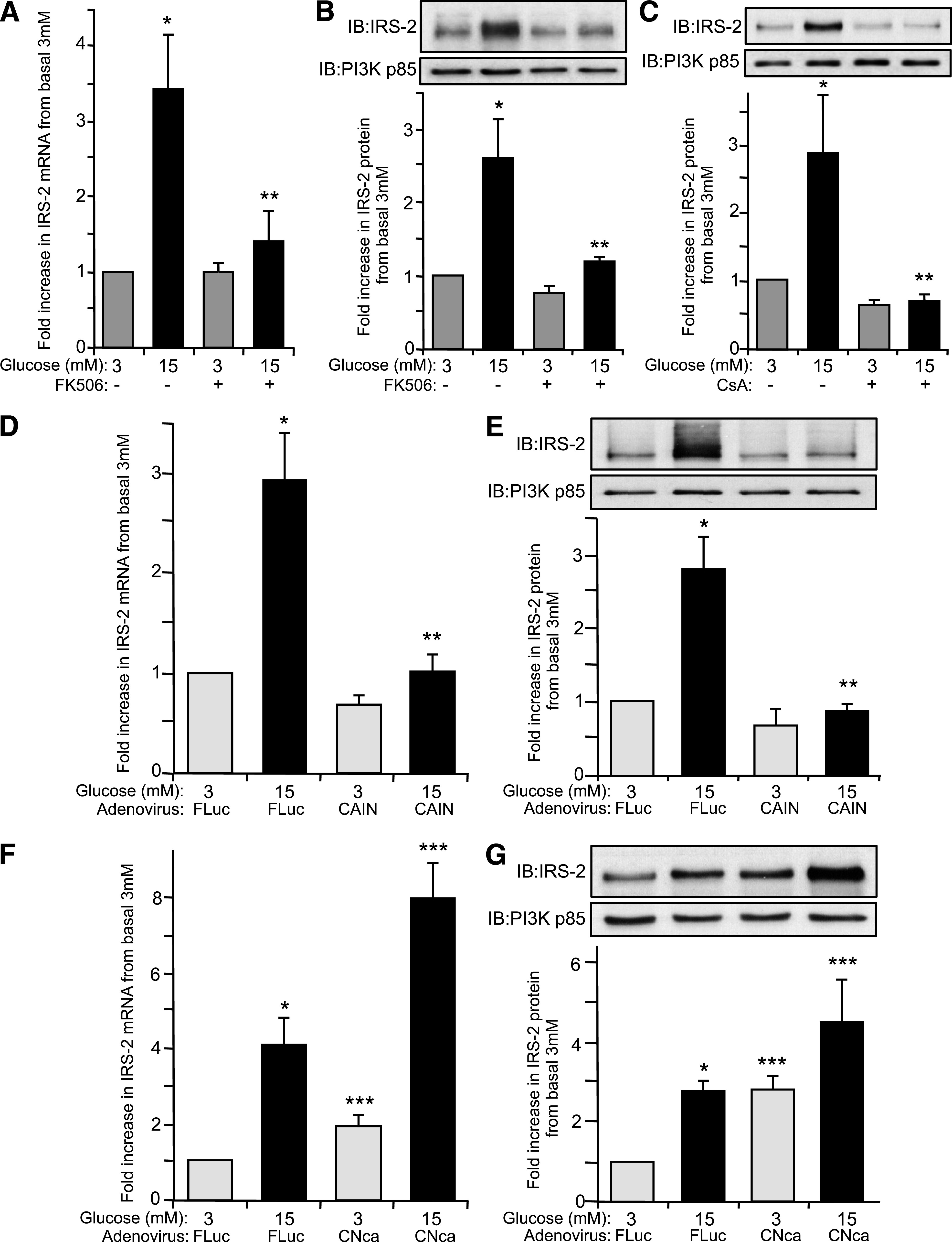
Glucose regulation of IRS-2 expression in β-cells is mediated by calcineurin. Isolated rat pancreatic islets were cultured overnight at normal 5.6 mmol/L glucose and then incubated at either basal 3 mmol/L or stimulatory 15 mmol/L glucose concentration for 6 h in presence or absence of calcineurin inhibitors FK506 (10 µmol/L) (A and B) or CsA (10 µmol/L) (C). A: IRS-2 and β-actin (internal reference control) mRNA expression levels were measured by real-time fluorescence-based quantitative RT-PCR. B and C: IRS-2 and PI3K(p85) (control) protein expression levels were analyzed by immunoblotting, where an example immunoblot (IB) is shown. Quantitative measurements are also shown as a mean ± SE above basal 3 mmol/L glucose control, where * indicates a statistically significant increase at 15 mmol/L glucose and ** indicates a statistically significant difference at 15 mmol/L glucose in the presence of FK506 or CsA versus control (P ≤ 0.05; n ≥ 4) (A–C). Isolated rat pancreatic islets were also infected with a recombinant AdV-CAIN (D and E), AdV-CNca (F and G), or AdV-FLuc and cultured overnight at normal 5.6 mmol/L glucose. Afterward, the adenovirally infected islets were then incubated at either basal 3 mmol/L or stimulatory 15 mmol/L glucose concentration for 6 h. D and F: IRS-2 and β-actin (internal reference control) mRNA expression levels were measured by real-time fluorescence-based quantitative RT-PCR. E and G: IRS-2 and PI3K(p85) (control) protein expression levels were analyzed by immunoblotting, where an example immunoblot is shown. The data are shown as a mean ± SE above basal 3 mmol/L glucose control, where * indicates a statistically significant increase at 15 mmol/L glucose above basal 3 mmol/L glucose, ** indicates a statistically significant inhibition with AdV-CAIN versus AdV-FLuc–infected control islets at 15 mmol/L glucose, and *** indicates a statistically significant increase in AdV-CNca–infected versus AdV-FLuc–infected control islets at the respective 3 and 15 mmol/L glucose concentrations (P ≤ 0.05; n ≥ 4) (D–G).
Glucose regulation of IRS-2 expression in β-cells is mediated by NFAT.
The NFAT family of transcription factors is known to be activated by the Ca2+/calcineurin pathway (28). To assess whether NFAT plays a role in IRS-2 expression, AdV-VIVIT, a synthetic peptide preventing NFAT activation by calcineurin without affecting phosphatase activity of calcineurin, was used (29). In AdV-VIVIT–infected rat islets the specific glucose-induced increase in IRS-2 mRNA levels was significantly decreased by ∼75% compared with control AdV-FLuc–infected islets (P ≤ 0.05) (Fig. 4A). Likewise, the glucose-induced increase in IRS-2 protein levels was effectively blocked in AdV-VIVIT–infected rat islets compared with AdV-FLuc–infected control islets (P ≤ 0.05) (Fig. 4B). VIVIT had no effect on IRS-2 mRNA or protein levels at basal 3 mmol/L glucose (Fig. 4A and B), indicating that this was specific for glucose-induced regulation of IRS-2 mRNA/protein expression in islet β-cells. To further investigate NFAT-mediated control of IRS-2 expression, the effect of increasing ectopic expression of NFATc1 isoform on IRS-2 expression in rat islets using an adenovirus AdV-NFATc1 was examined. In AdV-NFATc1–infected INS-1 cells (Fig. 5A) and rat islets (Fig. 5C), there was an approximate twofold increase in specific IRS-2 mRNA expression at basal 3 mmol/L glucose compared with respective AdV-FLuc–infected control INS-1 or rat islets (P ≤ 0.05), which was further enhanced at 15 mmol/L glucose (Fig. 5A and C). A complementary increase in IRS-2 protein expression was observed at basal 3 mmol/L glucose in AdV-NFATc1–infected INS-1 cells (Fig. 5B) and rat islets (Fig. 5D) compared relative to AdV-FLuc–infected control rat islets. Taken together, these results demonstrate that NFATc1 is implicated in the regulation of glucose-induced IRS-2 expression in β-cells.
FIG. 4.

Glucose regulation of IRS-2 expression in β-cells is decreased when NFAT is specifically inhibited. Isolated rat pancreatic islets were infected with a recombinant adenovirus expressing a specific peptide inhibitor of NFAT, AdV-VIVIT, or with AdV-FLuc and cultured overnight at normal 5.6 mmol/L glucose. Afterward, the adenovirally infected islets were then incubated at either basal 3 mmol/L or stimulatory 15 mmol/L glucose concentration for 6 h. A: IRS-2 and β-actin (internal reference control) mRNA expression levels were measured by real-time fluorescence-based quantitative RT-PCR. B: IRS-2 and PI3K(p85) (control) protein expression levels were analyzed by immunoblotting, where an example immunoblot (IB) and quantitative measurements are shown. The data are shown as a mean ± SE above basal 3 mmol/L glucose control, where * indicates a statistically significant increase at 15 mmol/L glucose above basal 3 mmol/L glucose and ** indicates statistically significant decrease at 15 mmol/L glucose in AdV-VIVIT–infected versus AdV-FLuc–infected control islets (P ≤ 0.05; n ≥ 4) (A and B).
FIG. 5.
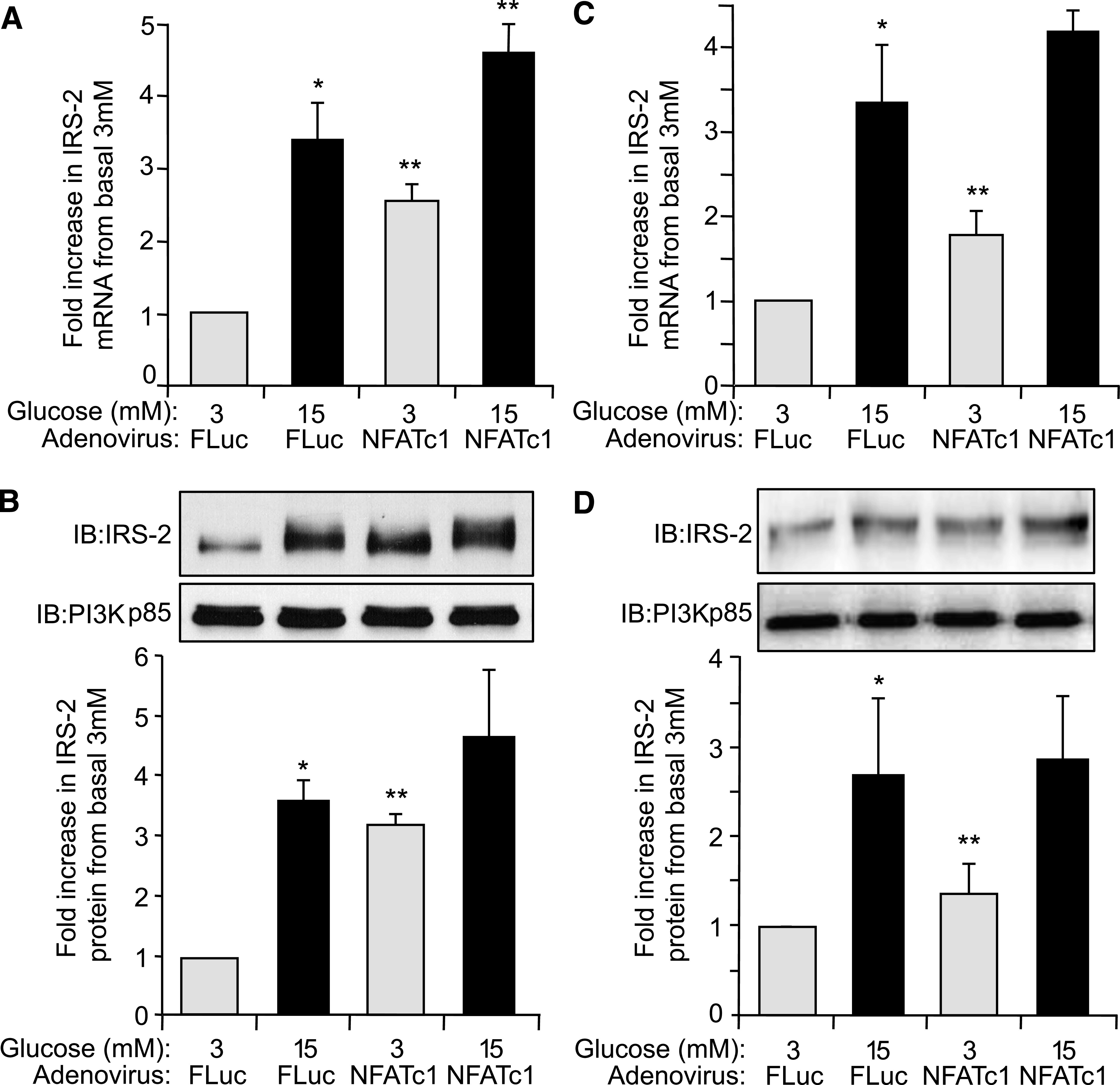
Glucose regulation of IRS-2 expression in β-cells is enhanced by increased expression of NFATc1. INS-1 β-cells (A and C) or isolated rat pancreatic islets (B and D) were infected with AdV-NFATc1 or with AdV-FLuc and cultured overnight at normal 5.6 mmol/L glucose. Afterward, the adenovirally infected INS-1 β-cells or islets were then incubated at either basal 3 mmol/L or stimulatory 15 mmol/L glucose concentration for 6 h. A and C: IRS-2 and β-actin (internal reference control) mRNA expression levels were measured by real-time fluorescence-based quantitative RT-PCR. The data are shown as a mean ± SE, where * indicates a statistically significant increase at 15 mmol/L glucose above basal 3 mmol/L glucose and ** indicates a significant increase in AdV-NFATc1–infected islets versus AdV-FLuc–infected control islets at respective glucose concentrations (P ≤ 0.05; n ≥ 4). B and D: IRS-2 and PI3K(p85) (control) protein expression levels were analyzed by immunoblotting, where example immunoblots (IBs) are shown. Quantified data are also shown as a mean ± SE, where * indicates a statistically significant increase at 15 mmol/L glucose above basal 3 mmol/L glucose and ** indicates a significant increase in AdV-NFATc1–infected islets versus AdV-FLuc–infected control islets at basal 3 mmol/L glucose (P ≤ 0.05; n ≥ 4).
NFATc1 binds to rat IRS-2 gene promoter in β-cells.
NFAT transcription factors bind to a consensus sequence (GGAAA) in target genes promoter to transactivate gene expression. Bioinformatic analysis of the first 3,020 bp of rat IRS-2 promoter (using a basic local alignment search tool [BLAST] search) indicated the presence of nine potential NFAT binding consensus sequences (Fig. 6A). To test whether NFATc1 binds to these consensus sequences, we conducted a ChIP assay using chromatin from INS-1 β-cells expressing NFATc1 or FLuc as a control, using AdV-NFATc1 and AdV-FLuc, respectively. Oligonucleotide DNA primers (Supplementary Fig. 1) were used to amplify specific regions containing NFAT binding sites on the IRS-2 promoter as indicated: sites A, sites B and C together, site D, site E, or sites F through to I together (Fig. 6A). Significant NFATc1 binding to sequences containing NFAT binding sites A, B/C, and D in the IRS-2 promoter was increased in AdV-NFATc1–infected cells compared with AdV-FLuc–infected control cells (P ≤ 0.05) (Fig. 6B and C). This binding was specific for NFATc1, since nothing was detected in rabbit IgG immunoprecipitated controls (Fig. 6B). These results suggest that the four NFAT consensus binding sequences proximal to the transcriptional start site on the endogenous IRS-2 promoter (sites A–D) are indeed binding sites for NFATc1 to control IRS-2 expression. The putative NFAT binding motifs more distal to the transcriptional start site on the IRS-2 promoter (sites E–I) (Fig. 6A) are not specifically recognized by NFATc1. Intriguingly, the NFAT binding sites A and D in the IRS-2 promoter are highly conserved across mammalian species, including human (Supplementary Fig. 1).
FIG. 6.
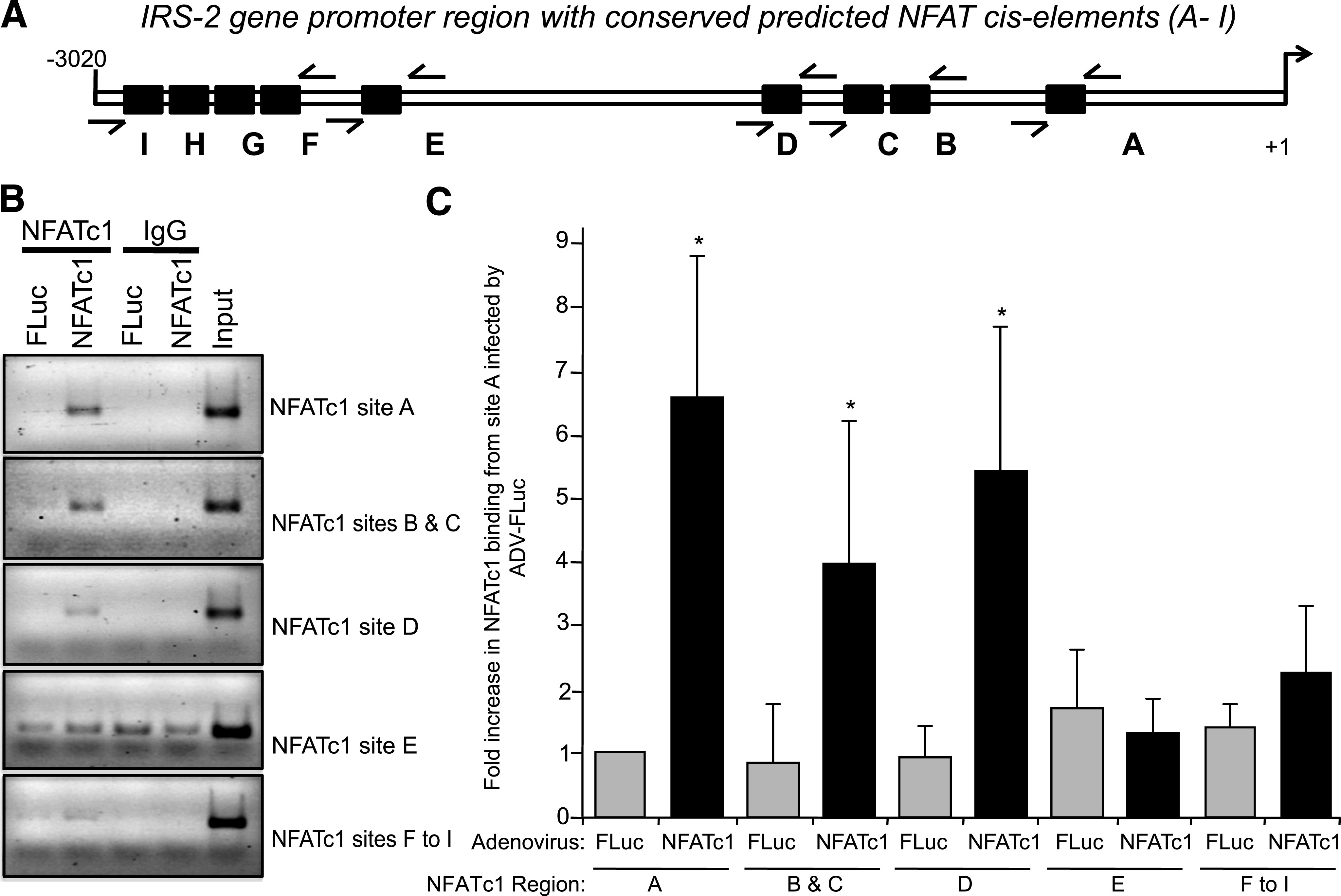
NFATc1 binds to rat IRS-2 gene promoter in β-cells. A: Predicted NFAT binding site elements on rat IRS-2 gene promoter. Examination of the rat IRS-2 gene promoter from −3,020 to +1 bp (transcription start point) showed multiple NFATc1 consensus sequences (GGAAA). Black boxes (A–I) represent NFAT consensus binding sites, and arrows indicates the five primer pairs used for the ChIP assay. B and C: Specific binding of NFATc1 to the endogenous rat IRS-2 promoter. INS-1 β-cells were infected with AdV-NFATc1, or AdV-Fluc as control, and the ChIP assay was performed using anti-NFATc1 antibody or Mouse IgG as a negative control. The input (B) corresponds to cross-linked and sheared chromatin. DNA fragments were then submitted to PCR amplification using primers to amplify five regions in the IRS-2 promoter containing the NFAT binding sites A, B and C together, D, E, or F through I as indicated above. PCR products were loaded onto an agarose gel, where an example is depicted (B). A series of ChIP assays were conducted (n = 6), and the combined data are shown as a mean ± SD above AdV-FLuc control for region A, where * indicates a significant increase (P ≤ 0.05) in AdV-NFATc1–infected cells versus control AdV-FLuc–infected cells (C).
Glucose induces NFATc1 translocation from cytosol to nucleus in β-cells.
NFAT activity is tightly regulated by calcineurin, which induces its activation by dephosphorylation, leading to NFAT translocation from the cytosol to the nucleus (30). By contrast, phosphorylated NFAT has negligible transcriptional activity and is retained in the cytosol. It was found that a 15 mmol/L stimulatory glucose concentration caused specific accumulation of NFATc1 to the nucleus of isolated rat islets and INS-1 cells, and this glucose-induced translocation of NFATc1 to the nucleus was prevented in the presence of FK506 (Fig. 7A and B). In complementary experiments in INS-1 cells, immunofluorescent staining of NFATc1 revealed that NFATc1 was mainly localized in the cytosolic compartment at basal 3 mmol/L glucose (Fig. 7C). In contrast, NFATc1 was mostly localized in the nucleus at 15 mmol/L stimulatory glucose concentration (Fig. 7C). Moreover, this glucose-induced translocation of NFATc1 from the cytosol to the nucleus of INS-1 cells was completely blocked in the presence of FK506 (Fig. 7C). Collectively, these data indicate that 15 mmol/L stimulatory glucose induces the translocation of NFATc1 from the cytosolic compartment to the nucleus of islet β-cell in a calcineurin-dependent manner.
FIG. 7.
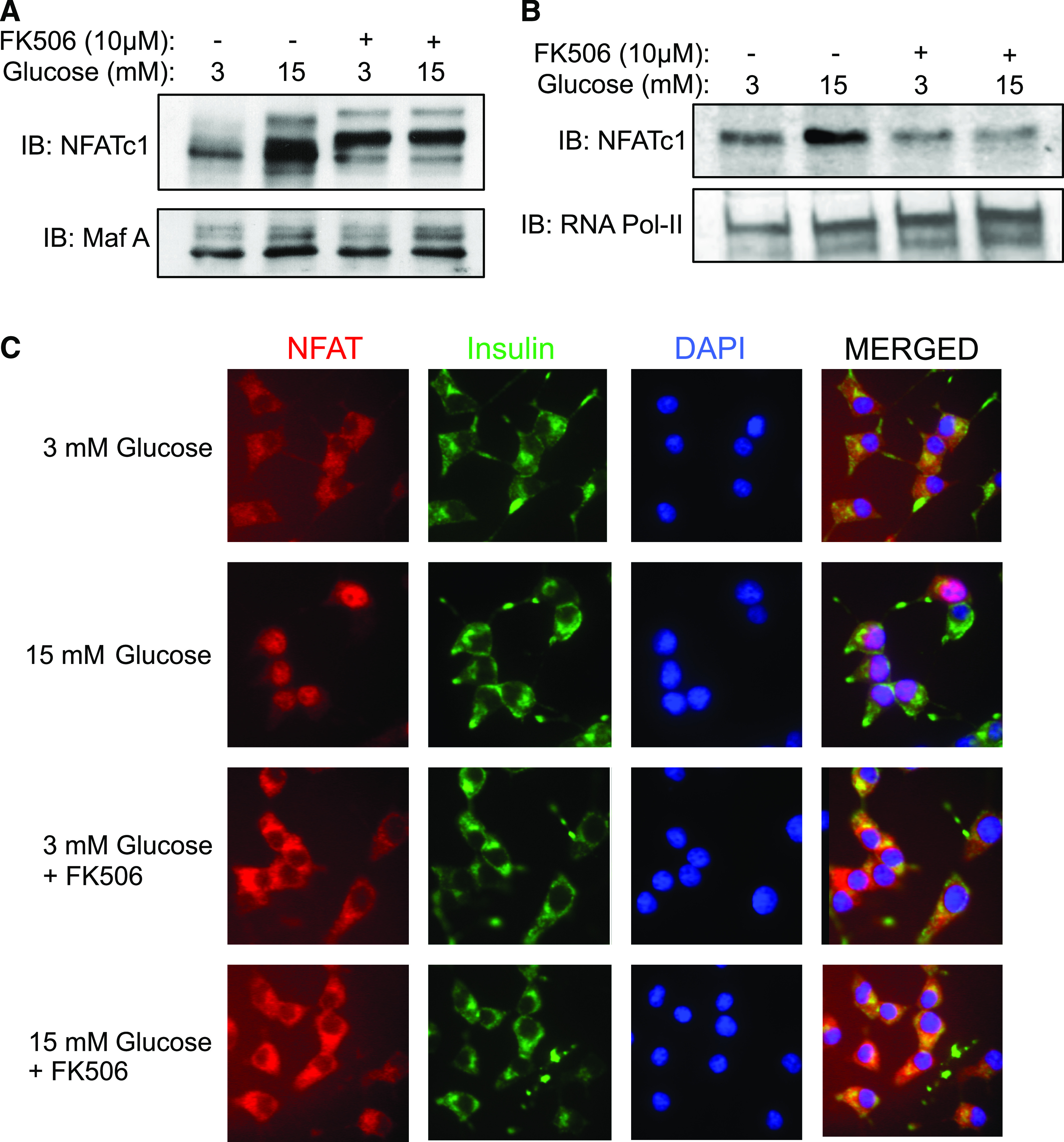
Glucose induces NFATc1 translocation from cytosol to nucleus in β-cells. INS-1 β-cells (A and C) or isolated rat islets (B) were cultured overnight at 5.6 mmol/L glucose and then incubated at either basal 3 mmol/L or stimulatory 15 mmol/L glucose concentration for 6 h ± FK506 (10 µmol/L). A and B: Nuclear proteins were extracted and nuclear NFATc1 protein expression levels were analyzed by immunoblotting relative to a Maf-A (A) or RNA polymerase-II (Pol-II) (B) protein expression as loading controls. Example immunoblots (IBs) of three individual experiments are shown. C: Cellular localization of NFATc1 in INS-1 pancreatic β-cells. NFATc1 (red) localization was analyzed by immunofluorescence and confocal microscopy relative to insulin immunostaining (green) and nuclei staining by DAPI (blue). Example images of three individual experiments are shown. (A high-quality digital representation of this figure is available in the online issue.)
DISCUSSION
Pancreatic β-cells are highly regulated by certain nutrients, peptide hormones, neurotransmitters, and pharmacological compounds, but the most physiologically relevant of these is glucose (31). Glucose metabolism is required to generate secondary signals that in turn control many of the normal functions of β-cells including insulin secretion, (pro)insulin production, adaptive β-cell growth, and survival (31). IRS-2 also is key to β-cell survival and compensatory growth in response to an increased metabolic load (4,6,7). IRS-2 turns over relatively rapidly in β-cells (10), but this is compensated for by IRS-2 expression being highly regulated at the transcriptional level, especially by glucose (10) and a glucose-dependent potentiation by incretins like GLP-1 (12). This glucose-mediated regulation of IRS-2 expression in islet β-cells occurs in vivo and is possibly unique to β-cells, since it is not found in other nutrient-sensing organs such as the liver (Fig. 1).
Here we show that the mechanism behind specific glucose-induced transcriptional regulation of IRS-2 expression in pancreatic β-cells (10) is predominately mediated by a Ca2+/calcineurin/NFAT signaling pathway. It is well established that an elevation in circulating blood glucose leads to increased β-cell glucose metabolism, which then raises the ATP-to-ADP ratio. This results in closing ATP-sensitive K+ channels, depolarization of the β-cell plasma membrane, opening of voltage-sensitive L-type Ca2+ channels, and an influx of Ca2+ into the β-cell that rapidly raises cytosolic [Ca2+]i (32). This is the principal signaling mechanism that leads to triggering insulin secretion from β-cells in response to elevated glucose (33,34). However, increased cytosolic [Ca2+]i can also activate other signal transduction pathways in β-cells, including the Ca2+/calmodulin-dependent activation of calcineurin (also known as protein phosphatase-2B). There are two isoforms of calcineurin, but only the β-isoform of calcineurin is expressed in pancreatic islet β-cells (25). Once calcineurin-β is activated in β-cells, it can dephosphorylate NFAT transcription factors, which are then activated and translocate from the cytosol to the nucleus where they bind to consensus NFAT binding elements (GGAAA) on certain gene promoters to specifically enhance their transcription (28). Here, glucose-induced increases in β-cell IRS-2 mRNA were paralleled by a complementary increase in IRS-2 protein levels, reaffirming this regulation was mediated at the transcriptional level (10). Moreover, our data are consistent with Ca2+/calcineurin/NFAT signaling being required for specific glucose-induced increase in IRS-2 expression in β-cells. Depolarization of the β-cell enhanced islet β-cell IRS-2 mRNA/protein expression, whereas blocking the rise in β-cell cytosolic [Ca2+]i with voltage-sensitive l-type Ca2+-channel inhibitors prevented the glucose-induced increase in IRS-2 mRNA/protein expression. Calcineurin inhibitors (FK506, CsA, and CAIN) specifically prevented glucose-induced increase in IRS-2 mRNA/protein expression and, in contrast, a constitutively activated calcineurin variant increased islet β-cell IRS-2 mRNA/protein expression. Calcineurin inhibitors also prevented a glucose-induced translocation of NFAT from the β-cell cytosol to the nucleus. Inhibition of NFAT by VIVIT prevented the specific increase in β-cell IRS-2 mRNA/protein expression by glucose, and adenoviral-mediated increase in NFATc1 expression enhanced islet β-cell IRS-2 mRNA/protein expression. NFATc1 was also shown to specifically associate to certain NFAT binding elements in the β-cell endogenous IRS-2 promoter, proximal to the transcriptional start site.
Specific inhibition of Ca2+ influx, calcineurin, and/or NFAT activation had minimal effect on IRS-2 mRNA/protein expression at basal 3 mmol/L glucose, despite effectively blocking glucose-induced IRS-2 expression in islet β-cells. But because a need for β-cell glucose metabolism was bypassed, such as causing depolarization of the β-cell, expression of activated calcineurin, or forced expression of NFATc1, an increased expression of IRS-2 mRNA/protein at basal 3 mmol/L glucose could be observed. However, under normal circumstances glucose does not control basal IRS-2 expression. Indeed, we have also shown that basal IRS-2 expression in β-cells is independent of glucose and mostly controlled by forkhead box class O (FoxO) transcription factors (35). The Ca2+/calcineurin/NFAT signaling is only directed at glucose-induced control of IRS-2 expression in β-cells. This has important implications for the glucose-dependent aspect of GLP-1–mediated transcriptional regulation of IRS-2 expression in β-cells via the cAMP-response element binding protein (CREB) (12). The CREB transcription factor interacts with CREB-regulated transcriptional coactivator-1 (CRTC-1), which is also activated by Ca2+/calcineurin–mediated dephosphorylation (36). Thus Ca2+/calcineurin can coordinate CRTC-1/CREB activation, in parallel to that for NFAT, rendering the glucose-dependent aspect for potentiating IRS-2 expression in β-cells by incretins, such as GLP-1 (13).
We have found that NFATc1 can specifically associate to conserved NFAT binding elements in the IRS-2 promoter. However, it should be noted that NFATc1 was used only as a model member of the NFAT family of transcription factors to implicate Ca2+/calcineurin/NFAT signaling in glucose-induced control of IRS-2 expression in β-cells. Although NFATc1 has been implicated in glucose-mediated transcriptional control of important β-cell genes, such as Pdx-1, glucokinase, and insulin (17,37), it cannot be ruled out that other NFAT isoforms are involved in glucose-induced regulation of β-cell IRS-2 gene transcription. Moreover, NFAT-mediated control of IRS-2 gene transcription is likely to be complex and require additional trans-acting factor partners to help efficiently drive IRS-2 gene transcription, which may include activation protein-1 (AP-1), CCAAT-enhancer–binding protein (C/EBP), myocyte enhancer factor-2 (MEF2), musculoaponeurotic fibrosarcoma oncogene homolog A (MafA), or peroxisome proliferator–activated receptor-γ (PPARγ) (30). However, very few NFAT transcriptional partners have been described in islet β-cells, and additional experimentation will be required to identify which are the key NFAT coactivators required for glucose-induced control of IRS-2 expression specific to β-cells.
The Ca2+/calcineurin/NFAT signaling in islet β-cells has been implicated to be important for β-cell survival, at least in part, via control of IRS-2 expression (18). Here, we show that Ca2+/calcineurin/NFAT activation is required for the glucose-induced increases in IRS-2 expression in β-cells that correlates with previous observations where normal physiological fluctuations in glucose have positive effects on β-cell survival (38). In addition, Ca2+/calcineurin/NFAT signaling has been implicated as an important contributor to the regulation of β-cell proliferation and function (17,39). Moreover, Ca2+/calmodulin activation of calcineurin is critical for important β-cell functions, such as insulin secretory granule transport and the second phase of glucose-induced insulin secretion (25). It is likely that Ca2+/calcineurin/NFAT signaling, together with regulation of IRS-2 expression in β-cells, makes an important contribution for the upregulation of β-cell functional mass to compensate for insulin resistance and increased metabolic load found in nondiabetes obesity (1). Intriguingly, calcineurin inhibitors (e.g., tacrolimus/FK506 and CsA) are commonly used as immunosuppressants in organ transplantation, and a frequent complication of their use is acquired post-transplant diabetes mellitus (PTDM). It is thought that PTDM is symptomatic of the long-term use of calcineurin inhibitors that compromises β-cell function, growth, and survival (40). This idea is corroborated by the findings of this study that show Ca2+/calcineurin/NFAT signaling is required for glucose-induced control of IRS-2 expression specifically in β-cells, and without this regulatory mechanism β-cell survival is compromised (18). However, it should also be noted that Ca2+/calcineurin/NFAT signaling must be tightly controlled in β-cells since uncontrolled chronic activation of calcineurin in β-cells can also lead to decreased β-cell mass and hyperglycemia (41).
In summary, we have found the mechanism behind glucose-induced transcriptional control of IRS-2 gene expression in islet β-cells is mediated by the Ca2+/calcineurin/NFAT pathway. Calcineurin/NFAT is an important axis in the functional adaptation of the β-cell to changes in metabolic demand. Considering the pivotal role that control of IRS-2 expression plays β-cell growth and survival (1,7), the identification of this pathway behind a regulation of IRS-2 expression in β-cells may represent a new therapeutic means of maintaining a functional β-cell mass that could avoid the onset of type 2 diabetes. For example, it introduces the concept that calcineurin activators could be beneficial to β-cells via upregulating IRS-2 expression and maintaining their survival. However, such hypothetical compounds would have to be relatively short acting in vivo to prevent chronic immune side effects (40) or overstimulate the β-cell (41). On the other hand, considering that calcineurin inhibitors are particularly detrimental to β-cell function and survival in PTDM (18,25), and glucose did not increase IRS-2 expression in the liver (Fig. 1), short-acting calcineurin activators have potential to be relatively selective toward β-cells. However, for the moment, this is speculative, and only future studies will reveal whether such an approach that exploits the mechanism of glucose-induced IRS-2 expression in β-cells emerges and proves to be effective therapeutically.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health Grant DK-55267 (to C.J.R.) and the Brehm Coalition. D.D. has been supported in part by a postdoctoral fellowship from the Fondation pour la Recherche Médicale.
No potential conflicts of interest relevant to this article were reported.
D.D. researched data, contributed to the experimental design and discussion, and wrote and edited the manuscript. S.T. researched data, contributed to the experimental design and discussion, and edited the manuscript. I.B. researched data and contributed to the experimental design. R.S. contributed to the experimental design and researched data. C.J.R. contributed to the experimental design and discussion and edited and reviewed the manuscript.
The authors thank Dr. C.M. Norris from the University of Kentucky, Lexington, Kentucky, for kindly providing AdV-VIVIT and Dr. J.D. Molkentin from the University of Cincinnati, Cincinnati, Ohio, for providing AdV-CAIN and AdV-CNca.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db11-0341/-/DC1.
See accompanying original article, p. 2883.
REFERENCES
- 1.Rhodes CJ. Type 2 diabetes-a matter of β-cell life and death? Science 2005;307:380–384 [DOI] [PubMed] [Google Scholar]
- 2.Chang-Chen KJ, Mullur R, Bernal-Mizrachi E. Beta-cell failure as a complication of diabetes. Rev Endocr Metab Disord 2008;9:329–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Butler PC, Meier JJ, Butler AE, Bhushan A. The replication of beta cells in normal physiology, in disease and for therapy. Nat Clin Pract Endocrinol Metab 2007;3:758–768 [DOI] [PubMed] [Google Scholar]
- 4.Withers DJ, Gutierrez JS, Towery H, et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature 1998;391:900–904 [DOI] [PubMed] [Google Scholar]
- 5.Withers DJ, Burks DJ, Towery HH, Altamuro SL, Flint CL, White MF. Irs-2 coordinates Igf-1 receptor-mediated beta-cell development and peripheral insulin signalling. Nat Genet 1999;23:32–40 [DOI] [PubMed] [Google Scholar]
- 6.Terauchi Y, Takamoto I, Kubota N, et al. Glucokinase and IRS-2 are required for compensatory beta cell hyperplasia in response to high-fat diet-induced insulin resistance. J Clin Invest 2007;117:246–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.White MF. Regulating insulin signaling and beta-cell function through IRS proteins. Can J Physiol Pharmacol 2006;84:725–737 [DOI] [PubMed] [Google Scholar]
- 8.Rhodes CJ, White MF. Molecular insights into insulin action and secretion. Eur J Clin Invest 2002;32(Suppl. 3):3–13 [DOI] [PubMed] [Google Scholar]
- 9.Hennige AM, Burks DJ, Ozcan U, et al. Upregulation of insulin receptor substrate-2 in pancreatic beta cells prevents diabetes. J Clin Invest 2003;112:1521–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lingohr MK, Briaud I, Dickson LM, et al. Specific regulation of IRS-2 expression by glucose in rat primary pancreatic islet beta-cells. J Biol Chem 2006;281:15884–15892 [DOI] [PubMed] [Google Scholar]
- 11.Amacker-Françoys I, Mohanty S, Niessen M, Spinas GA, Trüb T. The metabolisable hexoses D-glucose and D-mannose enhance the expression of IRS-2 but not of IRS-1 in pancreatic beta-cells. Exp Clin Endocrinol Diabetes 2005;113:423–429 [DOI] [PubMed] [Google Scholar]
- 12.Jhala US, Canettieri G, Screaton RA, et al. cAMP promotes pancreatic beta-cell survival via CREB-mediated induction of IRS2. Genes Dev 2003;17:1575–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Drucker DJ. The biology of incretin hormones. Cell Metab 2006;3:153–165 [DOI] [PubMed] [Google Scholar]
- 14.Weir GC, Bonner-Weir S. Five stages of evolving β-cell dysfunction during progression to diabetes. Diabetes 2004;53(Suppl. 3):S16–S21 [DOI] [PubMed] [Google Scholar]
- 15.Takamoto I, Terauchi Y, Kubota N, Ohsugi M, Ueki K, Kadowaki T. Crucial role of insulin receptor substrate-2 in compensatory beta-cell hyperplasia in response to high fat diet-induced insulin resistance. Diabetes Obes Metab 2008;10(Suppl. 4):147–156 [DOI] [PubMed] [Google Scholar]
- 16.Alonso LC, Yokoe T, Zhang P, et al. Glucose infusion in mice: a new model to induce β-cell replication. Diabetes 2007;56:1792–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heit JJ, Apelqvist AA, Gu X, et al. Calcineurin/NFAT signalling regulates pancreatic beta-cell growth and function. Nature 2006;443:345–349 [DOI] [PubMed] [Google Scholar]
- 18.Soleimanpour SA, Crutchlow MF, Ferrari AM, et al. Calcineurin signaling regulates human islet beta-cell survival. J Biol Chem 2010;285:40050–40059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lingohr MK, Dickson LM, Wrede CE, et al. Decreasing IRS-2 expression in pancreatic beta-cells (INS-1) promotes apoptosis, which can be compensated for by introduction of IRS-4 expression. Mol Cell Endocrinol 2003;209:17–31 [DOI] [PubMed] [Google Scholar]
- 20.Alarcón C, Lincoln B, Rhodes CJ. The biosynthesis of the subtilisin-related proprotein convertase PC3, but no that of the PC2 convertase, is regulated by glucose in parallel to proinsulin biosynthesis in rat pancreatic islets. J Biol Chem 1993;268:4276–4280 [PubMed] [Google Scholar]
- 21.Asfari M, Janjic D, Meda P, Li G, Halban PA, Wollheim CB. Establishment of 2-mercaptoethanol-dependent differentiated insulin-secreting cell lines. Endocrinology 1992;130:167–178 [DOI] [PubMed] [Google Scholar]
- 22.Schreiber E, Matthias P, Müller MM, Schaffner W. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res 1989;17:6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang SN, Berggren PO. Beta-cell CaV channel regulation in physiology and pathophysiology. Am J Physiol Endocrinol Metab 2005;288:E16–E28 [DOI] [PubMed] [Google Scholar]
- 24.Klee CB, Ren H, Wang X. Regulation of the calmodulin-stimulated protein phosphatase, calcineurin. J Biol Chem 1998;273:13367–13370 [DOI] [PubMed] [Google Scholar]
- 25.Donelan M, Julyan R, Kajio H, et al. Ca2+-dependent dephosphorylation of kinesin heavy chain on beta-granules in pancreatic beta-cells. Implications for regulated beta granule transport and insulin exocytosis. J Biol Chem 2002;277:24232–24242 [DOI] [PubMed] [Google Scholar]
- 26.Liu J, Farmer JDJ, Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991;66:807–815 [DOI] [PubMed] [Google Scholar]
- 27.Lai MM, Burnett PE, Wolosker H, Blackshaw S, Snyder SH. Cain, a novel physiologic protein inhibitor of calcineurin. J Biol Chem 1998;273:18325–18331 [DOI] [PubMed] [Google Scholar]
- 28.Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev 2003;17:2205–2232 [DOI] [PubMed] [Google Scholar]
- 29.Aramburu J, Yaffe MB, López-Rodríguez C, Cantley LC, Hogan PG, Rao A. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science 1999;285:2129–2133 [DOI] [PubMed] [Google Scholar]
- 30.Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol 2005;5:472–484 [DOI] [PubMed] [Google Scholar]
- 31.Rhodes CJ. Processing of the insulin molecule. In Diabetes Mellitus. A Fundamental and Clinical Text. 3rd ed. LeRoith D, Taylor SI, Olefsky JM, Eds. Philadelphia, PA, Lippincott-Raven Publishers, 2004, p. 27–50 [Google Scholar]
- 32.Deeney JT, Prentki M, Corkey BE. Metabolic control of β-cell function. Semin Cell Dev Biol 2000;11:267–275 [DOI] [PubMed] [Google Scholar]
- 33.Easom RA. β-granule transport and exocytosis. Semin Cell Dev Biol 2000;11:253–266 [DOI] [PubMed] [Google Scholar]
- 34.Wang Z, Thurmond DC. Mechanisms of biphasic insulin-granule exocytosis— roles of the cytoskeleton, small GTPases and SNARE proteins. J Cell Sci 2009;122:893–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsunekawa S, Demozay D, Briaud I, McCuaig J, Accili D, Stein R, Rhodes CJ. FoxO feedback control of basal IRS-2 expression in pancreatic β-cells is distinct from that in hepatocytes. Diabetes 2011;60:2883–2891. [DOI] [PMC free article] [PubMed]
- 36.Screaton RA, Conkright MD, Katoh Y, et al. The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell 2004;119:61–74 [DOI] [PubMed] [Google Scholar]
- 37.Lawrence MC, Bhatt HS, Watterson JM, Easom RA. Regulation of insulin gene transcription by a Ca2+-responsive pathway involving calcineurin and nuclear factor of activated T cells. Mol Endocrinol 2001;15:1758–1767 [DOI] [PubMed] [Google Scholar]
- 38.Wrede CE, Dickson LM, Lingohr MK, Briaud I, Rhodes CJ. Protein kinase B/Akt prevents fatty acid-induced apoptosis in pancreatic beta-cells (INS-1). J Biol Chem 2002;277:49676–49684 [DOI] [PubMed] [Google Scholar]
- 39.Nir T, Melton DA, Dor Y. Recovery from diabetes in mice by beta cell regeneration. J Clin Invest 2007;117:2553–2561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Penfornis A, Kury-Paulin S. Immunosuppressive drug-induced diabetes. Diabetes Metab 2006;32:539–546 [DOI] [PubMed] [Google Scholar]
- 41.Bernal-Mizrachi E, Cras-Méneur C, Ye BR, Johnson JD, Permutt MA. Transgenic overexpression of active calcineurin in beta-cells results in decreased beta-cell mass and hyperglycemia. PLoS ONE 2010;5:e11969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yaekura K, Julyan R, Wicksteed BL, et al. Insulin secretory deficiency and glucose intolerance in Rab3A null mice. J Biol Chem 2003;278:9715–9721 [DOI] [PubMed] [Google Scholar]