

Abstract
Quetiapine is an established drug for treatment of schizophrenia, bipolar disorder, and major depressive disorder. While initially manufactured as an immediate-release (IR) formulation, an extended-release (XR) formulation has recently been introduced. Pharmacokinetic studies show that quetiapine XR provides a lower peak and more stable plasma concentration than the IR formulation. This study investigated if the pharmacokinetic differences translate into different time curves for central D2 dopamine receptor occupancy. Eleven control subjects were examined with positron emission tomography (PET) and the radioligand [11C]raclopride. Eight subjects underwent all of the scheduled PET measurements. After baseline examination, quetiapine XR was administered once-daily for 8 d titrated to 300 mg/d on days 5–8, followed by 300 mg/d quetiapine IR on days 9–12. PET measurements were repeated after the last doses of quetiapine XR and IR at predicted times of peak and trough plasma concentrations. Striatal D2 receptor occupancy was calculated using the simplified reference tissue model. Peak D2 receptor occupancy was significantly higher with quetiapine IR than XR in all subjects (50±4% and 32±11%, respectively), consistent with lower peak plasma concentrations for the XR formulation. Trough D2 receptor occupancy was similarly low for both formulations (IR 7±7%, XR 8±6%). The lower peak receptor occupancy associated with quetiapine XR may explain observed pharmacodynamic differences between the formulations. Assuming that our findings in control subjects are valid for patients with schizophrenia, the study supports the view that quetiapine, like the prototype atypical antipsychotic clozapine, may show antipsychotic effect at lower D2 receptor occupancy than typical antipsychotics.
Keywords: D2 dopamine receptor, PET, quetiapine
Introduction
Quetiapine is an atypical antipsychotic drug with efficacy demonstrated in clinical trials for the treatment of schizophrenia, bipolar disorder, major depressive disorder, and generalized anxiety disorder. The clinical effects are thought to be mediated by both quetiapine and its main active human metabolite, norquetiapine (Jensen et al. 2008). Quetiapine in its original immediate-release (IR) formulation has a short half-life (7 h) (Nemeroff et al. 2002) and twice-daily administration is recommended for the treatment of schizophrenia and bipolar mania. To allow for once-daily dosing, an extended-release (XR) formulation has more recently been developed. Pharmacokinetic studies have shown that, when given in equivalent daily doses, the exposure in terms of area under the curve (AUC) of the two compounds is comparable. Whereas the XR formulation has a slightly lower peak plasma concentration (Cmax) than the IR formulation, it has a longer half-life (Figueroa et al. 2009).
To explore if there may be clinically important pharmacodynamic differences between the two formulations, it is of interest to establish if the difference in pharmacokinetic profile translates into different brain exposure and time-course of central receptor occupancy. Positron emission tomography (PET) studies of D2 dopamine receptor occupancy have been well validated and demonstrated to have high test–retest reproducibility (Nyberg et al. 1996), with D2 receptor occupancy showing a curvilinear relationship with plasma drug concentrations. In patients treated with quetiapine, PET studies have confirmed occupancy of the D2 receptor in the human brain. A marked fluctuation in D2 receptor occupancy during a dosing interval has been shown in patients treated with quetiapine IR at clinically relevant doses. Several studies have reported moderate D2 receptor occupancy (range 29–74%) at peak plasma levels, whereas D2 receptor occupancy has been low or undetectable at trough plasma levels (Gefvert et al. 1998; Kapur et al. 2000b; Mamo et al. 2008; Tauscher-Wisniewski et al. 2002). To date, quetiapine in the new XR formulation has been examined in a single study (Mamo et al. 2008). In the cross-over study by Mamo and colleagues 12 patients with schizophrenia or schizoaffective disorder were assigned to one of three dose groups of quetiapine (300, 600 or 800 mg/d). D2 occupancy was compared at estimated times of peak and trough plasma concentrations (Tmax and Tmin, respectively) of equivalent daily doses of quetiapine XR and IR. There was no statistically significant difference in D2 receptor occupancy between the formulations. However, the study cannot be viewed as conclusive, since the lack of a reported difference could be due to a large spread in plasma concentrations and the small sample size in each dose group (n=4). Moreover, the PET measurements at expected IR peak were performed 1 h post-administration and the Tmax of the IR formulation has been reported to be 2 h (Figueroa et al. 2009). Consequently, the D2 occupancy at IR peak in this study might have been underestimated, as plasma concentrations were not measured throughout the dosing interval to confirm that PET measurements were performed at true peak concentration.
The primary aim of the present study was to compare striatal D2 receptor occupancy with quetiapine IR and XR in equal doses. A secondary purpose was to examine the temporal relationship between the plasma concentration of quetiapine and D2 receptor occupancy. Eleven control subjects were examined with PET and the radioligand [11C]raclopride, before and after treatment with 300 mg/d quetiapine XR and IR, respectively. The D2 receptor occupancy values of the present study are discussed in relation to literature values for classical (typical) and atypical antipsychotic drugs.
Method
Study design
The study was conducted as a research collaboration between AstraZeneca, Södertälje, Sweden and the Department of Clinical Neuroscience, Karolinska Institutet, Stockholm, Sweden, and was approved by the local Ethics and Radiation Safety Committees and the Medical Products Agency in Sweden. The study was designed as an open label, one-sequence cross-over study, where each subject was his own control.
Study subjects
Eleven males aged between 21 and 29 yr (25±2.5, mean±s.d.) were recruited from an AstraZeneca database of subjects who expressed an interest in participating in clinical studies. The subjects were healthy according to medical history including psychiatric symptoms and physical examination including ECG, routine blood tests, and magnetic resonance imaging (MRI) examination of the brain. A negative urine drug screen was required for inclusion. No prescribed drugs were allowed from 7 d before the first PET measurement and throughout the study. The use of nicotine products was forbidden and caffeine intake was restricted to a maximum of five cups per day. The intake of these substances was monitored at an AstraZeneca study unit, where subjects were supervised throughout the study period. All subjects gave verbal and written consent after receiving a description of the study.
Administration of study drug and PET measurement schedule
Quetiapine was administered orally, once-daily, in the morning for 12 d. After 4 d of dose titration of quetiapine XR from 50 mg up to 300 mg, each subject received 300 mg/d quetiapine XR for 4 d. Treatment was then directly switched to 300 mg/d quetiapine IR for 4 d.
Each subject participated in five PET measurements in total. The measurements were performed at drug-free conditions (baseline), at expected maximum plasma concentration (Cmax) of quetiapine – which has been reported to be at 2 h after the quetiapine IR dose and 5 h after the XR dose (Figueroa et al. 2009) – and at trough plasma concentration (Cmin). The PET measurements at Cmax were performed on the fourth day of administration of XR and IR, respectively, whereas the measurements at Cmin were performed on the morning after the last dose of each formulation.
Determination of plasma concentrations
Blood samples for determination of plasma concentrations of quetiapine and norquetiapine were collected on the last day of treatment with the respective formulations. For quetiapine XR, samples were drawn pre-dose and at 0.5, 1, 2, 3, 4, 5, 5.5, 6, 8, 12, 24 and 25 h post-dose. For quetiapine IR, samples were obtained pre-dose and at 0.5, 1, 1.5, 2, 2.5, 5, 11, 24 and 25 h post-dose. The differences in sampling times for the two formulations were to allow for dense sampling at expected Tmax. The samples were analysed by BASi (Bioanalytical Systems Inc., USA) by a liquid chromatography–tandem mass spectrometry (LC–MS/MS) method described in detail elsewhere (Davis et al. 2010). The lower limit of quantification is <0.70 ng/ml (corresponding to <1.8 nmol/l quetiapine) (Davis et al. 2010).
MRI and PET measurements
MRI T1-weighted images were acquired using a 1.5 T GE Signa system (GE Medical Systems, USA). PET examinations were performed using high-resolution research tomography (HRRT; Siemens Molecular Imaging, USA) with a spatial resolution of ~1.5 mm full-width-half-maximum (FWHM) (Varrone et al. 2009). A plastic helmet made individually for each subject was used during each PET examination in order to minimize head movements and to ensure maintenance of the same head position in all experiments.
[11C]raclopride was prepared by methylation of the corresponding desmethyl precursor using [11C]methyl triflate, as described previously (Langer et al. 1999). At each examination, a saline solution containing [11C]raclopride with a radioactivity of 201–236 MBq was injected in the antecubital vein as a bolus over 2 s. The specific radioactivity at the time of injection exceeded 1900 Ci/mmol and the injected ligand mass was on all occasions <5 μg. This low mass does not cause any substantial mass effect of the ligand (Farde et al. 1995). Radioactivity in the brain was measured continuously over 63 min. The radioactivity from each PET measurement was reconstructed in a series of time-frames consisting of four 15-s, four 30-s, six 1-min, six 3-min and six 6-min frames.
Regions of interest (ROIs)
The MR images were realigned to the anterior– posterior commissure plane. ROIs were manually delineated on the realigned MR images for each subject using software originally developed for the Human Brain Atlas (Roland et al. 1994). Left and right putamina were drawn on 6–8 (number depending on individual anatomy) sagittal sections of the brain in concordance with the anatomical limits. The cerebellum was depicted on six adjacent central horizontal sections. MR images were co-registered to averaged PET images using statistical parametric mapping software (SPM5, Wellcome Department of Cognitive Neuroscience, UK). ROIs were then transferred to the series of PET images to generate time–activity curves.
Calculation of binding potential (BP) and D2 receptor occupancy
The BP in each PET measurement was calculated using the simplified reference tissue model, with the putamen as ROI and the cerebellum as reference region (Lammertsma & Hume, 1996). BP refers in this context to BPND, which represents the ratio at equilibrium between the concentration of specifically bound radioligand and that of the non-displaceable radioligand in tissue (Innis et al. 2007).
The D2 receptor occupancy at peak and trough concentration was calculated for each subject according to the equation:
| (1) |
where Occ is the D2 receptor occupancy, BPbaseline is the BP in the drug-free state, and BPdrug is the BP measured at the respective time-points (peak and trough) of drug treatment.
Calculation of apparent inhibition constant (Ki,app)
When assuming a linear relationship between drug concentration in brain and drug concentration in plasma, the relationship between D2 receptor occupancy and plasma drug concentration can be described by a curvilinear function according to the expression:
| (2) |
where Occmax is the maximal D2 receptor occupancy induced by the drug and Cp is the plasma concentration of the drug (Karlsson et al. 1995). Ki,app is an affinity constant corresponding to the drug plasma concentration required to occupy 50% of available receptors. D2 receptor occupancy values were plotted vs. the corresponding plasma concentrations of quetiapine at the PET measurements. From the measured values of occupancy and concentration, Ki,app was determined by fitting equation (2) to experimental data using a nonlinear least-squares minimization procedure. Occmax was fixed to 100%, as it has been previously demonstrated that 100% D2 receptor occupancy is approached for [11C]raclopride binding during treatment with high doses of antipsychotic drugs (Nyberg et al. 1998).
The ‘active moiety’
To allow comparisons with previous studies of D2 receptor occupancy after quetiapine treatment, the initial calculation of Ki,app and the estimated time curve for D2 receptor occupancy were based on the plasma concentration values for quetiapine only. The affinity of the main metabolite, norquetiapine, for the D2 receptor in vitro is of the same order as that for quetiapine (Jensen et al. 2008). Assuming similar affinity and similar passage across the blood–brain barrier in human subjects in vivo, the plasma concentration values of quetiapine and norquetiapine were summed to obtain a value for the ‘active moiety’. This approach has previously been applied for risperidone and its active metabolite, 9-OH risperidone (Cohen, 1994; Nyberg et al. 1993, 1999). In the calculation of Ki,app, the explanatory power of the active moiety was compared to quetiapine alone.
Estimation of the time curve for D2 receptor occupancy during a dosing interval
To obtain a time curve for D2 receptor occupancy throughout a dosing interval, the calculated Ki,app was entered in equation (2) together with the mean plasma concentration of quetiapine at each time-point. To enable comparisons between the two formulations, results were plotted separately for quetiapine XR and IR.
Statistics
A two-tailed, two-sample t test was used to compare the mean plasma concentrations achieved at peak and trough. The same test was used to compare mean D2 receptor occupancy with quetiapine IR and XR at Cmax and Cmin. Neuroreceptor occupancy is not normally distributed, due to the curvilinear relationship between plasma concentration and occupancy, and occupancy values can thus not be expected to be normally distributed. However, at low concentrations, the curve is approximately linear, which may allow the use of a parametric test. In the present study, the maximal mean D2 receptor occupancy measured at peak was 59% and therefore the t test was also applied to test differences at peak occupancies.
To ensure that quetiapine plasma concentration at Tmax was not significantly different from quetiapine plasma concentration during peak PET measurements, the t test was also applied to compare mean Cmax with mean concentration during peak PET (Cav,PET,peak) for the respective formulations.
The statistical analysis was performed using SAS statistical software JMP 8 (SAS Institute, USA).
Results
Of the 11 subjects enrolled in the study, eight underwent all of the scheduled PET examinations according to protocol. One subject discontinued before the fourth PET measurement due to an adverse event (panic attack, anxiety), and PET measurements and plasma concentrations during treatment with the IR formulation were therefore not obtained from this subject. Due to synthesis failure of the radioligand, PET at XR trough was not performed for one subject and PET at IR peak was delayed 3.5 h for another subject. D2 receptor occupancy and plasma concentration values from the delayed PET measurement were not included in the analysis, as the timing of this measurement did not correspond to the time of peak concentration.
Plasma concentrations
The plasma concentrations of quetiapine were maximal at about 1.7 h after treatment with the IR formulation and about 5 h after administration of the XR formulation (Table 1, Fig. 1). Cmax for quetiapine (mean±s.d.) was 1850±820 nmol/l and 894±380 nmol/l for the IR and XR formulations, respectively. The difference between formulations was statistically significant (d.f.=11.0, t=3.18, p<0.01). At Tmax, the concentration of norquetiapine was 624±100 nmol/l for IR and 346±110 nmol/l for XR (d.f.=17.0, t=5.55, p<0.0001).
Table 1.
Summary statistics of pharmacokinetic parameters of quetiapine and norquetiapine (PK analysis set)
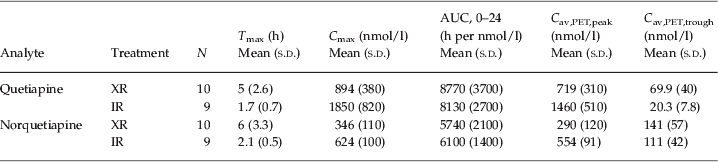
Tmax, Time of peak plasma concentration; Cmax, peak plasma concentration; AUC, area under the curve; Cav,PET,peak, average concentration of drug in plasma during PET examination at expected peak; Cav,PET,trough, average concentration of drug in plasma during PET examination at expected trough.
Fig. 1.

Arithmetic mean plasma concentration of quetiapine and norquetiapine during quetiapine IR (left) and XR (right) administration.
For both formulations, the mean trough concentrations of quetiapine during PET were significantly lower than the mean peak concentrations (IR: d.f.=8.0, t=6.66, p=0.0002; XR: d.f.=9.2, t=6.82, p<0.0001) (Fig. 1). Concentrations of quetiapine were 20.3±7.8 nmol/l for IR and 69.9±40 nmol/l for XR during the second (trough) PET measurement. The difference was statistically significant (d.f.=9.7, t=3.82, p<0.005). Trough concentrations of norquetiapine were 111±42 and 141±57 nmol/l for IR and XR, respectively. This difference was not statistically significant (d.f.=16.4, t=1.32, p=0.2).
D2 receptor occupancy
The PET measurements were performed close to the observed peak concentrations of quetiapine and norquetiapine, respectively. The time difference between the start time of the PET peak measurements and Tmax of quetiapine were: IR (0.14±0.43 h, range −0.55 to 0.9) and XR (0.12±2.6 h, range −6.76 to 2.24). Thus, a larger range was noted for the XR formulation, but the more blunted plasma concentration curve with this formulation makes the timing of the PET measurement less sensitive to deviations from Tmax. The difference between quetiapine Cmax and the plasma concentration measured during the peak PET measurements (Cav,PET,peak) was not statistically significant for either formulation (IR: Cmax 1850±820 vs. Cav,PET,peak 1460±510, d.f.=13.3, t=1.20, p=0.25; XR: Cmax 894±380 vs. Cav,PET,peak 719±310, d.f.=17.2, t=1.13, p=0.27). In all subjects, D2 receptor occupancy was higher at IR peak compared to XR peak (Figs 2 and 3). The mean D2 receptor occupancy in the putamen at peak concentration was 50±4% for quetiapine IR (n=9) and 32±11% for XR (n=11). The difference was statistically significant (d.f.=13.5, t=4.89, p=0.0003). There was no significant difference in D2 receptor occupancy at trough levels between the two formulations [IR 7±7% (n=10), XR 8±6% (n=10); d.f.=17.6, t=0.41, p=0.7]. In two subjects, trough occupancy even reached negative values (−1% at XR trough for one subject and −3% at IR trough for another subject). However, this minor negativity was within the range for test–retest reliability.
Fig. 2.

Parametric horizontal PET images through the caudate putamen level showing the binding potential after intravenous injection of [11C]raclopride in a human subject at baseline conditions and after administration of quetiapine IR and XR, respectively.
Fig. 3.

D2 dopamine receptor occupancy (%) in 11 subjects after administration of quetiapine IR and XR, respectively.
Apparent inhibition constant (Ki,app)
When using all PET measurements and the concentration of quetiapine measured after administration of IR and XR together, Ki,app was calculated to be 1369 nmol/l (Fig. 4). Ki,app calculated for quetiapine IR only was 1373 nmol/l and for XR only was 1365 nmol/l. Assuming that quetiapine and norquetiapine have the same affinity for the D2 receptor, the sum of the two concentrations was used as an estimate of the active moiety. Ki,app calculated for the active moiety using measurements for both formulations was 2004 nmol/l, and Ki,app calculated for IR and XR separately was 1968 nmol/l and 2041 nmol/l, respectively. The explanatory value of quetiapine and norquetiapine alone or in combination (interaction model) was also tested using nonlinear mixed-effects modelling. Goodness of fit was not improved using the interaction model (results not shown).
Fig. 4.

D2 dopamine receptor occupancy (%) vs. quetiapine plasma concentration (IR and XR formulations).
The mean peak D2 receptor occupancy measured after treatment with 300 mg of the XR formulation was 32% (range 13–45%). As the relationship between dose and plasma concentration of quetiapine is linear (DeVane & Nemeroff, 2001), it is possible to use equation (2) (see Methods section) to predict the dose required to occupy 50% of the receptor. The calculated Ki,app value corresponded to a dose of 638 mg at steady state following once-daily dosing. For the IR formulation, the mean peak D2 receptor occupancy measured was 50%, thus corresponding to the administered dose of 300 mg at steady state following once-daily dosing.
Estimated D2 receptor occupancy over time
The time curve for estimated D2 receptor occupancy during a dosing interval was calculated for both formulations using equation (2) (Fig. 5). After administration of quetiapine IR, a high and transient peak D2 receptor occupancy was reached within 2 h, after which D2 receptor occupancy slowly declined. For the XR formulation, peak D2 receptor occupancy was less pronounced and remained at a higher level compared to IR.
Fig. 5.

Estimated D2 dopamine receptor occupancy (%) vs. time for quetiapine IR (left) and XR (right) formulations. The solid lines describe the occupancy time curve for a typical individual. The grey areas represent the 95% prediction intervals.
Discussion
In the present PET study, we compared the D2 dopamine receptor occupancy obtained after administration of 300 mg quetiapine XR and IR once-daily at steady-state conditions. Peak D2 receptor occupancy was significantly higher (p=0.0003) with the IR formulation (Fig. 3), whereas there was no significant difference in trough D2 receptor occupancy between quetiapine IR (7±7%) and XR (8±6%). The study confirms that the previously reported pharmacokinetic differences between the two formulations (Figueroa et al. 2009) translate into different time curves for D2 receptor occupancy in the human brain.
The 300 mg daily dose used in the present study is at the lower end of the dose interval recommended for the treatment of schizophrenia, i.e. 150–750 mg for IR and 400–800 mg for XR (Seroquel® Prescribing Information, 2010a; Seroquel® XR Prescribing Information, 2010b). As for other CNS drugs, previous pharmacokinetic studies of quetiapine IR have shown inter-individual variability in the relationship between dose and plasma concentration (DeVane & Nemeroff, 2001; Hasselstrom & Linnet, 2004).
In clinical practice, quetiapine IR is predominantly administered twice-daily for the treatment of schizophrenia and bipolar mania according to approved labels. However, once-daily dosing of quetiapine IR has shown efficacy in bipolar depression (Calabrese et al. 2005). As the primary aim of this study was to achieve a direct comparison of the XR and IR formulations, IR was administered only once-daily. The plasma concentration and corresponding D2 receptor occupancy can therefore not be directly translated to the more commonly used twice-daily dosing. The peak D2 receptor occupancy associated with standard 150 mg twice-daily treatment with the IR formulation would, in all probability, be lower than the receptor occupancy obtained in the present study.
The dose of quetiapine XR required to occupy 50% of the D2 receptor was estimated to be 638 mg (continuous once-daily dosing). The recommended dose of quetiapine XR for the treatment of schizophrenia (i.e. 400–800 mg) would, according to the same equation, correspond to 39–56% occupancy of the D2 receptor. This estimation is in agreement with the literature, since the average peak D2 receptor occupancy measured after dosing with 800 mg quetiapine XR has been reported to be 56% (Mamo et al. 2008).
For typical antipsychotic drugs, it has been suggested that D2 receptor occupancy of at least 65–70% is required for an antipsychotic effect, and that the risk of extrapyramidal side-effects (EPS) is high when D2 receptor occupancy exceeds 80% (Farde et al. 1992; Kapur et al. 2000a). A therapeutic window of 65–80% D2 receptor occupancy has therefore been suggested. In PET studies with recommended doses of the second-generation antipsychotic drugs, risperidone and olanzapine, D2 receptor occupancy has been within this window (Kapur et al. 1999; Nordström et al. 1998; Nyberg et al. 1999; Tauscher et al. 2002). However, the atypical antipsychotic drug, clozapine, has an antipsychotic effect at lower D2 receptor occupancy (20–67%), despite an unsurpassed efficacy in the treatment of schizophrenia (Kane, 1989; Nordström et al. 1995). Previous PET studies have shown that quetiapine induces low to moderate D2 receptor occupancy (29–74% at peak, 0–27% at trough) when given at clinical doses (Gefvert et al. 1998; Kapur et al. 2000b; Mamo et al. 2008; Tauscher-Wisniewski et al. 2002). The results in the present study corroborate these previous observations. Based on calculations using equation (2) (see Methods section), 1185 mg quetiapine XR would be required to achieve 65% D2 occupancy at peak. It can thus not be excluded that quetiapine, like clozapine, induces an antipsychotic effect at low striatal D2 receptor occupancy (for review see Nord & Farde, 2010). However, whether the antipsychotic effect is related to D2 occupancy alone, or if additional mechanisms are involved remains to be elucidated.
The Ki,app value can also be used to calculate the quetiapine dose required to exceed 80% of D2 receptor occupancy, above which the risk of EPS is increased (Farde et al. 1992; Kapur et al. 2000a). Interestingly, quetiapine XR doses exceeding 2550 mg (4×Ki,app) would be needed to reach this level. Thus, during treatment with clinically recommended doses, it is unlikely that D2 receptor occupancy reaches the 80% threshold suggested for EPS. This is in agreement with clinical studies showing a low incidence of EPS in quetiapine-treated patients (Baldwin & Scott, 2009; Weiden, 2007).
A strength of this study is that individual baseline values of BP were used to calculate D2 receptor occupancy. Using this methodology, errors based on individual variability in receptor densities are minimized. However, a critical experimental condition is that D2 receptor occupancy is indeed measured at Cmax. As the peak is sharp, peak PET measurements with IR are particularly sensitive to errors if Tmax and the start of PET measurement are not exactly matched. A comparison of the plasma concentration curve with the time of the peak PET measurement for individual subjects shows that the PET measurements in most cases started at Tmax. In one subject the PET measurement started just after Tmax and in another subject PET measurement started just before Tmax, but the peak concentrations were reached within the time-frame of the PET measurements. Peak D2 receptor occupancy values in these two subjects were 45% and 52%, and were thus within the range of other assessments. However, the possibility that D2 receptor occupancy might have been underestimated in these two subjects cannot be excluded. Regarding the XR formulation, peak PET measurements are less sensitive as the time curve for plasma concentration is more blunted.
Pharmacokinetic studies have demonstrated dose equivalence of the two formulations based on the calculated AUC for standardized dosing intervals (Figueroa et al. 2009). The present pharmacokinetic findings confirm similar AUC and consistent Ki values between the formulations (Table 1). However, our study also demonstrates more pronounced fluctuations in D2 receptor occupancy with quetiapine IR than XR. It cannot be excluded that this difference may result in different pharmacodynamics. Indeed, a recent experimental study comparing quetiapine XR and IR, titrated from 50 mg up to 300 mg once-daily, in 63 healthy subjects demonstrated that quetiapine XR produced significantly less sedation than quetiapine IR during the initial treatment phase (Datto et al. 2009).
The findings in this study might seem discrepant to the findings in a previous study of Mamo et al. (2008), where no statistical difference in D2 occupancy was shown between quetiapine IR and XR. An important difference between the studies is that quetiapine IR was given twice-daily in the study of Mamo et al. whereas we used a once-daily administration. As already described, the peak occupancy after a twice-daily administration of quetiapine IR would in all likelihood be lower than the peak occupancy after the same dose administered once-daily. The Ki value received from our data was similar for both formulations, indicating that the difference in occupancy between the formulations depends on differences in plasma concentration. As the treatment regimen in the study by Mamo et al. did not provide any statistically different peak plasma concentrations of quetiapine IR and XR, this condition could explain the lack of difference in the peak occupancy measured.
Conclusion
Central D2 dopamine receptor occupancy differentiates quetiapine IR and XR. The more sustained D2 receptor occupancy after administration of quetiapine XR vs. IR makes this formulation appropriate for the once-daily treatment regimen. The findings in the present study support the view that quetiapine may have an antipsychotic effect at moderate and intermediate striatal D2 receptor occupancy. Further studies are needed to evaluate if the antipsychotic effect of quetiapine depends on D2 receptor occupancy alone or if, intriguingly, there are additional antipsychotic mechanisms related to other receptor systems.
Acknowledgements
The study was sponsored by AstraZeneca, Södertälje, Sweden. The authors thank the members of the PET group at the Karolinska Institutet and the AstraZeneca Clinical Pharmacology Unit, Huddinge, for kind assistance during this study. The funding of this paper for Open Access publication was provided by AstraZeneca, Södertälje, Sweden. Bill Wolvey from PAREXEL provided medical writing support funded by AstraZeneca. The authors are entirely responsible for the scientific content of the paper.
[Clinical trial registration information: Positron emission tomography (PET) study with [11C]raclopride to determine central D2 dopamine occupancy of SEROQUEL (registration identification no.: NCT00832221) (http://www.clinicaltrials.gov/ct2/show/NCT00832221?term=quetiapine&rank=42)].
Statement of Interest
M.N. was temporarily employed at AstraZeneca at the time when the clinical part of the study was performed. S.N., J.B., A.J., and L.F. are full- or part-time employees of AstraZeneca.
References
- Baldwin CM, Scott LJ. Quetiapine extended release: in schizophrenia. CNS Drugs. 2009;23:261–269. doi: 10.2165/00023210-200923030-00007. [DOI] [PubMed] [Google Scholar]
- Calabrese JR, Keck PE, Macfadden W, Minkwitz M. et al. A randomized, double-blind, placebo controlled trial of quetiapine in the treatment of bipolar I or II depression. American Journal of Psychiatry. 2005;162:1351–1360. doi: 10.1176/appi.ajp.162.7.1351. [DOI] [PubMed] [Google Scholar]
- Cohen LJ. Risperidone. Pharmacotherapy. 1994;14:253–265. [PubMed] [Google Scholar]
- Datto C, Berggren L, Patel JB, Eriksson H. Self-reported sedation profile of immediate-release quetiapine fumarate compared with extended-release quetiapine fumarate during dose initiation: a randomized, double-blind, crossover study in healthy adult subjects. Clinical Therapeutics. 2009;31:492–502. doi: 10.1016/j.clinthera.2009.03.002. [DOI] [PubMed] [Google Scholar]
- Davis PC, Bravo O, Gehrke M, Azumaya CT. Development and validation of an LC-MS/MS method for the determination of quetiapine and four related metabolites in human plasma. Journal of Pharmaceutical and Biomedical Analysis. 2010;51:1113–1119. doi: 10.1016/j.jpba.2009.11.018. [DOI] [PubMed] [Google Scholar]
- DeVane CL, Nemeroff CB. Clinical pharmacokinetics of quetiapine: an atypical antipsychotic. Clinical Pharmacokinetics. 2001;40:509–522. doi: 10.2165/00003088-200140070-00003. [DOI] [PubMed] [Google Scholar]
- Farde L, Hall H, Pauli S, Halldin C. Variability in D2-dopamine receptor density and affinity: a PET study with [11C]raclopride in man. Synapse. 1995;20:200–208. doi: 10.1002/syn.890200303. [DOI] [PubMed] [Google Scholar]
- Farde L, Nordström AL, Wiesel FA, Pauli S. et al. Positron emission tomographic analysis of central D1 and D2 dopamine receptor occupancy in patients treated with classical neuroleptics and clozapine. Relation to extrapyramidal side effects. Archives of General Psychiatry. 1992;49:538–544. doi: 10.1001/archpsyc.1992.01820070032005. [DOI] [PubMed] [Google Scholar]
- Figueroa C, Brecher M, Hamer-Maansson JE, Winter H. Pharmacokinetic profiles of extended release quetiapine fumarate compared with quetiapine immediate release. Progress in Neuropsychopharmacology and Biological Psychiatry. 2009;33:199–204. doi: 10.1016/j.pnpbp.2008.09.026. [DOI] [PubMed] [Google Scholar]
- Gefvert O, Bergström M, Långström B, Lundberg T. et al. Time course of central nervous dopamine-D2 and 5-HT2 receptor blockade and plasma drug concentrations after discontinuation of quetiapine (Seroquel) in patients with schizophrenia. Psychopharmacology (Berlin) 1998;135:119–126. doi: 10.1007/s002130050492. [DOI] [PubMed] [Google Scholar]
- Hasselstrom J, Linnet K. Quetiapine serum concentrations in psychiatric patients: the influence of comedication. Therapeutic Drug Monitoring. 2004;26:486–491. doi: 10.1097/00007691-200410000-00005. [DOI] [PubMed] [Google Scholar]
- Innis RB, Cunningham VJ, Delforge J, Fujita M. et al. Consensus nomenclature for in vivo imaging of reversibly binding radioligands. Journal of Cerebral Blood Flow and Metabolism. 2007;27:1533–1539. doi: 10.1038/sj.jcbfm.9600493. [DOI] [PubMed] [Google Scholar]
- Jensen NH, Rodriguiz RM, Caron MG, Wetsel WC. et al. N-desalkylquetiapine, a potent norepinephrine reuptake inhibitor and partial 5-HT1A agonist, as a putative mediator of quetiapine's antidepressant activity. Neuropsychopharmacology. 2008;33:2303–2312. doi: 10.1038/sj.npp.1301646. [DOI] [PubMed] [Google Scholar]
- Kane JM. The current status of neuroleptic therapy. Journal of Clinical Psychiatry. 1989;50:322–328. [PubMed] [Google Scholar]
- Kapur S Zipursky R Jones C Remington G et al. 2000aRelationship between dopamine D2 occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia American Journal of Psychiatry 157514–520. [DOI] [PubMed] [Google Scholar]
- Kapur S Zipursky R Jones C Shammi CS et al. 2000bA positron emission tomography study of quetiapine in schizophrenia: a preliminary finding of an antipsychotic effect with only transiently high dopamine D2 receptor occupancy Archives of General Psychiatry 57553–559. [DOI] [PubMed] [Google Scholar]
- Kapur S, Zipursky RB, Remington G. Clinical and theoretical implications of 5-HT2 and D2 receptor occupancy of clozapine, risperidone, and olanzapine in schizophrenia. American Journal of Psychiatry. 1999;156:286–293. doi: 10.1176/ajp.156.2.286. [DOI] [PubMed] [Google Scholar]
- Karlsson P, Farde L, Halldin C, Sedvall G. et al. Oral administration of NNC 756 – a placebo controlled PET study of D1-dopamine receptor occupancy and pharmacodynamics in man. Psychopharmacology (Berlin) 1995;119:1–8. doi: 10.1007/BF02246046. [DOI] [PubMed] [Google Scholar]
- Lammertsma AA, Hume SP. Simplified reference tissue model for PET receptor studies. Neuroimage. 1996;4:153–158. doi: 10.1006/nimg.1996.0066. [DOI] [PubMed] [Google Scholar]
- Langer O, Nagren K, Dolle F, Lundkvist C. et al. Precursor synthesis and radiolabelling of the dopamine D2 receptor ligand [11C]raclopride from [11C]methyl triflate. Journal of Labelled Compounds and Radiopharmaceuticals. 1999;42:1183–1193. [Google Scholar]
- Mamo DC, Uchida H, Vitcu I, Barsoum P. et al. Quetiapine extended-release vs. immediate-release formulation: a positron emission tomography study. Journal of Clinical Psychiatry. 2008;69:81–86. doi: 10.4088/jcp.v69n0111. [DOI] [PubMed] [Google Scholar]
- Nemeroff CB, Kinkead B, Goldstein J. Quetiapine: preclinical studies, pharmacokinetics, drug interactions, and dosing. Journal of Clinical Psychiatry. 2002;63:5. (Suppl. 13), [PubMed] [Google Scholar]
- Nord M, Farde L. Antipsychotic occupancy of dopamine receptors in schizophrenia. CNS Neuroscience & Therapeutics. 2010;17:97. doi: 10.1111/j.1755-5949.2010.00222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordström AL, Farde L, Nyberg S, Karlsson P. et al. D1, D2, and 5-HT2 receptor occupancy in relation to clozapine serum concentration: a PET study of schizophrenic patients. American Journal of Psychiatry. 1995;152:1444–1449. doi: 10.1176/ajp.152.10.1444. [DOI] [PubMed] [Google Scholar]
- Nordström AL, Nyberg S, Olsson H, Farde L. Positron emission tomography finding of a high striatal D2 receptor occupancy in olanzapine-treated patients. Archives of General Psychiatry. 1998;55:283–284. doi: 10.1001/archpsyc.55.3.283. [DOI] [PubMed] [Google Scholar]
- Nyberg S, Dencker SJ, Malm U, Dahl ML. et al. D2- and 5-HT2 receptor occupancy in high-dose neuroleptic-treated patients. International Journal of Neuropsychopharmacology. 1998;1:95. doi: 10.1017/S1461145798001229. [DOI] [PubMed] [Google Scholar]
- Nyberg S, Eriksson B, Oxenstierna G, Halldin C. et al. Suggested minimal effective dose of risperidone based on PET-measured D2 and 5-HT2A receptor occupancy in schizophrenic patients. American Journal of Psychiatry. 1999;156:869–875. doi: 10.1176/ajp.156.6.869. [DOI] [PubMed] [Google Scholar]
- Nyberg S, Farde L, Eriksson L, Halldin C. et al. 5-HT2 and D2 dopamine receptor occupancy in the living human brain. A PET study with risperidone. Psychopharmacology (Berlin) 1993;110:265–272. doi: 10.1007/BF02251280. [DOI] [PubMed] [Google Scholar]
- Nyberg S, Farde L, Halldin C. Test-retest reliability of central [11C]raclopride binding at high D2 receptor occupancy. A PET study in haloperidol-treated patients. Psychiatry Research. 1996;67:163–171. doi: 10.1016/0925-4927(96)02921-6. [DOI] [PubMed] [Google Scholar]
- Roland PE, Graufelds CJ, Wåhlin J, Ingelman L. et al. Human brain atlas for high-resolution functional and anatomical mapping. Human Brain Mapping. 1994;1:173–184. doi: 10.1002/hbm.460010303. [DOI] [PubMed] [Google Scholar]
- Seroquel® Prescribing Information 2010a). Quetiapine fumarate (http://www1.astrazeneca-us.com/pi/Seroquel.pdf). Accessed November 2010.
- Seroquel® XR Prescribing Information 2010b). Quetiapine fumarate (http://www1.astrazeneca-us.com/pi/seroquelxr.pdf). Accessed November 2010.
- Tauscher J, Jones C, Remington G, Zipursky RB. et al. Significant dissociation of brain and plasma kinetics with antipsychotics. Molecular Psychiatry. 2002;7:317–321. doi: 10.1038/sj.mp.4001009. [DOI] [PubMed] [Google Scholar]
- Tauscher-Wisniewski S, Kapur S, Tauscher J, Jones C. et al. Quetiapine: an effective antipsychotic in first-episode schizophrenia despite only transiently high dopamine-2 receptor blockade. Journal of Clinical Psychiatry. 2002;63:992–997. doi: 10.4088/jcp.v63n1106. [DOI] [PubMed] [Google Scholar]
- Varrone A, Sjöholm N, Eriksson L, Gulyás B. et al. Advancement in PET quantification using 3D-OP-OSEM point spread function reconstruction with the HRRT. European Journal of Nuclear Medicine and Molecular Imaging. 2009;36:1639–1650. doi: 10.1007/s00259-009-1156-3. [DOI] [PubMed] [Google Scholar]
- Weiden PJ. EPS profiles: the atypical antipsychotics are not all the same. Journal of Psychiatric Practice. 2007;13:13–24. doi: 10.1097/00131746-200701000-00003. [DOI] [PubMed] [Google Scholar]