

Abstract
OBJECTIVE
Sleep deprivation is associated with increased risk of adult type 2 diabetes mellitus (T2DM). It is uncertain whether sleep deprivation and/or altered sleep architecture affects glycemic regulation or insulin sensitivity or secretion. We hypothesized that in obese adolescents, sleep disturbances would associate with altered glucose and insulin homeostasis.
RESEARCH DESIGN AND METHODS
This cross-sectional observational study of 62 obese adolescents took place at the Clinical and Translational Research Center and Sleep Laboratory in a tertiary care children’s hospital. Subjects underwent oral glucose tolerance test (OGTT), anthropometric measurements, overnight polysomnography, and frequently sampled intravenous glucose tolerance test (FSIGT). Hemoglobin A1c (HbA1c) and serial insulin and glucose levels were obtained, indices of insulin sensitivity and secretion were calculated, and sleep architecture was assessed. Correlation and regression analyses were performed to assess the association of total sleep and sleep stages with measures of insulin and glucose homeostasis, adjusted for confounding variables.
RESULTS
We found significant U-shaped (quadratic) associations between sleep duration and both HbA1c and serial glucose levels on OGTT and positive associations between slow-wave sleep (N3) duration and insulin secretory measures, independent of degree of obesity, pubertal stage, sex, and obstructive sleep apnea measures.
CONCLUSIONS
Insufficient and excessive sleep was associated with short-term and long-term hyperglycemia in our obese adolescents. Decreased N3 was associated with decreased insulin secretion. These effects may be related, with reduced insulin secretory capacity leading to hyperglycemia. We speculate that optimizing sleep may stave off the development of T2DM in obese adolescents.
Sleep deprivation is endemic; 9.3% of U.S. adults sleep <6 h per night (1), and 75% of high-school seniors report getting insufficient sleep (2). This cumulative societal sleep curtailment is significant, as sleep deprivation is associated with a number of metabolic consequences: increased predisposition to obesity (3) and insulin resistance (IR) (4) in both adults and children, increased risk of type 2 diabetes mellitus (T2DM) in adults (5), and higher fasting glucose in young adults with preexisting diabetes (6). The metabolic consequences of insufficient sleep may be the result of a lack of total sleep or insufficiency of a certain sleep component. The American Academy of Sleep Medicine recognizes four different sleep stages indicated as follows: stage 1 (N1), a brief transition between wake and sleep; stage 2 (N2); stage 3 (N3), “slow-wave” or “deep” sleep; and rapid eye movement (REM) (dream) sleep. In adult studies, cerebral glucose utilization declines (7) and plasma glucose rises (8) in N3 sleep. One pediatric study found a negative association between REM sleep duration and obesity (9), but there is little pediatric data on sleep architecture and glucose and insulin homeostasis. A potential confounding factor is obstructive sleep apnea (OSA), a syndrome more common in obesity in which upper airway obstruction leads to sleep fragmentation and desaturation (10). OSA has been associated with T2DM risk in adults (10) and with IR in children (11,12). We hypothesized that in obese adolescents (who are at risk for T2DM), altered sleep architecture is associated with abnormalities of insulin secretion and sensitivity and of glucose homeostasis independently of confounding factors (e.g., degree of obesity, presence of OSA, sex, and pubertal stage). Therefore, the aim of our study was to investigate the relationship between sleep architecture and insulin secretion and sensitivity and overall glycemia in this population.
RESEARCH DESIGN AND METHODS
This was a cross-sectional study of obese (BMI, >95th percentile for age and sex) pubertal adolescents recruited from an obesity clinic in The Children’s Hospital of Philadelphia. Exclusion criteria included having previously diagnosed diabetes or sleep disorders, genetic syndromes affecting glucose tolerance or sleep, or major organ system illness, or taking medications affecting insulin or glucose metabolism. The protocol was approved by The Children’s Hospital of Philadelphia Institutional Review Board; informed consent was obtained from the parents or guardians, and assent was obtained from the participants.
Anthropometrics
Demographic data and medical history were obtained from guardians and participants. Physical examination, including pubertal (Tanner) staging, was performed by a study investigator. Weight was measured using a digital scale (Scaletronix, White Plains, NY). Height was measured using a wall-mounted stadiometer (Holtain Inc., Crymych, U.K.). BMI was calculated as weight (kilograms) divided by height (meters) squared. BMI percentiles and z scores were assessed using age- and sex-specific reference data (13).
Glucose and metabolic testing
After a 12-h overnight fast, an oral glucose tolerance test (OGTT) was performed: subjects ingested oral glucose solution (1.75 g/kg, maximum 75 g), and blood samples for glucose and insulin were obtained at –10, 0, 10, 30, 60, 90, 12, 150, and 180 min. Hemoglobin A1c (HbA1c) was also measured. The following morning, after an overnight fast, subjects underwent a frequently sampled intravenous glucose tolerance test (FSIGT): infusion of 0.25 g/kg of 25% dextrose intravenously over 30 s, infusion of regular human insulin (0.015 units/kg i.v.) over 5 min at t = 20 min, and drawing of blood samples for glucose and insulin at t = −5, 2, 4, 8, 19, 22, 30, 40, 50, 70, 100, and 180 min. Plasma glucose levels were measured by the glucose dehydrogenase method (Hemocue Analyzer; Hemocue Inc., Cypress, CA). Plasma insulin levels were measured by radioimmunoassay (LINCO, St. Charles, MO). The MINMOD Millenium software program (14) was used to estimate indices of glucose and insulin dynamics from the FSIGT.
Calculated insulin sensitivity and secretion parameters
A. OGTT.
- Homeostasis model assessment of IR (HOMA-IR) is a validated measure of insulin sensitivity (15):

- Insulinogenic index (IGI) is a measure of insulin secretion that has been validated in children against the hyperglycemic clamp (16):
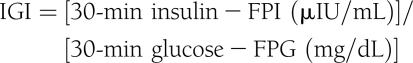
- Whole-body insulin sensitivity index (WBISI) is an insulin sensitivity measure that has been validated in obese children and adolescents (15):
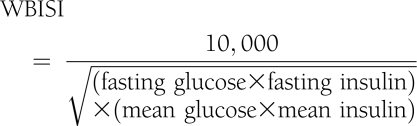
Higher WBISI levels indicate greater insulin sensitivity.
B. FSIGT.
Acute insulin response to glucose (AIRg) is a parameter of early pancreatic response to glucose, calculated as the mean incremental plasma insulin concentration over baseline in the first 8 min of the FSIGT (14).
Sensitivity to insulin (SI) is a parameter calculated from serial insulin and glucose values during the FSIGT (14).
Overnight polysomnography
Overnight polysomnography (PSG) was performed the night between the OGTT and FSIGT. Signals were recorded on a computerized system (Rembrandt; Rescare, Buffalo, NY). The following parameters were recorded: electroencephalogram (C3/A2, C4/A1, O1/A2, and O2/A1); right and left electro-oculograms; submental electromyogram; tibial electromyogram; electrocardiogram; chest and abdominal wall motion by inductance plethysmography; oronasal pressure/airflow (nasal pressure cannula with oral thermistor bead; Pro-Tech, Woodinville, WA); end-tidal PCO2, measured at the nose by infrared capnometry (Nellcor N-1000); arterial oxygen saturation (SaO2) by pulse oximetry, and oximeter pulse waveform. Studies were reviewed by a single sleep Board-certified investigator (L.J.B.), who had no knowledge of subjects’ metabolic status. Sleep architecture (N1, N2, N3, and REM sleep stages) and respiratory disturbances (including the apnea-hypopnea index [AHI], arousal index, and lowest oxyhemoglobin saturation [lowest SaO2]) were scored using standard pediatric criteria (17).
Statistical analysis
Statistical analysis was performed using SPSS Statistics 17.0 analysis software. Histograms and one-sample Kolmogorov-Smirnov tests were used to assess normality of distribution of continuous variables. Distributions of seriously skewed variables were normalized via logarithmic transformation. Pearson or Spearman correlations were used to examine associations between sleep architecture or OSA measures and parameters of glucose homeostasis and insulin secretion and sensitivity. Hierarchical linear regression procedures were used to evaluate the aforementioned relationships while controlling for potential confounding variables (e.g., degree of obesity and OSA). Covariate selection for the stepwise regression stages was guided by correlation analyses. Assumptions of linearity were tested by examining plots of the standardized residuals as a function of standardized predicted values. Where curve estimation procedures uncovered curvilinear relationships, polynomial regressions were conducted. Analysis of covariance (ANCOVA) models were used to examine differences in the outcome variables between sexes and among different pubertal stages, controlling for covariates. As we tested three underlying hypotheses relating to the relationship between sleep architecture and insulin secretion and sensitivity and overall glycemia, we used an adjusted P value of <0.017 (0.05/3) for statistical significance.
RESULTS
Study subjects
Seventy obese adolescents were screened for participation; seven cancelled prior to the study date and one did not undergo PSG, leaving 62 participants for analysis. Baseline subject characteristics are presented in Table 1. Insulin and glucose values, calculated indices, and PSG results are presented in Table 2.
Table 1.
Subject characteristics
| Characteristic | Mean ± SD (range) or number (%) |
|---|---|
| Age (years) | 14.4 ± 2.1 (8–17.5) |
| Sex | |
| Male | 28 (45%) |
| Female | 34 (55%) |
| Race | |
| White | 23 (37.1%) |
| African American | 34 (54.8%) |
| Asian American | 1 (1.6%) |
| >1 race or other | 4 (6.4%) |
| Ethnicity | |
| Hispanic | 8 (17.7%) |
| Non-Hispanic | 54 (82.3%) |
| Tanner stage (breast or genitalia) | |
| Tanner 2 | 5 (8.1%) |
| Tanner 3 | 12 (19.4%) |
| Tanner 4 | 14 (22.6%) |
| Tanner 5 | 31 (50%) |
| BMI (kg/m2) | 36.76 ± 6.82 (26.84–56.33) |
| BMI z score | 2.37 ± 0.38 (1.53–3.21) |
Table 2.
Glucose tolerance testing and PSG results
| Characteristic | Mean ± SD (range) |
|---|---|
| Insulin and glucose measures | |
| Fasting plasma glucose (mg/dL) | 92 ± 10 (74–130) |
| Fasting plasma insulin (μIU/mL) | 26.7 ± 16.1 (6.6–66) |
| 2-h plasma glucose (mg/dL) | 130 ± 31 (90–237) |
| 2-h plasma insulin (μIU/mL) | 194.4 ± 250.6 (6.5–1,541.5) |
| HbA1c (%) | 5.4 ± 0.4 (4.6–6.4) |
| HOMA-IR | 6.1 ± 4.0 (1.4–17.6) |
| IGI | 3.89 ± 3.08 (0.38–12.32) |
| AIRg | 1,787.48 ± 1,635.72 (30.33–7,433.40) |
| WBISI | 2.54 ± 1.57 (0.42–7.37) |
| SI | 2.01 ± 1.39 (0.031–6.29) |
| Sleep architecture and OSA measures | |
| Sleep latency (minutes) | 20.9 ± 18.7 (0.5–91.0) |
| TST (minutes) | 424.6 ± 57.8 (291.5–552) |
| %TST in N1 (%) | 9.2 ± 5.7 (1.5–32.4) |
| %TST in N2 (%) | 49.2 ± 7.2 (31.2–62.5) |
| %TST in N3 (%) | 21.2 ± 4.9 (10.4–32.5) |
| %TST in REM (%) | 20.3 ± 5.2 (8.4–31.3) |
| AHI | 4.7 ± 10.7 (0.0–68.5) |
| Distribution | |
| AHI <5 | N = 49 |
| AHI 5–10 | N = 7 |
| AHI >10 | N = 6 |
| Arousal index (%) | 14.8 ± 9.7 (6.8–72.4) |
| Lowest SaO2 (%) | 92 ± 4 (82–100) |
Sleep architecture (durations are given in minutes; percentages are denoted as %). %TST in N = percentage of total sleep time spent in a given sleep stage (e.g., %TST in N1=% total sleep time in N1).
Sleep and glucose homeostasis
Total sleep time (TST) was significantly or near-significantly associated with both short- and long-term measures of glucose homeostasis (Table 3). Curve estimation modeling and regression statistics showed that these relationships were U shaped (quadratic) (Fig. 1A–C). There was no association between any measure of OSA and measures of glucose homeostasis (Supplementary Table 1).
Table 3.
Correlations of sleep architecture with measures of glucose homeostasis
| TST | N1 duration‡ | N1 (%TST)‡ | N2 duration | N2 (%TST) | N3 duration | N3 (%TST) | REM duration | REM (%TST) | |
|---|---|---|---|---|---|---|---|---|---|
| Fasting plasma glucose (mg/dL) | −0.291 * | 0.129 | 0.195 | −0.105 | 0.130 | −0.328 † | −0.160 | −0.305 ‖ | −0.233 |
| Glu 1 h (mg/dL) | −0.293 * | −0.169 | −0.082 | −0.205 | −0.009 | −0.084 | 0.041 | −0.106 | 0.000 |
| Glu 2 h (mg/dL)‡ | −0.366 † | 0.054 | 0.172 | −0.236 | 0.086 | −0.313 ‖ | −0.103 | −0.221 | −0.071 |
| HbA1c (%) | −0.357 † | 0.146 | 0.225 | −0.185 | 0.050 | −0.235 | −0.037 | −0.350 † | −0.279 * |
All numbers represent correlation coefficients. Glu 1 h, glucose level 1 h after oral glucose ingestion on OGTT; Glu 2 h, glucose level 2 h after oral glucose ingestion on OGTT. Sleep durations are given in minutes (percentages are denoted as %). Numbers in boldface indicate significant association, and numbers in italics indicate near-significant association (P value between 0.017 and 0.05).
*P < 0.05.
‡Spearman correlation analysis.
†P < 0.01.
‖P < 0.017.
Figure 1.

Sleep duration and glucose homeostasis measures. A: Association between sleep duration and fasting plasma glucose levels on OGTT. B: Association between 2-h glucose level on OGTT and total sleep duration (minutes) that evening. C: Association between HbA1c and total sleep duration (minutes) that evening. In all three panels, the U-shaped relationships suggest that a sleep duration of 420–510 min (7–8.5 h) is associated with optimal glucose homeostasis.
On regression analysis, TST was the most significant predictor of glucose homeostasis measures. Individual sleep stages, pubertal stage, and sex were not significant predictors of any glucose homeostasis measure; BMI z score was a significant contributor, and Tanner stage was a marginal contributor, to the overall 2-h glucose model only (not to the overall fasting glucose or HbA1c models). Sex did not contribute significantly to any glucose model.
For the overall regression models mentioned above, adjusted R2 and P values were as follows: 0.201 (P = 0.002) for fasting glucose, 0.442 (P < 0.0005) for 2-h glucose, and 0.200 (P = 0.002) for HbA1c.
Sleep and insulin secretory measures
N3 sleep, both total duration and the percentage of total sleep time in N3 (%TST in N3), correlated significantly or with marginal significance (P value between 0.017 and 0.05) with several insulin secretory measures (Table 4) in bivariate analysis. Curve estimation modeling uncovered a cubic relationship between N3 and AIRg (r2 = 0.286; P = 0.001), with inflection points at approximately 65 and 98 min. OSA measures did not associate significantly with any measure of insulin secretion (Supplementary Table 2). A marginally significant negative association was seen between pubertal stage and both N3 duration (r = −0.282; P = 0.028) and %TST in N3 (r = −0.252; P = 0.050), but there was no association between pubertal stage and any of the insulin secretory measures examined, or between sex and insulin secretory measures.
Table 4.
Correlation of sleep architecture with measures of insulin secretion and sensitivity
| TST | N1 duration (min)‡ | N1 (%TST)‡ | N2 duration (min) | N2 (%TST) | N3 duration (min) | N3 (%TST) | REM duration (min) | REM (%TST) | |
|---|---|---|---|---|---|---|---|---|---|
| Sleep architecture and measures of insulin secretion | |||||||||
| 1-h insulin (μIU/mL)‡ | −0.063 | −0.143 | −0.116 | −0.170 | −0.201 | 0.277 * | 0.288 * | 0.055 | 0.110 |
| 2-h insulin (μIU/mL)‡ | −0.039 | −0.082 | −0.034 | −0.214 | −0.280 * | 0.246 (P = 0.058) | 0.348 † | 0.046 | 0.107 |
| IGI‡ | 0.179 | −0.10 | −0.042 | 0.007 | −0.185 | 0.288 * | 0.265 * | 0.041 | −0.060 |
| AIRg‡ | 0.180 | −0.117 | −0.144 | −0.104 | −0.272 (P = 0.051) | 0.367 † | 0.375 † | 0.137 | 0.081 |
| Sleep architecture and measures of insulin sensitivity | |||||||||
| Fasting insulin (μIU/mL)‡ | −0.005 | 0.115 | 0.137 | −0.267 * | −0.235 | 0.130 | 0.172 | 0.055 | 0.077 |
| HOMA-IR‡ | −0.056 | 0.137 | 0.170 | −0.282 * | −0.202 | 0.083 | 0.148 | −0.008 | 0.021 |
| WBISI | 0.157 | −0.101 | −0.152 | 0.178 | 0.124 | 0.016 | −0.096 | 0.094 | 0.054 |
| SI | 0.139 | −0.101 | −0.139 | 0.321 * | 0.337 * | −0.146 | −0.213 | −0.028 | −0.070 |
Sleep architecture (durations are given in minutes; percentages are denoted as %). %TST in N = percentage of total sleep time spent in a given sleep stage. All numbers represent correlation coefficients. Numbers in boldface indicate significant association, and numbers in italics indicate near-significant association (P value between 0.017 and 0.05).
‡Spearman correlation analysis.
*P < 0.05.
†P < 0.01.
N3 duration remained the strongest predictor of insulin secretory measures on stepwise regression analysis. Other sleep stages, TST, OSA measures, and sex were not significant predictors of insulin secretory measures in the final regression model. BMI z score contributed significantly to the AIRg final model but not to the IGI model, and pubertal stage contributed significantly to the final IGI model but not the AIRg model. Adjusted R2 and P values for the overall models were 0.161 (P = 0.002) for IGI and 0.383 (P < 0.0005) for AIRg.
Sleep and insulin sensitivity
Correlation analysis showed a marginally significant negative association between N2 sleep and several insulin sensitivity measures (Table 4). A marginally significant negative correlation was seen between AHI and SI (r = −0.338; P = 0.025; see Supplementary Table 3). Pubertal stage and sex did not associate significantly with any insulin sensitivity measure. However, on regression analysis, no strong associations were seen between sleep architecture or OSA and OGTT-derived insulin sensitivity measures. We found a marginal relationship between N2 duration and HOMA-IR (overall R2 = 0.088; P = 0.040), and although the relationship between %TST in N2 and SI was stronger, BMI z score (i.e., degree of obesity) was the strongest predictor in that model (overall model R2 = 0.400; P < 0.0005).
CONCLUSIONS
In this multiethnic group of obese adolescents, we found strong relationships between sleep, hyperglycemia, and insulin secretion. Specifically, we found U-shaped relationships between total sleep duration and measures of both short- and long-term glycemia, and positive or cubic associations between N3 and insulin secretory measures, even after adjusting for potential confounders such as degree of obesity, OSA, sex, and pubertal stage. Our sleep duration data suggest that glucose metabolism is optimal when 7.5–8.5 h of sleep is achieved. This is consistent with adult data noting U-shaped associations between self-reported sleep duration and T2DM risk (5).
Adults with T2DM have also been reported to have shorter N3 duration than nondiabetic adults (18). Although one postulated mechanism suggests that N3 loss increases IR (19), our results instead demonstrated a relationship between N3 and insulin secretion. This relationship, which appeared to be a function of N3 itself rather than of total sleep duration, suggested that an absolute N3 duration of 1 h may be needed to achieve a stable amount of insulin secretion, and that increasing N3 might greatly improve insulin secretion. Our results may help explain the aforementioned association between N3 lack and T2DM, as a loss in first-phase insulin secretion is both an early marker of T2DM and part of its pathogenesis (20). Parasympathetic activity, which is increased in N3 sleep (21), stimulates glucose-induced insulin secretion (22); we speculate that increased parasympathetic activity may be responsible for our observed associations. Although growth hormone (GH) secretion occurs largely during the N3 sleep (23), GH is unlikely to be a factor in the associations, as GH increases insulin resistance (24) rather than insulin secretion. In addition, insulin-like growth factor-1 levels in our subjects did not correlate significantly with either sleep duration or any individual sleep stage, further corroborating that GH secretion is unlikely to explain the observed associations.
Finally, we found a positive association between N2 sleep and insulin sensitivity, with varying contribution from degree of obesity. Although N2 duration was also negatively associated with at least one marker of OSA, the AHI, indicating possible confounding, the AHI did not consistently associate with insulin sensitivity measures; thus, the observed association may represent an intrinsic relationship between N2 sleep and insulin sensitivity.
A night in the sleep laboratory does not necessarily reflect sleep at home, a possible limitation of our study. However, in our study, the OGTT preceded the PSG. Thus, any observed associations between sleep parameters and OGTT-derived measures could not have been short-term effects of a night in the laboratory. The association between TST and HbA1c, which reflects long-term glycemia, also supported our findings. Also, families completed post-PSG surveys asking whether sleep in the laboratory was typical of home sleep. Only 32% indicated worse sleep in the laboratory; these subjects had no significant demographic, anthropometric, or glucose/insulin parameter differences compared with the remaining subjects, and of the sleep parameters, the only difference seen was a lower absolute REM duration in the “poorer sleep” group.
Our subjects’ mean TST of 7.1 h was similar to the mean 7.2-h sleep duration reported in the 2006 National Sleep Foundation adolescent poll (2). A recent study showed an association between short sleep on actigraphy and metabolic dysregulation in school-age children (25). Although home-based actigraphy studies could be useful, actigraphy is less precise and cannot discriminate between sleep stages.
In conclusion, we found significant relationships between sleep duration and sleep architecture and measures of glucose homeostasis and insulin secretion. To our knowledge, this is the first report of an association between N3 sleep and changes in β-cell function and of a U-shaped association between sleep duration and glucose levels in a pediatric population. We speculate that inadequate sleep duration and altered sleep architecture (relative suppression of N3 sleep) may play a role in T2DM development. Ensuring adequate sleep might reduce the risk of T2DM in at-risk obese adolescents.
Supplementary Material
Acknowledgments
This study was funded by the Pennsylvania State Tobacco Settlement Fund and Grant UL1-RR-24134 from the National Center for Research Resources (support for the University of Pennsylvania Clinical and Translational Research Center).
No potential conflicts of interest relevant to this article were reported.
D.K. and L.E.L.K. researched data, contributed to discussion, wrote the manuscript, and reviewed and edited the manuscript. P.C.B. researched data, contributed to discussion, and reviewed and edited the manuscript. P.R.G. contributed to discussion, wrote the manuscript (statistician), and reviewed and edited the manuscript. R.I.B. contributed to discussion and reviewed and edited the manuscript. L.J.B. researched data, contributed to discussion, wrote the manuscript, and reviewed and edited the manuscript.
The authors would like to acknowledge the contributions of Carole Marcus (Division of Pediatric Pulmonary Medicine, The Children’s Hospital of Philadelphia, and Department of Pediatrics, Perelman School of Medicine at the University of Pennsylvania), Andrea Kelly (Division of Pediatric Endocrinology and Diabetes, The Children’s Hospital of Philadelphia, and Department of Pediatrics, Perelman School of Medicine at the University of Pennsylvania), and Diva D. De Leon-Crutchlow (Division of Pediatric Endocrinology and Diabetes, The Children’s Hospital of Philadelphia, and Department of Pediatrics, Perelman School of Medicine at the University of Pennsylvania).
Footnotes
This article contains Supplementary Data online at http://care.diabetesjournals.org/lookup/suppl/doi:10.2337/dc11-1093/-/DC1.
The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
References
- 1.Knutson KL, Van Cauter E, Rathouz PJ, DeLeire T, Lauderdale DS. Trends in the prevalence of short sleepers in the USA: 1975-2006. Sleep 2010;33:37–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carskadon MA, Mindell J, Drake C. Contemporary sleep patterns of adolescents in the USA: results of the 2006 National Sleep Foundation Sleep in America Poll. Presented at the Annual Meeting of the European Sleep Research Society, Innsbruck, Switzerland, 14 September 2006 [Google Scholar]
- 3.Cappuccio FP, Taggart FM, Kandala NB, et al. Meta-analysis of short sleep duration and obesity in children and adults. Sleep 2008;31:619–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buxton OM, Pavlova M, Reid EW, Wang W, Simonson DC, Adler GK. Sleep restriction for 1 week reduces insulin sensitivity in healthy men. Diabetes 2010;59:2126–2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yaggi HK, Araujo AB, McKinlay JB. Sleep duration as a risk factor for the development of type 2 diabetes. Diabetes Care 2006;29:657–661 [DOI] [PubMed] [Google Scholar]
- 6.Knutson KL, Van Cauter E, Zee P, Liu K, Lauderdale DS. Cross-sectional associations between measures of sleep and markers of glucose metabolism among subjects with and without diabetes: the Coronary Artery Risk Development in Young Adults (CARDIA) sleep study. Diabetes Care 2011;34:1171–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maquet P, Dive D, Salmon E, et al. Cerebral glucose utilization during sleep-wake cycle in man determined by positron emission tomography and [18F]2-fluoro-2-deoxy-d-glucose method. Brain Res 1990;513:136–143 [DOI] [PubMed] [Google Scholar]
- 8.Boyle PJ, Scott JC, Krentz AJ, Nagy RJ, Comstock E, Hoffman C. Diminished brain glucose metabolism is a significant determinant for falling rates of systemic glucose utilization during sleep in normal humans. J Clin Invest 1994;93:529–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu X, Forbes EE, Ryan ND, Rofey D, Hannon TS, Dahl RE. Rapid eye movement sleep in relation to overweight in children and adolescents. Arch Gen Psychiatry 2008;65:924–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tasali E, Mokhlesi B, Van Cauter E. Obstructive sleep apnea and type 2 diabetes: interacting epidemics. Chest 2008;133:496–506 [DOI] [PubMed] [Google Scholar]
- 11.Hannon TS, Lee S, Chakravorty S, Lin Y, Arslanian SA. Sleep-disordered breathing in obese adolescents is associated with visceral adiposity and markers of insulin resistance. Int J Pediatr Obes 2011;6:157–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kelly A, Dougherty S, Cucchiara A, Marcus CL, Brooks LJ. Catecholamines, adiponectin, and insulin resistance as measured by HOMA in children with obstructive sleep apnea. Sleep 2010;33:1185–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ogden CL, Kuczmarski RJ, Flegal KM, et al. Centers for Disease Control and Prevention 2000 growth charts for the United States: improvements to the 1977 National Center for Health Statistics version. Pediatrics 2002;109:45–60 [DOI] [PubMed] [Google Scholar]
- 14.Boston RC, Stefanovski D, Moate PJ, Sumner AE, Watanabe RM, Bergman RN. MINMOD Millennium: a computer program to calculate glucose effectiveness and insulin sensitivity from the frequently sampled intravenous glucose tolerance test. Diabetes Technol Ther 2003;5:1003–1015 [DOI] [PubMed] [Google Scholar]
- 15.Yeckel CW, Weiss R, Dziura J, et al. Validation of insulin sensitivity indices from oral glucose tolerance test parameters in obese children and adolescents. J Clin Endocrinol Metab 2004;89:1096–1101 [DOI] [PubMed] [Google Scholar]
- 16.Sinha R, Fisch G, Teague B, et al. Prevalence of impaired glucose tolerance among children and adolescents with marked obesity. N Engl J Med 2002;346:802–810 [DOI] [PubMed] [Google Scholar]
- 17.Iber C, American Academy of Sleep Medicine. The AASM Manual for the Scoring of Sleep and Associated Events: Rules, Terminology and Technical Specifications. Westchester, IL, American Academy of Sleep Medicine, 2007 [Google Scholar]
- 18.Pallayova M, Donic V, Gresova S, Peregrim I, Tomori Z. Do differences in sleep architecture exist between persons with type 2 diabetes and nondiabetic controls? J Diabetes Sci Tech 2010;4:344–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tasali E, Leproult R, Ehrmann DA, Van Cauter E. Slow-wave sleep and the risk of type 2 diabetes in humans. Proc Natl Acad Sci USA 2008;105:1044–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weyer C, Bogardus C, Mott DM, Pratley RE. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J Clin Invest 1999;104:787–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baharav A, Kotagal S, Gibbons V, et al. Fluctuations in autonomic nervous activity during sleep displayed by power spectrum analysis of heart rate variability. Neurology 1995;45:1183–1187 [DOI] [PubMed] [Google Scholar]
- 22.Gilon P, Henquin JC. Mechanisms and physiological significance of the cholinergic control of pancreatic beta-cell function. Endocr Rev 2001;22:565–604 [DOI] [PubMed] [Google Scholar]
- 23.Van Cauter E, Kerkhofs M, Caufriez A, Van Onderbergen A, Thorner MO, Copinschi G. A quantitative estimation of growth hormone secretion in normal man: reproducibility and relation to sleep and time of day. J Clin Endocrinol Metab 1992;74:1441–1450 [DOI] [PubMed] [Google Scholar]
- 24.Krag MB, Gormsen LC, Guo Z, et al. Growth hormone-induced insulin resistance is associated with increased intramyocellular triglyceride content but unaltered VLDL-triglyceride kinetics. Am J Physiol Endocrinol Metab 2007;292:E920–E927 [DOI] [PubMed] [Google Scholar]
- 25.Spruyt K, Molfese DL, Gozal D. Sleep duration, sleep regularity, body weight, and metabolic homeostasis in school-aged children. Pediatrics 2011;127:e345–e352 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.