

Abstract
Single-molecule measurements are a valuable tool for revealing details of enzyme mechanisms by enabling observation of unsynchronized behavior. However, this approach often requires immobilizing the enzyme on a substrate, a process which may alter enzyme behavior. We apply a microfluidic trapping device to allow, for the first time, prolonged solution-phase measurement of single enzymes in solution. Individual redox events are observed for single molecules of a blue nitrite reductase and are used to extract the microscopic kinetic parameters of the proposed catalytic cycle. Changes in parameters as a function of substrate concentration are consistent with a random sequential substrate binding mechanism.
Keywords: ABEL trap, enzymology, electron transfer, fluorescence, catalysis
Measurements made on the activity of single enzymes have revolutionized the picture of these molecular machines’ mode of operation by providing evidence for static and dynamic heterogeneity, enabling the observation of transient intermediates, and allowing determination of the microscopic rate constants comprising complex catalytic cycles under turnover conditions (1–4). However, conclusions regarding enzyme dynamics require the observation of multiple turnovers of a single molecule with a requisite observation window of several seconds or more. Because this window is several orders of magnitude longer than the typical dwell time of a solution-phase molecule diffusing through a confocal detection volume, molecules that naturally exist in solution are often immobilized on a solid support (1–3, 5). Although gross comparison between properties measured on immobilized and freely diffusing biomolecules tends to show good agreement (1, 3), more subtle aspects of biomolecule behavior such as property distribution widths and shapes (6, 7) and conformational dynamics (6, 8) have been shown to be affected by immobilization, and in some cases, immobilization has been suspected of contributing to observed enzyme behavioral heterogeneity (4). In this report we take an important step toward viewing the richness of single-enzyme dynamics without immobilization by making observations of multiple turnover events of single solution-phase molecules of a blue nitrite reductase (bNiR), an important redox-active enzyme in bacterial denitrification (9).
Prolonged solution-phase measurements are enabled through use of a specialized microfluidic trapping device that cancels the Brownian motion of a single emissive solution-phase object through the use of directed electroosmotic forces, the anti-Brownian electrokinetic (ABEL) trap (10, 11). The ABEL trap estimates particle position via a revolving laser spot phase-locked in a homebuilt analogue circuit to a restoring force gated by photon detection and applied via two orthogonal pairs of electrodes (11). This scheme results in the confinement of the object in a spatially homogeneous excitation volume for multiple seconds without immobilization and can be applied to single biomolecules substantially smaller than the large dielectric beads that are trapped with optical tweezers. We have previously used the ABEL trap to study conformational dynamics of single photosynthetic antenna proteins (6) and fluorescently labeled DNA molecules (12).
bNiR is a multicopper enzyme that carries out the one-electron reduction of nitrite,  , to nitric oxide, NO, in the presence of a suitable reductant (9). It is a trimer, 10 nm in diameter, and contains two different Cu sites (13). The type 1 (T1) Cu, coordinated by two His, a Cys, and a Met residue (13), is the initial site of reduction by an electron donor. Intramolecular electron transfer (ET) (14) over 12.6 Å results in reduction of the proximal type 2 (T2) Cu, which is located at the monomer-monomer interface and is coordinated by three His residues (13). One of these T2 Cu ligands is adjacent to the Cys residue at the T1 site, and this covalent network has been proposed as the pathway for ET (15–17). Nitrite reduction occurs at the T2 Cu with protons for water formation being provided by a hydrogen bond network composed of a bound water, and the second coordination sphere Asp92 and His249 residues (18). Recently, it has been suggested that the intramolecular ET event is coupled to proton transfer (19). Considerable mechanistic interest on nitrite reductases (NiRs) has focused on whether intramolecular ET proceeds consistently before or after nitrite binding (ordered mechanisms) or via a mixture of both pathways (random-sequential mechanism) (20, 21). Much seemingly conflicting evidence has appeared for both mechanisms, with ET without nitrite binding occurring via pulse radiolysis (17), ET shown to be blocked in the absence of nitrite via X-ray absorption spectroscopy (22), and both pathways shown to be operating simultaneously (a random-sequential mechanism) via activity assays and protein film voltammetry (21).
, to nitric oxide, NO, in the presence of a suitable reductant (9). It is a trimer, 10 nm in diameter, and contains two different Cu sites (13). The type 1 (T1) Cu, coordinated by two His, a Cys, and a Met residue (13), is the initial site of reduction by an electron donor. Intramolecular electron transfer (ET) (14) over 12.6 Å results in reduction of the proximal type 2 (T2) Cu, which is located at the monomer-monomer interface and is coordinated by three His residues (13). One of these T2 Cu ligands is adjacent to the Cys residue at the T1 site, and this covalent network has been proposed as the pathway for ET (15–17). Nitrite reduction occurs at the T2 Cu with protons for water formation being provided by a hydrogen bond network composed of a bound water, and the second coordination sphere Asp92 and His249 residues (18). Recently, it has been suggested that the intramolecular ET event is coupled to proton transfer (19). Considerable mechanistic interest on nitrite reductases (NiRs) has focused on whether intramolecular ET proceeds consistently before or after nitrite binding (ordered mechanisms) or via a mixture of both pathways (random-sequential mechanism) (20, 21). Much seemingly conflicting evidence has appeared for both mechanisms, with ET without nitrite binding occurring via pulse radiolysis (17), ET shown to be blocked in the absence of nitrite via X-ray absorption spectroscopy (22), and both pathways shown to be operating simultaneously (a random-sequential mechanism) via activity assays and protein film voltammetry (21).
The T2 Cu possesses no significant absorption in the visible range (23). However, the oxidized T1 Cu possesses an intense S(Cys) → Cu(II) charge transfer transition giving rise to strong visible absorption (9). This absorption vanishes upon reduction of the T1 Cu (23). This spectral change was recently employed to make a proximal dye molecule a fluorescent reporter of the T1 Cu oxidation state (24). The oxidized Cu significantly quenches the dye fluorescence via Förster resonant energy transfer (FRET) due to overlap between the dye’s emission and the colored Cu’s absorption, whereas the reduced colorless Cu leaves the dye unquenched and maximally emissive (24), Fig. 1. Single green NiR enzymes (gNiR) (25) from Alcaligenes faecalis were immobilized and fluorescence intensity fluctuations were shown to be indicative of enzyme turnovers (24), although individual turnovers could not be resolved. This approach is similar to early single-molecule fluorescence spectroscopy studies on cholesterol oxidase (1) but does not require an intrinsically fluorescent enzyme. We now extend this paradigm to a solution-phase measurement where the spectral properties of bNiR enable single turnover resolution.
Fig. 1.

bNiR structure. Each monomer of bNiR contains a T1 Cu (red sphere) and a T2 Cu (green sphere). Molecules of bNiR are site-specifically labeled at low dye concentration to generate a majority singly labeled population. Redox cycling at the T1 Cu results in a time-varying quenching rate of the proximal label, Atto647N (blue aromatic structure), as the FRET rate changes due to the T1 Cu’s changing electronic absorption spectrum. Image generated from Protein Data Bank (PDB) ID code 1OE1 (13).
Results
Single molecules of labeled bNiR were observed to quickly pass in and out of the detection volume of the ABEL trap in a few milliseconds when the feedback control was switched off, but were held for multiple seconds when feedback was on, with emission intensity plateaus observed as the trapped solution-phase molecule experienced a spatially flat excitation profile (Fig. 2). Trapped molecules were observed to undergo fluctuations between two discrete intensity states (Fig. 2). These fluctuations represent changes in oxidation state of one T1 Cu of a single solution-phase enzyme. The ratio of intensities, 0.23, corresponds well with previously observed values (26). A histogram of intensity levels shows a high degree of separation between the upper and lower state (Fig. 3). The relative amplitude of the lower peak, which corresponds to the oxidized T1 Cu population, rises as the nitrite concentration is increased. When the reductant and electron shuttle are added without substrate (red curve), the upper peak is dominant, consistent with assignment of this peak to the reduced state of the T1 Cu. Some residual lower peak intensity remains due to incomplete reduction and consequent presence of a small population of singly reduced bNiR molecules. When the substrate is added without reductant and shuttle, the lower peak is dominant, consistent with assignment of the lower peak to the oxidized state of the T1 Cu. Some residual upper peak remains, likely due to incomplete oxidation. When a non-redox-active protein is labeled with Atto647N and trapped under the above conditions, no lower peak is observed (SI Appendix, Fig. 1), confirming that the intensity fluctuations are due to changes in enzyme properties and not artifactual dye photophysics.
Fig. 2.

Intensity-time traces of six single molecules of labeled bNiR caught in the ABEL trap. Colored (blue/red/black) traces show intensities derived from applying a change-point-finding algorithm as a filter on time-tagged data. The gray trace is 1-ms binned data. The intensity is observed to fluctuate between two discrete intensity levels, an upper state (blue) and a lower state (red) whereas background is colored black. A variety of behaviors are observed, with a diversity of transitions per trace (zero, few, or many) and of dwell times in each intensity state (short or long). Conditions: [ASC], 5 mM; [PES], 100 nM; 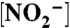 , 5 mM (A–E) or 0.2 mM (F).
, 5 mM (A–E) or 0.2 mM (F).
Fig. 3.

Histogram of intensities compiled from many molecules under varying conditions at 10 ms binning normalized to the central peak near 25 counts/ms. The amplitude of the lower intensity peak is seen to rise as a function of substrate concentration. The lower peak is seen to nearly disappear when no substrate is added, whereas the upper intensity peak is seen to nearly disappear when no reductant is added. The off-scale peak at lowest intensity is from background.
To quantitatively treat enzyme kinetics, we measured the dwell times in the upper and lower states. States were distinguished via application of a bin-free state identification procedure consisting of first using an intensity-change-point-finding algorithm (27) as a smart low-pass filter directly on time-tagged data followed by the use of an intensity thresholding algorithm. The procedure and selection criteria are described in detail in SI Appendix. Dwell time distributions compiled from many molecules are seen to conspicuously change as a function of nitrite concentration, Fig. 4 A and B, for upper and lower states, respectively. Each histogram is constructed from data from between 150 and 2,000 individually probed molecules.
Fig. 4.

Fits for dwell times of the upper intensity state (A), lower intensity state (B), and population ratio (C), at multiple substrate concentrations for one representative dataset. In A and B, dwell times in 20-ms bins (dark blue bars) are fit with red lines that follow the function forms derived from Fig. 6A. In C, population ratios calculated from the determined parameters (red squares) are seen to fall within the error bars of the measured ratios (blue circles). Increasing substrate concentration results in a shift toward shorter dwell times in the upper state, a shift to longer dwell times in the lower state, and a larger fraction of time spent in the lower state.
Discussion
To model changes in dwell time distributions, we began with an ordered kinetic scheme (Fig. 5). Kinetic equations for single-molecule dynamics are expressed in terms of probabilities rather than concentrations. Expected dwell time distributions are determined by differentiating the integrated rate law with respect to time (SI Appendix) (28, 29).
Fig. 5.

A simple kinetic scheme and extracted intramolecular ET rate constants. (A) A kinetic scheme where the left side box represents the oxidation state of the T1 Cu (O/R, oxidized/reduced), whereas the right side describes the T2 Cu. Only molecules with a reduced T1 Cu are fluorescent (red highlight). (B) The rate constant of intramolecular ET, k23, is seen to increase as a function of substrate concentration. Error bars are from four independent measurements, each comprised of between one thousand and several thousand individual intensity traces.
Assuming the validity of the scheme in Fig. 5, dwell times in the upper state, RO (where the first letter, R, describes the oxidation state of the T1 Cu and the second letter, O, describes the oxidation state of the T2 Cu, see Fig. 5), can be modeled as a simple monoexponential decay to directly extract the rate constant of intramolecular ET, k23. These fits are shown in SI Appendix, Fig. 3. Surprisingly, k23 is not a simple rate constant, as it is observed to increase as a function of nitrite concentration (Fig. 5B). Previously, conflicting effects of nitrite binding on intramolecular ET have been reported. ET was inhibited by the absence of nitrite binding in crystallized gNiR variants at 100 K (22), although in our data, state shifts in singly reduced bNiR molecules at zero nitrite concentration, summarized in Fig. 4 A and B, suggest that k23 is not blocked by the absence of substrate under these conditions. Electron-nuclear double resonance measurements (30) and crystallographic studies (31) on the T2 Cu suggest a greater thermodynamic driving force for ET after nitrite bonding due to weaker coordination of His ligands, and a consequent increase in rate constant. In contrast, pulse radiolysis measurements showed a sharp decrease in rate upon nitrite binding (16). Bulk activity assays and electrochemical measurements have suggested that an increase in ET rate constant upon nitrite binding is expected at higher pH values, whereas a decrease upon binding is expected at lower pH, depending on whether the nitrite displaces a water molecule or hydroxide ion (21). Our direct observation of an ET rate constant increase at pH 7.0 is consistent with a shift between a mechanism where ET precedes nitrite binding to one where ET occurs after nitrite binding with a consequent increase in ET rate (21). These changes are in agreement with a random-sequential mechanism whereby ET can occur before or after binding with the relative contributions of each determined by environmental variables such as substrate concentration and pH.
If the intramolecular ET rate constant is affected by nitrite binding via allosteric regulation, where the nitrite interacts with the protein at some site other than the active site, the scheme in Fig. 5A remains valid. However, if as suggested, the ET rate constant is altered by conformational and energetic changes as a result of nitrite binding to the active site, then the scheme in Fig. 5A is too simplistic and a more complex scheme is required that explicitly considers substrate binding. Such an alternative kinetic scheme (24) is shown in Fig. 6A, with the upper right branch explicitly including substrate binding before ET (see caption).
Fig. 6.

Advanced kinetic scheme and population dynamics. (A) A kinetic scheme where the left side box represents the oxidation state of the T1 Cu (O/R, oxidized/reduced), the middle describes the T2 Cu, and the right side box indicates substrate binding (s or empty, substrate bound/unbound). Only molecules with a reduced T1 Cu are strongly fluorescent (red highlight). (B) Equilibrium populations of each kinetic microstate as a function of substrate concentration, where circle area is directly proportional to the fractional population. Each color corresponds to a particular nitrite concentration, as shown on the color bar. Population is seen to be dominated by RO and OR at low concentrations and OO, RO, and ROs at high concentrations. (C) Population dynamics of each kinetic microstate in the bright upper manifold. (D) Population dynamics of each kinetic microstate in the dark lower manifold.
Dwell time distributions in the upper states, RO and ROs (where the first letter, R, refers to the oxidation state of the T1 Cu, the second letter, O, refers to the oxidation state of the T2 Cu, and the presence/absence of the third letter, s, indicates that the substrate is bound/unbound), and lower states (all other cases) can be expressed in terms of the ten microscopic rate constant parameters in the scheme, as described in SI Appendix. To further constrain the data fitting procedure, we measured the relative amounts of time spent in the upper and lower states for many molecular events (Fig. 4C). This quantity can also be expressed as a function of the microscopic rate constant parameters. To complete the analysis, all three data plots in Fig. 4, comprised of dwell time distributions (upper and lower) and population ratios, were fit simultaneously. This global analysis allowed determination of the complete set of microscopic rate constant parameters in the kinetic scheme of Fig. 6. Determined rate constants are exhibited in Table 1, with confidence intervals determined from four independently acquired datasets. Limitations to the global analysis are discussed in SI Appendix.
Table 1.
Microscopic rate parameters, values, and standard deviations
| Parameter | Value | σExpt* |
| kA | 5.4† | 0.2 |
| kC | 6.7† | 1.7 |
| kC′ | 9.9† | 1.5 |
| kD | 13† | 2.3 |
|
kE |
8.4‡ |
5.4 |
| kB | 141‡ | 36 |
| kB′ | 255† | 151 |
| kD′ | 181† | 126 |
| kE′ | 29† | 25 |
| kF | 167† | 51 |
The top half of the table includes parameters that can be estimated well from our global analysis, whereas the lower half includes parameters that are difficult to measure from our global analysis, as determined by calculating the Cramer–Rao bound on the information available. Details of this calculation are given in SI Appendix.
*Standard deviations, σ, are from four independent measurements.
†In units of s-1.
‡In units of s-1 mM-1.
Determination of all of the rate constants in the kinetic scheme via data acquired entirely under one set of turnover conditions allows a unique view into the dynamics of this enzyme. Moreover, these dynamics were measured in a solution-phase environment and were therefore minimally affected by surface interactions. To further describe the enzyme’s behavior, we have developed alternate representations of the system parameters and dynamics. The extracted rate constants were used to calculate the steady-state populations of each of the five states (Fig. 6B) as well as the probabilities of occupying a particular state as a function of time after entering the upper and lower manifolds (Fig. 6 C and D, respectively). These probabilities sum to one at time zero due to a normalization condition and decay to zero at long times due to the vanishing probability of staying in the manifold indefinitely, a consequence of the overall cyclic nature of the enzyme mechanism. The shift to higher populations in the lower reduced intensity manifold with increasing [ ], as shown in Fig. 4C and Fig. 6B, is consistent with the increase in the number of individual bNiR molecules spending more time in the lower state observed while making fluorescence lifetime imaging measurements on immobilized bNiR molecules (26).
], as shown in Fig. 4C and Fig. 6B, is consistent with the increase in the number of individual bNiR molecules spending more time in the lower state observed while making fluorescence lifetime imaging measurements on immobilized bNiR molecules (26).
Several of the steps in the kinetic schemes correspond to intramolecular ET events. Rather than requiring five individual forward ET rate constants (one value of k23 for each value of [ ]) as in the scheme in Fig. 5A, by using the scheme in Fig. 6A the dwell time distributions can be understood reasonably well with only two forward ET rate constants, one exhibited without bound substrate, kC, and one exhibited with bound substrate, kD (21, 32). As shown in Fig. 6B, at low substrate concentration the majority of population escapes the upper state manifold (consisting of RO and ROs) via RO with a rate constant kC. However, as substrate concentration increases, the majority exits through the state ROs with a rate constant kD (21), with the observed rate constant interpolating between the two limiting values. These dynamics are further shown in Fig. 6C, as population is shown to rapidly shift from RO to ROs at high concentrations of substrate before leaving the bright upper state manifold.
]) as in the scheme in Fig. 5A, by using the scheme in Fig. 6A the dwell time distributions can be understood reasonably well with only two forward ET rate constants, one exhibited without bound substrate, kC, and one exhibited with bound substrate, kD (21, 32). As shown in Fig. 6B, at low substrate concentration the majority of population escapes the upper state manifold (consisting of RO and ROs) via RO with a rate constant kC. However, as substrate concentration increases, the majority exits through the state ROs with a rate constant kD (21), with the observed rate constant interpolating between the two limiting values. These dynamics are further shown in Fig. 6C, as population is shown to rapidly shift from RO to ROs at high concentrations of substrate before leaving the bright upper state manifold.
Population in the lower state manifold is initially concentrated in the T1 Cu oxidized, T2 Cu reduced (OR) state at vanishing substrate concentration, Fig. 6B. However, this population rapidly decreases with increasing substrate concentration, with the majority of the population in the manifold residing in the OO state, before the bottleneck of slow reduction via the solution phase electron donor. As shown in Fig. 6D, population can enter the lower state manifold at either OR or ORs (both show nonzero values at time zero). As the population in the ROs state increases, the majority of the population is shown to enter via ORs. However, population is quickly shifted to the OO state before leaving the manifold after reduction, kA.
The scheme in Fig. 6 and parameters in Table 1 describe a system that proceeds with an ordered mechanism at low substrate concentrations where intramolecular ET precedes substrate binding. However, at intermediate concentrations both pathways in the scheme in Fig. 6 are active. In this regime, individual molecules will stochastically follow one or the other pathway with some characteristic partitioning probability, i.e., a random-sequential mechanism. At high substrate concentrations, the system approaches the opposite ordered mechanism, where substrate binding precedes intramolecular ET. In this picture, only the microscopic rate constants in the kinetic scheme determine the partitioning between the two ordered mechanisms.
In contrast to some other single-enzyme experiments (2, 3), product formation was not directly measured. The rate of substrate consumption,  , can be calculated from the kinetic scheme. The complex scheme in Fig. 6A will exhibit a slight deviation from Michaelis–Menten (MM) kinetics at high substrate concentration (32–34) (SI Appendix, Fig. 5). Nevertheless, approximation by the MM functional form yields kcat, 2.9 s-1 and Km, 860 μM. This value of kcat is well within the range of kcat values established from multiple bulk experiments. A more detailed comparison is presented in SI Appendix.
, can be calculated from the kinetic scheme. The complex scheme in Fig. 6A will exhibit a slight deviation from Michaelis–Menten (MM) kinetics at high substrate concentration (32–34) (SI Appendix, Fig. 5). Nevertheless, approximation by the MM functional form yields kcat, 2.9 s-1 and Km, 860 μM. This value of kcat is well within the range of kcat values established from multiple bulk experiments. A more detailed comparison is presented in SI Appendix.
Though measured from observations on individual bNiR molecules, the rate constants shown in Table 1 are ensemble-averaged values. Determination of these parameters for a single enzyme requires a substantially longer viewing window than can be achieved with the current implementation of the ABEL trap and future designs hold promise of extending this window (35). Molecule-to-molecule variability is suggested by the diversity of behavior seen in Fig. 2 but at the moment is not required to explain the observed behavior. Molecule-to-molecule variability has begun to be quantified in immobilized NiR molecules (24, 26).
Individual kinetic parameters can be measured via bulk experiments by tuning reaction conditions to contrived configurations so as to make a specific step rate limiting and thus experimentally accessible for measurement. However, single-molecule measurements allow observation of unsynchronized reaction intermediates, the analysis of which enables simultaneous determination of many kinetic parameters under relevant turnover conditions. The use of the ABEL trap to make these measurements on single solution-phase enzymes opens a window of unprecedented detail into the dynamics of single enzymes in solution.
In conclusion, we extracted kinetic parameters from single molecules of bNiR probed individually in solution. Molecules trapped for multiple seconds and discrete intensity levels corresponding to oxidized and reduced states of the T1 Cu were observed. Extracted parameters lend further support to a random-sequential mechanism and offer direct evidence that an increase in substrate concentration leads to higher rate constants of intramolecular ET under these conditions. Further experiments are ongoing to include effects of pH and to enable long enough trapping windows to allow parameter estimation for individual enzymes.
Materials and Methods
Protein Isolation and Labeling.
The Lys329Cys mutant of the bNiR from Alcaligenes xylosoxidans, which introduces a Cys residue 2 nm from the T1 Cu within the same monomer, was isolated and purified as described previously (25). The Cys was modified with Atto647N-maleimide yielding protein that was 9% labeled (26).
ABEL Trap Loading Procedure.
A 700-nm deep quartz microfluidic cell (11) was cleaned in a UV-ozone cleaner (Jelight) for 30 min, sonicated for 10 min in 1 M potassium hydroxide, thoroughly rinsed with water, and dried with a stream of dry N2. Following a previously published procedure (6, 36), a drop of positively charged polyethylene imine (PEI, 1 g/L, pH 8.0, passed through a 0.02-μm filter), was deposited at the microfluidic input port and allowed to enter and coat the cell via capillary action. The cell was allowed to sit for 10 min with the fluid inside. The cell was then blown dry with N2, and filled with ultrapure water the same way. The cell was then blown dry, and a drop of negatively charged polyacrylic acid (PAA, 1 g/L, pH 8.0, passed through a 0.02-μm filter) was allowed to coat the inside in the same manner as PEI. The cell was allowed to sit for 10 min, blown dry, and rinsed with water as above. The entire coating procedure was repeated one more time, leaving the cell coated with two alternating pairs of layers, SiO2/PEI/PAA/PEI/PAA. If a bare silica cell is used, rampant sticking of bNiR to the surface was observed. With the above coating procedure, sticking was nearly completely eliminated.
Following deposition of the polyelectrolyte multilayer, the dried cell was filled via capillary action with freshly prepared buffer, 1∶1 20 mM Hepes, pH 7.0∶glycerol with sacrificial electron donor, 5 mM sodium ascorbate (ASC), electron shuttle, 100 nM phenazine ethosulfate (PES) (37), and varying amounts of substrate, sodium nitrite. This solution was allowed to sit for 5 min and then a freshly prepared solution of the same composition but including < 0.1 nM labeled bNiR was added via capillary action. The loaded ABEL trap cell was then allowed to stand for 5 min before beginning optical measurements.
Optical Setup and Trapping.
Excitation was provided by a continuous-wave 638-nm excitation laser (FiberTEC, Blue Sky), which was passed through a 515–640-band-pass filter to eliminate red spectral sidebands. The laser light was steered into the epiport of a Nikon TE-300 microscope and focused with an oil objective (N.A. = 1.3) to provide an average power of 15 kW/cm2 at the sample. Beam deflection to produce the revolving spot was achieved through the same apparatus described previously (11). The excitation spot is 0.6 μm in diameter in the sample plane, as measured by scanning a fluorescent bead through the focal volume, and is revolved at 40 kHz with a radius of 0.4 μm in the sample plane. Sample fluorescence is collected back through the same objective and passed through dichroic (645DRLP) and long pass (640ALP) filters, a 200-μm pinhole, and focused onto an Si avalanche photodiode detector (Perkin Elmer, SPCM-CD 2801). Time-correlated single photon counting is achieved using the PicoHarp 300 (Picoquant) timing module. Feedback direction and magnitude are calculated for each 25-μs cycle by phase-sensitive integration of photons detected in the previous cycle. A single detected photon produces a feedback pulse of 12-V vector magnitude lasting 25 μs, which is applied across the 20-μm transverse width of the shallow region of the cell. Voltages are produced by four PA-83 high-voltage operational amplifiers (Apex Microtechnologies) to generate forces in the appropriate direction to cancel Brownian motion. Only objects that maintained residency in the trap for over 200 ms were included in the analysis.
Data Analysis.
Time-tagged photon detection data was analyzed in Matlab 7.7. A change-point-finding algorithm (27) was applied with a low critical value and then a thresholding algorithm was used to identify shifts between the upper and lower intensity states and background. A full description of the analysis procedure can be found in SI Appendix.
Supplementary Material
Acknowledgments.
We thank Y. Jiang, Q. Wang, and A. E. Cohen for assistance with trap implementation, and A. Fürstenberg, S. F. Lee, M. A. Thompson, W. Duim, and S. Bockenhauer for helpful discussions. This work was funded by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the US Department of Energy through Grant DE-FG02-07ER15892 and the EdRox Marie Curie Research Training Network (MCRTN-035649-EdRox).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1113572108/-/DCSupplemental.
References
- 1.Lu HP, Xun L, Xie XS. Single-molecule enzymatic dynamics. Science. 1998;282:1877–1882. doi: 10.1126/science.282.5395.1877. [DOI] [PubMed] [Google Scholar]
- 2.Velonia K, et al. Single-enzyme kinetics of CALB-catalyzed hydrolysis. Angew Chem Int Ed Engl. 2005;44:560–564. doi: 10.1002/anie.200460625. [DOI] [PubMed] [Google Scholar]
- 3.English BP, et al. Ever-fluctuating single enzyme molecules: Michaelis–Menten equation revisited. Nat Chem Biol. 2006;2:87–94. doi: 10.1038/nchembio759. [DOI] [PubMed] [Google Scholar]
- 4.Smiley RD, Hammes GG. Single molecule studies of enzyme mechanisms. Chem Rev. 2006;106:3080–3094. doi: 10.1021/cr0502955. [DOI] [PubMed] [Google Scholar]
- 5.Rasnik I, McKinney SA, Ha T. Surfaces and orientations: Much to FRET about? Acc Chem Res. 2005;38:542–548. doi: 10.1021/ar040138c. [DOI] [PubMed] [Google Scholar]
- 6.Goldsmith RH, Moerner WE. Watching conformational- and photodynamics of single fluorescent proteins in solution. Nat Chem. 2010;2:179–186. doi: 10.1038/nchem.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Talaga DS, et al. Dynamics and folding of single two-stranded coiled-coil peptides studied by fluorescent energy transfer confocal microscopy. Proc Natl Acad Sci USA. 2000;97:13021–13026. doi: 10.1073/pnas.97.24.13021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedel M, Baumketner A, Shea JE. Effects of surface tethering on protein folding mechanisms. Proc Natl Acad Sci USA. 2006;103:8396–8401. doi: 10.1073/pnas.0601210103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Averill BA. Dissimilatory nitrite and nitric oxide reductases. Chem Rev. 1996;96:2951–2964. doi: 10.1021/cr950056p. [DOI] [PubMed] [Google Scholar]
- 10.Cohen AE, Moerner WE. Suppressing Brownian motion of individual biomolecules in solution. Proc Natl Acad Sci USA. 2006;103:4362–4365. doi: 10.1073/pnas.0509976103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cohen AE, Moerner WE. Controlling Brownian motion of single protein molecules and single fluorophores in aqueous buffer. Opt Express. 2008;16:6941–6956. doi: 10.1364/oe.16.006941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen AE, Moerner WE. Principal-components analysis of shape fluctuations of single DNA molecules. Proc Natl Acad Sci USA. 2007;104:12622–12627. doi: 10.1073/pnas.0610396104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellis MJ, Dodd FE, Sawers G, Eady RR, Hasnain SS. Atomic resolution structures of native copper nitrite reductase from Alcaligenes xylosoxidans and the active site mutant Asp92Glu. J Mol Biol. 2003;328:429–438. doi: 10.1016/s0022-2836(03)00308-5. [DOI] [PubMed] [Google Scholar]
- 14.Gray HB, Winkler JR. Electron flow through metalloproteins. Biochim Biophys Acta. 2010;1797:1563–1572. doi: 10.1016/j.bbabio.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 15.Messerschmidt A, et al. X-ray crystal-structure of the blue oxidase ascorbate oxidase from zucchini—analysis of the polypeptide fold and a model of the copper sites and ligands. J Mol Biol. 1989;206:513–529. doi: 10.1016/0022-2836(89)90498-1. [DOI] [PubMed] [Google Scholar]
- 16.Suzuki S, et al. Spectroscopic characterization and intramolecular electron transfer processes of native and type 2 Cu-depleted nitrite reductases. J Biol Inorg Chem. 1997;2:265–274. [Google Scholar]
- 17.Suzuki S, et al. Pulse-radiolysis studies on nitrite reductase from Achromobacter cycloclastes IAM 1013: Evidence for intramolecular electron-transfer from type-1 Cu to type-2 Cu. J Am Chem Soc. 1994;116:11145–11146. [Google Scholar]
- 18.Adman ET, Godden JW, Turley S. The structure of copper-nitrite reductase from Achromobacter cycloclastes at 5 pH values, with NO2- bound and with type-II copper depleted. J Biol Chem. 1995;270:27458–27474. doi: 10.1074/jbc.270.46.27458. [DOI] [PubMed] [Google Scholar]
- 19.Leferink NGH, et al. Proton-coupled electron transfer in the catalytic cycle of Alcaligenes xylosoxidans copper-dependent nitrite reductase. Biochemistry. 2011;50:4121–4131. doi: 10.1021/bi200246f. [DOI] [PubMed] [Google Scholar]
- 20.Zumft WG. Cell biology and molecular basis of denitrification. Microbiol Mol Biol Rev. 1997;61:533–616. doi: 10.1128/mmbr.61.4.533-616.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wijma HJ, Jeuken LJC, Verbeet MP, Armstrong FA, Canters GW. A random-sequential mechanism for nitrite binding and active site reduction in copper-containing nitrite reductase. J Biol Chem. 2006;281:16340–16346. doi: 10.1074/jbc.M601610200. [DOI] [PubMed] [Google Scholar]
- 22.Hough MA, Antonyuk SV, Strange RW, Eady RR, Hasnain SS. Crystallography with online optical and X-ray absorption spectroscopies demonstrates an ordered mechanism in copper nitrite reductase. J Mol Biol. 2008;378:353–361. doi: 10.1016/j.jmb.2008.01.097. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki S, Kataoka K, Yamaguchi K, Inoue T, Kai Y. Structure-function relationships of copper-containing nitrite reductases. Coord Chem Rev. 1999;190–192:245–265. [Google Scholar]
- 24.Kuznetsova S, et al. The enzyme mechanism of nitrite reductase studied at single-molecule level. Proc Natl Acad Sci USA. 2008;105:3250–3255. doi: 10.1073/pnas.0707736105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sato K, Dennison C. Active site comparison of Co-II blue and green nitrite reductases. Chem Eur J. 2006;12:6647–6659. doi: 10.1002/chem.200600036. [DOI] [PubMed] [Google Scholar]
- 26.Tabares LC, et al. Fluorescence lifetime analysis of nitrite reductase from Alcaligenes xylosoxidans at the single molecules level reveals enzyme mechanism. Chem Eur J. doi: 10.1002/chem.201102063. doi: 10.1002/chem.201102063. [DOI] [PubMed] [Google Scholar]
- 27.Watkins LP, Yang H. Detection of intensity change points in time-resolved single-molecule measurements. J Phys Chem B. 2005;109:617–628. doi: 10.1021/jp0467548. [DOI] [PubMed] [Google Scholar]
- 28.Van Kampen NG. Stochastic Processes in Physics and Chemistry. Amsterdam: Elsevier; 1992. pp. 44–47. [Google Scholar]
- 29.Xie XS. Single-molecule approach to enzymology. Single Mol. 2001;2:229–236. [Google Scholar]
- 30.Veselov A, Olesen K, Sienkiewicz A, Shapleigh JP, Scholes CP. Electronic structural information from Q-band ENDOR on the type 1 and type 2 copper liganding environment in wild-type and mutant forms of copper-containing nitrite reductase. Biochemistry. 1998;37:6095–6105. doi: 10.1021/bi971604r. [DOI] [PubMed] [Google Scholar]
- 31.Strange RW, et al. Structural and kinetic evidence for an ordered mechanism of copper nitrite reductase. J Mol Biol. 1999;287:1001–1009. doi: 10.1006/jmbi.1999.2648. [DOI] [PubMed] [Google Scholar]
- 32.Wijma HJ, Jeuken LJC, Verbeet MP, Armstrong FA, Canters GW. Protein film voltammetry of copper-containing nitrite reductase reveals reversible inactivation. J Am Chem Soc. 2007;129:8557–8565. doi: 10.1021/ja071274q. [DOI] [PubMed] [Google Scholar]
- 33.Ninio J. Alternative to the steady-state method—derivation of reaction-rates from 1st-passage times and pathway probabilities. Proc Natl Acad Sci USA. 1987;84:663–667. doi: 10.1073/pnas.84.3.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cao J. Michaelis–Menten equation and detailed balance in enzymatic networks. J Phys Chem B. 2011;115:5493–5498. doi: 10.1021/jp110924w. [DOI] [PubMed] [Google Scholar]
- 35.Wang Q, Moerner WE. Optimal strategy for trapping single fluorescent molecules in solution using the ABEL trap. Appl Phys B Lasers Opt. 2010;99:23–30. doi: 10.1007/s00340-009-3843-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kartalov E, Unger M, Quake SR. Polyelectrolyte surface interface for single-molecule fluorescence studies of DNA polymerase. BioTechniques. 2003;34:505–510. doi: 10.2144/03343st02. [DOI] [PubMed] [Google Scholar]
- 37.Ghosh R, Quayle JR. Phenazine ethosulfate as a preferred electron-acceptor to phenazine methosulfate in dye-linked enzyme assays. Anal Biochem. 1979;99:112–117. doi: 10.1016/0003-2697(79)90050-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.