

Abstract
Motivation: Most existing methods for DNA sequence analysis rely on accurate sequences or genotypes. However, in applications of the next-generation sequencing (NGS), accurate genotypes may not be easily obtained (e.g. multi-sample low-coverage sequencing or somatic mutation discovery). These applications press for the development of new methods for analyzing sequence data with uncertainty.
Results: We present a statistical framework for calling SNPs, discovering somatic mutations, inferring population genetical parameters and performing association tests directly based on sequencing data without explicit genotyping or linkage-based imputation. On real data, we demonstrate that our method achieves comparable accuracy to alternative methods for estimating site allele count, for inferring allele frequency spectrum and for association mapping. We also highlight the necessity of using symmetric datasets for finding somatic mutations and confirm that for discovering rare events, mismapping is frequently the leading source of errors.
Availability: http://samtools.sourceforge.net
Contact: hengli@broadinstitute.org
1 INTRODUCTION
The 1000 Genomes Project (1000 Genomes Project Consortium, 2010) sets an excellent example on how to design a sequencing project to get the maximum output pertinent to human populations. An important lesson from this project is to sequence many human samples at relatively low coverage instead of a few samples at high coverage. We adopt this strategy because with higher coverage, we will mostly reconfirm information from other reads, but with more samples, we will be able to reduce the sampling fluctuations, gain power on variants present in multiple samples and get access to many more rare variants. On the other hand, sequencing errors counteract the power in variant calling, which necessitates a minimum coverage. The optimal balancing point is broadly regarded to be in the 2–6 fold range per sample (Le and Durbin, 2010; Li et al., 2011), depending on the sequencing error rate, level of linkage disequilibrium (LD) and the purpose of the project.
A major concern with this design is that at 2–6 fold coverage per sample, non-reference alleles may not always be covered by sequence reads, especially at heterozygous loci. Calling variants from each individual and then combining the calls usually yield poor results. The preferred strategy is to enhance the power of variant discovery by jointly considering all samples (Depristo et al., 2011; Le and Durbin, 2010; Li et al., 2011; Nielsen et al., 2011). This strategy largely solves the variant discovery problem, but acquiring accurate genotypes for each individual remains unsolved. Without accurate genotypes, most of the previous methods [e.g. testing Hardy–Weinberg equilibrium (HWE) and association mapping] would not work.
To reuse the rich methods developed for genotyping data, the 1000 Genomes Project proposes to impute genotypes utilizing LD across loci (Browning and Yu, 2009; Howie et al., 2009; Li et al., 2009b, 2010a). Suppose at a site A, one sample has a low coverage. If some samples at A have high coverage and there exists a site B that is linked with A and has sufficient sequence support, we can transfer information across sites and between individuals, and thus make a reliable inference for the low-coverage sample at A. The overall genotype accuracy can be greatly improved.
However, imputation is not without potential concerns. First, imputation cannot be used to infer the regional allele frequency spectrum (AFS) because imputation as of now can only be applied to candidate variant sites, while we need to consider non-variants to infer AFS. Second, the effectiveness of imputation depends on the pattern of LD, which may lead to potential bias in population genetical inferences. Third, the current imputation algorithms are slow. For a thousand samples, the fastest algorithm may be slower than read mapping algorithms, which is frequently the bottleneck of analyzing NGS data (H.M.Kang, personal communication). Considering more samples and using more accurate algorithms will make imputation even slower.
These potential concerns make us reconsider if imputation is always preferred. We notice that we perform imputation mainly to reuse the methods developed for genotyping data, but would it be possible to derive new methods to solve classical medical and population genetical problems without precise genotypes?
Another application of NGS that requires genotype data is to discover somatic mutations or germline mutations between a few related samples (Conrad et al., 2011; Ley et al., 2008; Mardis et al., 2009; Pleasance et al., 2010a, b; Roach et al., 2010; Shah et al., 2009). For such an application, samples are often sequenced to high coverage. Although it is not hard to achieve an error rate one per 100 000 bases (Bentley et al., 2008), mutations occur at a much lower rate, typically of the order of 10−6 or even 10−7. Naively calling genotypes and then comparing samples frequently would not work well (Ajay et al., 2011), because subtle uncertainty in genotypes may lead to a bulk of errors. From another angle, however, when discovering rare mutations, we only care about the difference between samples. Genotypes are just a way of measuring the difference. Is it really necessary to go through the genotype calling step?
This article explores the answer to these questions. We will show in the following how to compute various statistics directly from sequencing data without knowing genotypes. We will also evaluate our methods on real data.
2 METHODS
This section presents the precise equations on how to infer various statistics such as the genotype frequency and AFS, and to perform various statistical test such as testing HWE and associations. Some of these equations have already been described in the existing literature, but for theoretical completeness, we give the equations using our notations. The last subsection reviews the existing methods and summarizes the differences between them, as well as between ours and the existing formulation.
In this section, we suppose there are n individuals with the i-th individual having mi ploidy. At a site, the sequence data for the i-th individual is represented as di and the genotype is gi which is an integer in [0, mi], equal to the number of reference alleles in the individual. Table 1 gives notations common across this section. The detailed derivation of the equations in this article is presented in an online document (http://bit.ly/stmath).
Table 1.
Common notations
| Symbol | Description |
|---|---|
| n | Number of samples |
| mi | Ploidy of the i-th sample (1≤i≤n) |
| M | Total number of chromosomes in samples: M=∑imi |
| di | Sequencing data (bases and qualities) for the i-th sample |
| gi | Genotype (the number of reference alleles) of the i-th sample (0≤gi≤mi)a |
| ϕk | Probability of observing k reference alleles (∑k=0Mϕk=1) |
| Pr{A} | Probability of an event A |
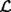 i(θ) i(θ) |
Likelihood function for the i-th sample: i(θ)=Pr{di|θ} |
aIn this article, we only consider biallelic variants.
2.1 Assumptions
2.1.1 Site independency
We assume data at different sites are independent. This may not be true in real data, because sequencing and mapping are context dependent; when there is an insertion or deletion (INDEL) error or INDEL polymorphism, sites nearby are also correlated in alignment. Nonetheless, most of the existing methods make this assumption for simplicity. The effect of site dependency may also be reduced by post-filtering and properly modeling the mapping and alignment errors (Li et al., 2008; Li, 2011).
2.1.2 Error independency and sample independency
We assume that at a site the sequencing and mapping errors of different reads are independent. As a result, the likelihood functions of different individuals are independent:
| (1) |
In real data, errors may be dependent of sequence context (Nakamura et al., 2011). The independency assumption may not hold. It is possible to model error dependency within an individual (Li et al., 2008), but the sample independency assumption is essential to all the derivations below.
2.1.3 Biallelic variants
We assume all variants are biallelic. In the human population, the fraction of triallelic SNPs is ~0.2% (Hodgkinson and Eyre-Walker, 2010). The biallele assumption does not have a big impact to the modeling of SNPs, though it may have a bigger impact to the modeling of INDELs at microsatellites.
2.2 Computing genotype likelihoods
For one sample at a site, the sequencing data d is composed of an array of bases on sequencing reads plus their base qualities. As we only consider biallelic variants, we may focus on the two most evident types of nucleotides and drop the less evident types if present. Thus, at any site we see at most two types of nucleotides. This treatment is not optimal, but sufficient in practice.
Suppose at a site there are k reads. Without losing generality, let the first l bases (l≤k) be identical to the reference and the rest be different. The error probability of the j-th read base is ϵj. Assuming error independency, we can derive that
| (2) |
where m is the ploidy.
2.3 Inferences from multiple samples
2.3.1 Estimating the site allele frequency
In this section, we estimate the per-site reference allele frequency ψ. For the i-th sample, let mi be the ploidy, gi the genotype and di the sequencing data. Assuming HWE, we can compute the likelihood of ψ:
| (3) |
where i(gi) is computed by Equation (2) and
| (4) |
is the probability mass function of the binomial distribution Binom(m, ψ).
Knowing the likelihood of ψ, we may numerically find the max-likelihood estimate with, for example, Brent's method (Brent, 1973). An alternative approach is to infer using an expectation–maximization algorithm (EM), regarding the sample genotypes as missing data. Given we know the estimate ψ(t) at the t-th iteration, the estimate at the (t+1)-th iteration is
| (5) |
where M=∑imi is the total number of chromosomes in samples.
When the signal from the data is strong, or equivalently for each i, one of i(g) is much larger than others, the EM algorithm converges faster than the direct numerical solution using Brent's method. However, when the signal from the data is weak, numerical method may converge faster than EM (Kim et al., 2011). In implementation, we apply 10 rounds of EM iterations. If the estimate does not converge after 10 rounds, we switch to Brent's method.
2.3.2 Estimating the genotype frequencies
In this section, we assume all samples have the same ploidy: m=m1=···=mn and aim to estimate ξg, the frequency of genotype g. The likelihood of {ξ0,…, ξm} is:
| (6) |
with the constraint ∑gξg=1. The EM iteration equation is
| (7) |
An important application of genotype frequencies is to test HWE for diploid samples (m=2). When genotypes are known, we can perform a one-degree χ2 test. This approach would not work for sequencing data as it does not account for the uncertainty in genotypes, especially when the average read depth of each individual is low. A proper solution is to perform a likelihood-ratio test (LRT). The test statistic is
| (8) |
where
| (9) |
is the max-likelihood estimate of the site allele frequency and similarly  ,
,  and
and 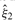 are the max-likelihood estimate of the genotype frequencies. Because
are the max-likelihood estimate of the genotype frequencies. Because 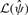 has one degree of freedom and
has one degree of freedom and 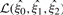 has two degrees of freedom, the De statistic approximately follows the one-degree χ2 distribution. For genotype data, De approaches the standard HWE test statistic computed from a 3×2 contingency table.
has two degrees of freedom, the De statistic approximately follows the one-degree χ2 distribution. For genotype data, De approaches the standard HWE test statistic computed from a 3×2 contingency table.
2.3.3 Estimating haplotype frequencies between loci
In this section, we assume all samples are diploid. Given k loci, let  be a haplotype where hj equals 1 if the allele at the j-th locus is identical to the reference, and equals 0 otherwise. Let
be a haplotype where hj equals 1 if the allele at the j-th locus is identical to the reference, and equals 0 otherwise. Let 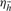 be the frequency of haplotype
be the frequency of haplotype  satisfying
satisfying 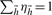 , where
, where
knowing the genotype likelihood at the j-th locus for the i-th individual (j)i(g), we can compute the haplotype frequencies iteratively with:
| (10) |
When sample genotypes are all certain, this EM iteration is reduced to the standard EM for estimating haplotype frequencies using genotype data (Excoffier and Slatkin, 1995).
The time complexity of computing Equation (10) is O(n · 4k) and thus it is impractical to estimate the haplotype frequency for many loci jointly. A typical use of Equation (10) is to measure LD between two loci.
2.3.4 Testing associations
Suppose we divide samples into two groups of size n1 and n−n1, respectively, and want to test if Group 1 significantly differs from Group 2. One possible test statistic could be (Kim et al., 2010, 2011)
| (11) |
where  is the max-likelihood estimate of the site allele frequency of all samples [Equation (9)], and
is the max-likelihood estimate of the site allele frequency of all samples [Equation (9)], and  and
and  are the estimates of allele frequency in Groups 1 and 2, respectively. Under the null hypothesis, D approximately follows the one-degree χ2 distribution.
are the estimates of allele frequency in Groups 1 and 2, respectively. Under the null hypothesis, D approximately follows the one-degree χ2 distribution.
A potential concern with the Da1 statistic is that the computation of (ψ) assumes HWE. When HWE is violated, false positives may arise (Nielsen et al., 2011). For diploid samples, a safer statistic is
| (12) |
which in principle follows the two-degree χ2 distribution under the null hypothesis. However, when both cases and controls are in HWE, the degree of freedom is reduced and this statistic is underpowered.
We have not found a powerful test statistic robust to HWE violation. For practical applications, we propose to take the P-value computed with Da1, while filtering candidates having a low Da2 to reduce false positives caused by HWE violation (see Section 3).
2.3.5 Estimating the number of non-reference alleles
In this section, we use the term site reference allele count to refer to the number of reference alleles at one single site. Allele count is a discrete number while allele frequency is contiguous.
For convenience, define random vector  to be a genotype configuration, and X=∑iGi to be the site reference allele count in all the samples. Assuming HWE, we have
to be a genotype configuration, and X=∑iGi to be the site reference allele count in all the samples. Assuming HWE, we have
where  is the total number of reference alleles in a genotype configuration
is the total number of reference alleles in a genotype configuration 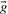 , and δkl is the Kronecker delta function which equals 1 if k=l and equals 0 otherwise. The likelihood of allele count is
, and δkl is the Kronecker delta function which equals 1 if k=l and equals 0 otherwise. The likelihood of allele count is
| (13) |
where  represents all sequencing data. To compute this probability efficiently, we define
represents all sequencing data. To compute this probability efficiently, we define
| (14) |
for 0≤l≤∑i=1jmi and zjl=0 otherwise. zjl can be calculated iteratively with
| (15) |
starting from z00=1. Comparing the definition of znk and Equation (15), we know that
| (16) |
which computes the likelihood of the allele count.
Although the computation of the likelihood function (k) is more complex than of (ψ), (k) is discrete, which is more convenient to maximize or sum over. This likelihood function establishes the foundation of the Bayesian inference.
2.3.6 Numerical stability of the allele count estimation
When computing zjl with Equation (15), floating point underflow may occur given large j. A numerically stable approach is to compute  instead, where Mj=∑i=1j mi. Thus
instead, where Mj=∑i=1j mi. Thus
| (17) |
and by replacing zjk with  in Equation (15), we can derive:
in Equation (15), we can derive:
 |
(18) |
However, we note that yjl may decrease exponentially with increasing j. Floating point underflow may still occur. An even better solution is to rescale yjl for each j, similar to the treatment of the forward algorithm for Hidden Markov Models (Durbin et al., 1998). In practical implementation, we compute
| (19) |
where tj is chosen such that  .
.
As another implementation note, most yjl are close to zero and thus ynk can be computed in a band rather than in a triangle. This may dramatically speed up the computation of the likelihood.
2.3.7 Calling variants
In variant calling, we have a strong prior knowledge that at most of the sites all samples are homozygous to the reference. To utilize the prior knowledge, we may adopt a Bayesian inference for variant calling. Let ϕk, k=1,…, M, be the probability of seeing k reference alleles among M chromosomes/haplotypes. For convenience, define Φ={ϕk}, which is in fact the sample AFS for M chromosomes. Recall that X is the number of reference alleles in the samples. The posterior of X is
| (20) |
where (k) is defined by Equation (13) and computed by Equation (17). In variant calling, we define variant quality as
and call the site as a variant if Qvar is large enough, because in deriving (k), we do not require the ploidy of each sample to be the same. The variant calling method described here are in theory applicable to pooled resequencing with unequal pool sizes.
2.3.8 Estimating the sample AFS
For variant calling [Equation (20)], we typically take the Wright–Fisher AFS as the prior. We can also estimate the sample AFS with the maximum-likelihood inference when the Wright-Fisher prior deviates from the data.
Suppose we have L sites of interest and we want to estimate the frequency spectrum across these sites. Let Xa, a=1,…, L, be a random variable representing the number of reference alleles at site a. We can use an EM algorithm to find Φ that maximizes Pr{d|Φ}, the probability of data across all samples and all sites conditional on AFS. The iteration equation is
| (21) |
We call this method of estimating AFS as EM-AFS. Alternatively, we may also acquire the max-likelihood estimate of the allele count at each site using Equation (16). The normalized histogram of these counts gives the AFS. We call this method as site-AFS. We will compare the two methods in Section 3.
2.4 Discovering somatic and germline mutations
One of the key goals in cancer resequencing is to identify the somatic mutations between a normal-tumor sample pair (Robison, 2010), which can be achieved by computing a likelihood ratio. Given a pair of samples, the following likelihood ratio is an informative score:
| (22) |
where [·](g) is computed by Equation (2),  maximizes [1](g)[2](g), and similarly
maximizes [1](g)[2](g), and similarly 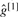 and
and  maximize [1](g) and [2](g), respectively.
maximize [1](g) and [2](g), respectively.
Note that in most practical cases, 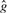 equals either
equals either 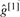 or
or  . When this stands, we have:
. When this stands, we have:
and then we can prove:
This equation has an intuitive interpretation: we are certain about a candidate somatic mutation only if both genotypes in both samples are clearly better than other possible genotypes.
A natural extension to discovering somatic mutations is to discover de novo and somatic mutations in a family trio (Conrad et al., 2011). To identify such mutations, we may compute the maximum likelihoods of genotype configurations without the family constraint and with the constraint, and then take the ratio between the two resulting likelihoods. The larger the ratio, the more confident the mutation. More exactly, the likelihood ratio is:
| (23) |
where c(gc), f(gf) and m(gm) are the child, father and mother genotype likelihoods, respectively, and 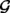 is the set of genotype configurations satisfying the Mendelian inheritance.
is the set of genotype configurations satisfying the Mendelian inheritance.
Although most of the derivation in this article assumes that variants are biallelic, we drop this assumption in the implementation for methods described in this subsection. We have observed false somatic/germline mutations caused by the mismodeling of triallelic variants (M.Depristo, personal communication). The biallelic assumption may lead to false positives.
2.5 Related works
During SNP calling, Thunder (Li et al., 2011) and glfMultiples (http://bit.ly/glfmulti) compute the site allele frequency by numerically maximizing the likelihood [Equation (2)]. Genome Analysis Toolkit (GATK; Depristo et al., 2011) infers the frequency with EM [Equation (5)]. Kim et al. (2011) infers the frequency with both the numerical and the EM algorithms. Li et al. (2010b) derived an alternative method to estimate the site allele frequency, which is not covered in this article. SeqEM (Martin et al., 2010) estimates the genotype frequency using EM [Equation (7)] with a different parameterization. Le and Durbin (2010) derived Equation (16). The conclusion is correct, but the derivation is not rigorous: the binomial coefficient in Equation (13) was left out. Yi et al. (2010) came to a similar set of equations to Equations (15) and (20), but the prior is taken from the estimated site allele frequency. To the best of our knowledge, Kim et al. (2010) is the first to use genotype likelihood-based LRT to compute P-value of associations [Equation (11)] with more thorough evaluation in a recent paper (Kim et al., 2011). Nielsen et al. (2011) further proposed to test associations with a score test (Schaid et al., 2002). Except Kim et al. (2010), all the previous works focus on diploid samples, while many equations in this article can be in theory applied to multiploidy samples and pooled samples.
In this article, our contribution includes testing HWE, estimating haplotype frequency, the proposal of two-degree association test, a simple but effective model for discovering somatic mutations, the rigorous derivation and numerically stable implementation of a discrete allele count estimator and an EM algorithm for inferring AFS.
3 RESULTS
3.1 Implementation
Most of equations for diploid samples (m=2) have been implemented in the SAMtools software package (Li et al., 2009a), which is distributed under the MIT open source license, free to both academic and commercial uses. The exact Equations (17)–(19) have also been implemented in GATK as the default SNP calling model.
The SAMtools package consists of two key components samtools and bcftools. The former computes the genotype likelihood (g) using an improved version of Equation (2) that considers error dependencies; the latter component calls variants and infers various statistics described in this article. To clearly separate the two steps, we designed a new Binary variant call format (BCF), which is the binary representation of the variant call format (VCF; Danecek et al., 2011) and is more compact and much faster to process than VCF. On real data, computing genotype likelihoods especially for INDELs is typically 10 times slower than variant calling. The separation of genotype likelihood computation and subsequent inferences enhances the flexibility and improves the efficiency for inferring AFS. Bcftools also directly works with VCF files, but is less efficient than with BCF files.
Table 2 shows how VCF information tags generated by SAMtools are related to the equations in this article. We refer to the SAMtools manual page for detailed description.
Table 2.
SAMtools specific VCF information
| INFOa | Equationb | Description |
|---|---|---|
| AF1 | 3, 5 | Non-reference site allele frequency |
| G3 | 7 | Diploid genotype frequency |
| HWE | 8 | P-value of Hardy–Weinberg equilibrium |
| NEIR | 10 | Neighboring r2 linkage disequilibrium statistic |
| LRT | 11 | One-degree association test P-value |
| LRT2 | 12 | Two-degree association test P-value |
| AC1 | 17, 18, 19 | Non-reference site allele count |
| FQ | 20 | Probability of the site being polymorphic among samples |
| CLR | 22, 23 | Log likelihood ratio score for de novo mutations |
aTag at the VCF additional information field (INFO).
bRelated, though not exact, equations for computing the values.
3.2 Inferring the allele count
We downloaded the chromosome 20 alignments of 49 Pilot-1 CEU samples sequenced by the 1000 Genomes Project using the Illumina technology only. We called the SNPs with SAMtools and imputed the genotypes with Beagle under the default settings. At 32 522 sites genotyped using the Omni genotyping chip and polymorphic in the 49 samples, the root-mean-square deviation (RMSD) between the allele count acquired from Omni genotypes and the estimate using Equation (16) equals 3.7, the same as the RMSD between the Omni and the Beagle-imputed genotypes. Not surprisingly, imputed genotypes are more accurate when there is a tightly linked SNP nearby, while the imputation-free estimate is less affected (Fig. 1).
Fig. 1.

Correlation of the site allele count accuracy with LD. The site allele count is estimated with Beagle imputation (solid line) and with Equation (16) (dashed line) at sites typed by the Omni genotyping chip. For each Omni SNP, the maximum r2 LD statistic between the SNP and 20 nearby SNPs called by SAMtools (10 upstream and 10 downstream) is computed from imputed genotypes. Omni SNPs are then ordered by the maximum r2 and approximately evenly divided into 15 bins. For each bin, the RMSD between the Omni allele count and the estimated allele count is computed as a measurement of the allele count accuracy.
However, on the unreleased European data from the 1000 Genomes Project consisting of 670 samples, Beagle imputation is better than our imputation-free method [RMSD(imput) = 12.7; RMSD (imput-free) = 15.0]. We conjure that this is because with more samples, it is more frequent for two samples to share a long haplotype. The LD plays a more important role in counteracting the lack of coverage. Nonetheless, we should beware that sites selected on the Omni genotyping chip may not be a good representative of all SNPs. For example, for the sites on the Omni chip, only 8% of SNPs do not have a nearby SNP with r2>0.05 in a 20-SNP window (the ‘nearby SNPs’ include all SNPs discovered in the 670 samples), but this percentage is increased to 30% for all SNPs. The large fraction of unlinked SNPs might hurt the accuracy of imputation-based methods.
We have also evaluated our method on an unpublished target reqsequencing dataset consisting of ~2000 samples (Haiman,C. and Henderson,B., personal communication). The imputation-based method does not perform well [RMSD(imput) = 54.8; RMSD(imput-free) = 42.5], probably due to the lack of linked SNPs around fragmented target regions.
3.3 Inferring the AFS
To evaluate the accuracy of the estimated AFS, we compared the AFS obtained from the low-coverage data produced by the 1000 Genomes Project and from the high-coverage data released by Complete Genomics (http://bit.ly/m7LzvF). Figure 2 reveals that we can infer a fairly accurate AFS using the EM-AFS method with 3-fold coverage per sample. On the other hand, the site-AFS estimate is less stable, though the overall trend looks right. To estimate properties across multiple sites, summing over the posterior distribution using EM-AFS is more appropriate.
Fig. 2.

The derived AFS conditional on heterozygotes discovered in the NA18507 genome (Bentley et al., 2008; AC:SRA000271). Heterozygotes were called with SAMtools on BWA (Li and Durbin, 2009) alignment. The ancestral sequences were determined from the Ensembl EPO alignment (Paten et al., 2008), with the requirement of the chimpanzee and orangutan sequences being identical. The AFS at these heterozygotes were computed in three ways: (i) from the nine independent Yoruba individuals sequenced by Complete Genomics (Drmanac et al., 2010) and analyzed using CGA Tools version 1.10.0; (ii) from nine random Pilot-1 Yoruba individuals released by the 1000 Genomes Project using the EM-AFS method and (iii) from the same 9 Pilot-1 individuals using site-AFS.
3.4 Performing association test
To evaluate the performance of the association test statistics Da1 [Equation (11)], we constructed a perfect negative control using the 1000 Genomes data and derived the empirical distribution of Da1. We expect to see no associations. Figure 3 shows that Da1 largely follows the one-degree χ2 distribution. However, this method also produces one false positive SNP (P<10−6). Closer investigation reveals that the SNP significantly violates HWE [P<10−6, computed with Equation (8)], and thus violates the assumption behind the derivation of Da1. In fact, if we test the association with Da2 which does not assume HWE, the false positive will be suppressed (P>0.001). To test association using the one-degree likelihood-ratio test statistic, it is important to control HWE.
Fig. 3.

QQ-plot comparing the association test statistics to the one-degree and the two-degree χ2 distribution. The 49 CEU samples sequenced by the 1000 Genomes Project using the Illumina technology were randomly assigned to two groups of size 24 and 25, respectively. (A) two association test statistics were computed on chromosome 20 between the two groups: one by the one-degree likelihood ratio test [Equation (11)] and the other by the canonical one-degree χ2 test based on Beagle imputed genotypes; (B) the two-degree likelihood rate test statistic [Equation (12)].
3.5 Comparing sequencing data from the same individual
3.5.1 Comparing datasets of similar characteristics
We acquired the NA12878 data used by Depristo et al. (2011). This sample was sequenced with HiSeq2000 using two libraries with each put on eight lanes and each sequenced to about 30-fold coverage. We split the data in two by library and computed Dp [Equation (22)] at each base on chromosome 20 to identify sites that are called differently between the two libraries. With a stringent threshold Dp≥30 and without any filtering, 32 differences are called between the libraries and most from the centromere. Since the libraries were made from the same DNA at almost the same time, we expect to see no difference between the libraries. Seeing 32 differences is very unlikely. To explore if this is due to mismapping, we extracted reads around the 32 sites and remapped them with BWA-SW (Li and Durbin, 2010). Four differences remain around the centromere, implying that most of the differences between libraries are caused by the variation in read mapping. We further mapped the reads around the four sites to a version of the human reference genome used by the 1000 Genomes Project for phase-2 mapping (http://bit.ly/GRCh37d). No differences are left. This exercise reveals that when we come to very rare events, mapping errors, instead of sequencing errors, lead to most of the artifacts.
3.5.2 Comparing datasets of different characteristics
We also did a harder version of the exercise above: comparing this 60-fold HiSeq data to the old Illumina data for the same individual obtained >2 years ago by the 1000 Genomes Project. We note that although DNA used in the two datasets was originated from the same individual, somatic mutations in cell lines, which are of the order of 1000 per diploid genome (Conrad et al., 2011), may be present. If the cell lines used in two studies have greatly diverged, we might see up to a dozen somatic mutations on chromosome 20.
This time with a threshold Dp≥30 and a maximum depth filter 150, we identified 667 single-base differences between the two datasets, far more than our expectation. Again we sought to reduce mapping errors by remapping reads with BWA-SW to the 1000 Genomes Project phase-2 reference genome. The number of differences between the HiSeq and the old Illumina data quickly drops to 33. If we further filter out clustered SNPs using a 100 bp window, 13 potential differences are left, 2 of the initial candidates. This exercise again proves that mismapping is the leading source of errors.
To see if the simple likelihood ratio [Equation (22)] is comparable to more sophisticated methods, we briefly tried SomaticSniper (Larson et al., in press) on our data. With a somatic score cutoff 65, which is about 30 in the ‘2log’ scale as in Dp, SomaticSniper identified 1826 differences. SAMtools called fewer, because it limits the mapping quality of reads with excessive mismatches and applies base alignment quality (Li, 2011) to fix alignment errors around INDELs. With the two features switched off, SAMtools called 1696 differences, half of which overlap the differences found by SomaticSniper. Calls unique to one method tend to have a mutation score close to the threshold.
4 DISCUSSIONS
We have proposed a statistical framework for SNP calling as well as analyzing sequencing data but without explicitly calling SNPs or their genotypes. With this framework, we can discover somatic and germline mutations with appropriate input data, efficiently estimate site allele frequency, allele frequency spectrum and linkage disequilibrium, and test Hardy–Weinberg equilibrium and association. On real data, we have demonstrated that our method is able to achieve comparable accuracy to the best alternative methods. We have also extensively evaluated the performance of our method on several unpublished datasets and got sensible results. Thus, we conclude that useful information can be obtained directly from sequencing data without SNP calling or imputation.
Here we also want to emphasize a few findings in our evaluation of the methods. First, we confirmed that imputation is a viable method for transferring our knowledges on genotyping data to low-coverage sequencing data. It is likely to have higher accuracy than our method given homogeneous whole-genome data consisting of many samples. Nonetheless, we showed that the accuracy of imputation depends on the LD nearby, which has long been speculated but without direct evidence from real data until our work. Second, our proposed EM-AFS method is able to accurately estimate AFS from low-coverage sequencing data. It is more appropriate than estimating the site frequency separately and then doing a histogram. Third, we observed that violation of HWE may cause false positives in association mapping with the one-degree likelihood ratio test (Kim et al., 2011). A two-degree likelihood ratio test is a conservative way to avoid such an artifact. At last, we highlighted the importance of using data of similar characteristics in the discovery of somatic mutations. We also want to put a particular emphasis on the necessity of controlling mapping errors when looking for very rare events such as somatic mutations, germline mutations and RNA editing. It may be necessary to use two distinct mapping algorithms to call variants and then take the intersection.
Frequently, we require to know the exact DNA sequences or genotypes only to estimate parameters or compute statistics. In these cases, the sequences and genotypes are just intermediate results. When the sequence itself is uncertain, mostly due to the uncertainty in sequencing and mapping, it may sometimes be preferred to directly work with the uncertain sequence, which may carry more information than an arbitrarily ascertained sequence. We have showed that many population genetical parameters and statistical tests can be adapted to work on uncertain sequences, and believe more existing methods can be adapted in a similar manner. Knowing the exact sequence is convenient, but not always indispensable.
ACKNOWLEDGEMENTS
We are grateful to Christopher Haiman for providing the unpublished dataset for assessing the performance, to Petr Danecek for evaluating the methods in this article on large-scale datasets, to Rasmus Nielsen for the observation on the occasional slow convergence of the EM algorithm and to Si Quang Le and Richard Durbin for the help on understanding the QCall model. We also thank the 1000 Genomes Project analysis subgroup and the GSA team at Broad Institute for various helpful discussions, and thank all the SAMtools users for evaluating the software package.
Funding: National Institutes of Health (1U01HG005208-01).
Conflict of Interest: none declared.
REFERENCES
- 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajay SS, et al. Accurate and comprehensive sequencing of personal genomes. Genome Res. 2011;21:1498–1505. doi: 10.1101/gr.123638.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brent RP. Algorithms for Minimization without Derivatives. Englewood Cliffs, New Jersey: Prentice-Hall; 1973. [Google Scholar]
- Browning BL, Yu Z. Simultaneous genotype calling and haplotype phasing improves genotype accuracy and reduces false-positive associations for genome-wide association studies. Am. J. Hum. Genet. 2009;85:847–861. doi: 10.1016/j.ajhg.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad D, et al. Variation in genome-wide mutation rates within and between human families. Nat. Genet. 2011;43:712–714. doi: 10.1038/ng.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, et al. The variant call format and vcftools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drmanac R, et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science. 2010;327:78–81. doi: 10.1126/science.1181498. [DOI] [PubMed] [Google Scholar]
- Durbin R, et al. Biological Sequence Analysis. Cambridge, UK: Cambridge University Press; 1998. [Google Scholar]
- Excoffier L, Slatkin M. Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol. Biol. Evol. 1995;12:921–927. doi: 10.1093/oxfordjournals.molbev.a040269. [DOI] [PubMed] [Google Scholar]
- Hodgkinson A, Eyre-Walker A. Human triallelic sites: evidence for a new mutational mechanism? Genetics. 2010;184:233–241. doi: 10.1534/genetics.109.110510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie BN, et al. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, et al. Design of association studies with pooled or un-pooled next-generation sequencing data. Genet. Epidemiol. 2010;34:479–491. doi: 10.1002/gepi.20501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, et al. Estimation of allele frequency and association mapping using next-generation sequencing data. BMC Bioinformatics. 2011;12:231. doi: 10.1186/1471-2105-12-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le SQ, Durbin R. SNP detection and genotyping from low-coverage sequencing data on multiple diploid samples. Genome Res. 2010;21:952–960. doi: 10.1101/gr.113084.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley TJ, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. The sequence alignment/map format and samtools. Bioinformatics. 2009a;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Improving SNP discovery by base alignment quality. Bioinformatics. 2011;27:1157–1158. doi: 10.1093/bioinformatics/btr076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, et al. Genotype imputation. Annu. Rev. Genomics Hum. Genet. 2009b;10:387–406. doi: 10.1146/annurev.genom.9.081307.164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, et al. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol. 2010a;34:816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat. Genet. 2010b;42:969–972. doi: 10.1038/ng.680. [DOI] [PubMed] [Google Scholar]
- Li Y, et al. Low-coverage sequencing: Implications for design of complex trait association studies. Genome Res. 2011;21:940–951. doi: 10.1101/gr.117259.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin ER, et al. SeqEM: an adaptive genotype-calling approach for next-generation sequencing studies. Bioinformatics. 2010;26:2803–2810. doi: 10.1093/bioinformatics/btq526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, et al. Sequence-specific error profile of illumina sequencers. Nucleic Acids Res. 2011;39:e90. doi: 10.1093/nar/gkr344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, et al. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 2011;12:443–451. doi: 10.1038/nrg2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paten B, et al. Enredo and pecan: genome-wide mammalian consistency-based multiple alignment with paralogs. Genome Res. 2008;18:1814–1828. doi: 10.1101/gr.076554.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleasance ED, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010a;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleasance ED, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010b;463:184–190. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach JC, et al. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 2010;328:636–639. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison K. Application of second-generation sequencing to cancer genomics. Brief. Bioinformatics. 2010;11:524–534. doi: 10.1093/bib/bbq013. [DOI] [PubMed] [Google Scholar]
- Schaid DJ, et al. Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am. J. Hum. Genet. 2002;70:425–434. doi: 10.1086/338688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SP, et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009;461:809–813. doi: 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- Yi X, et al. Sequencing of 50 human exomes reveals adaptation to high altitude. Science. 2010;329:75–78. doi: 10.1126/science.1190371. [DOI] [PMC free article] [PubMed] [Google Scholar]