

Abstract
Motivation: Sequencing projects increasingly target samples from non-clonal sources. In particular, metagenomics has enabled scientists to begin to characterize the structure of microbial communities. The software tools developed for assembling and analyzing sequencing data for clonal organisms are, however, unable to adequately process data derived from non-clonal sources.
Results: We present a new scaffolder, Bambus 2, to address some of the challenges encountered when analyzing metagenomes. Our approach relies on a combination of a novel method for detecting genomic repeats and algorithms that analyze assembly graphs to identify biologically meaningful genomic variants. We compare our software to current assemblers using simulated and real data. We demonstrate that the repeat detection algorithms have higher sensitivity than current approaches without sacrificing specificity. In metagenomic datasets, the scaffolder avoids false joins between distantly related organisms while obtaining long-range contiguity. Bambus 2 represents a first step toward automated metagenomic assembly.
Availability: Bambus 2 is open source and available from http://amos.sf.net.
Contact: mpop@umiacs.umd.edu
Supplementary Information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Metagenomics, the direct sequencing of DNA from all organisms in an environment without culturing, has recently emerged as a new scientific field that enables the discovery of novel organisms and genes (Yooseph et al., 2007)—as well as the study of population structure and dynamics (Arumugam et al., 2011; Koenig et al., 2011). Metagenomic studies have greatly expanded the understanding of microbial diversity. For example, viral quasi-species have been shown to affect pathogenicity in the poliovirus due to cooperation between differently adapted individuals in a population, as well as between coinfecting viruses (Vignuzzi et al., 2005). Other recent studies have relied on metagenomics to identify novel genes and uncultured microbes (Hess et al., 2011).
The assembly of metagenomic data is complicated by several factors such as: (i) widely different levels of representation for different organisms in a community; (ii) genomic variation between closely related organisms; (iii) conserved genomic regions shared by distantly related organisms; and (iv) repetitive sequences within individual genomes. Similar challenges occur in the assembly of polymorphic eukaryotes, a challenging domain for existing assembly algorithms. For example, the assembly of the sea squirt genome Ciona savignyi required extensive manual intervention and customized scripts despite the fact that this genome is fairly ‘simple’—there were only two haplotypes of roughly equal coverage (Vinson et al., 2005). Metagenomic data are considerably more complex. Due to the lack of assembly tools specifically targeted at metagenomic projects, studies rely on existing assemblers and attempt to mitigate some of the challenges posed by the data through iterative adjustment of assembly parameters and post-processing. Tuning is critical as existing assemblers make frequent errors even in simulated datasets with significantly lower complexity than true environments (Mavromatis et al., 2007). At the same time, current assemblers produce fragmented assemblies, hampering downstream analysis. For example, in the analysis of the Global Ocean Survey data, the Celera Assembler (Myers et al., 2000) was heavily modified to allow high error rates in order to account for strain variation, and to overcome the effects of varied coverage levels on the statistical repeat detection procedure (Rusch et al., 2007; Venter et al., 2004). Only two assemblers were developed specifically for metagenomic datasets (Laserson et al., 2011; Peng et al., 2011). However, neither utilizes mate-pairs, our focus in this work.
We present novel scaffolding algorithms optimized for non-clonal assembly. Though our algorithms are also applicable to polymorphic genomes, the primary focus of this article is on metagenomic analysis. These algorithms are implemented in a software tool called Bambus 2. Bambus 2 supersedes our previous scaffolder, Bambus (Pop et al., 2004), which was targeted at clonal Sanger data. We will show that, when applied to metagenomic datasets, Bambus 2 generates large scaffolds while avoiding false joins between distantly related organisms. Furthermore, our software can automatically identify genomic regions of variation that correspond to previously characterized polymorphic loci.
1.1 Metagenomic scaffolding
In our opinion, the main challenge in metagenomic assembly is to develop an assembler that can automatically generate contiguous assemblies yet accurately capture genomic variation information throughout the assembly process.
It is important to first define the basic concepts underlying genome scaffolding. Most modern genome assemblers start by reconstructing segments of the genome that are unambiguously defined by the set of reads. These segments, called unitigs, are sections of the genome entirely contained in either unique regions or repeats, i.e. they do not span the boundary between individual repeats or between repeats and unique regions. The nucleic acid sequence of unitigs can, therefore, be unambiguously reconstructed.
Irrespective of the assembly algorithm employed, the unitigs themselves are generally small and assembly software must use additional information to increase the size of the contigs produced. Commonly, assemblers leverage the information contained in mate-pairs—information constraining (in orientation, the DNA strand from which the sequence originated, and approximate distance) the pairwise position of reads along the genome. The process through which mate-pair information is used to increase contig sizes, as well as to determine a global arrangement of contigs along the genome, is called scaffolding. Note that longer contigs can also be constructed by careful analysis of the assembly graph without the use of mate-pair information (Kingsford et al., 2010; Nagarajan and Pop, 2009)—we broadly consider scaffolding to also include such analyses. Most existing genome assemblers contain dedicated scaffolding modules [e.g. Butler et al. (2008); Li et al. (2010); Myers et al. (2000); Zerbino et al. (2009)]. The unitig graph is output by a variety of modern assemblers such as Newbler (Margulies et al., 2005), Celera Assembler (Myers et al., 2000) and SOAPdenovo (Li et al., 2010), allowing scaffolding tools to operate as a stand-alone module post-assembly (Dayarian et al., 2010; Gao et al., 2011; Pop et al., 2004). Throughout the article, we will assume that the unitig graph is given and will demonstrate how this information can be used to effectively analyze metagenomic datasets.
Genomic repeats are the major challenge when assembling isolate genomes, and their effect is compounded in metagenomic datasets. Repeats link together disparate sections of the genome. As the number of reconstructions grows exponentially with the number of repeats (Kingsford et al., 2010), it is intractable to find the one correct reconstruction. Therefore, most assemblers start by masking out unitigs that appear to represent repetitive segments of a genome. Celera Assembler, for example, uses depth of coverage statistics to determine whether a particular unitig represents a repeat, then ignores these unitigs until the later stages of scaffolding (Myers et al., 2000). Coverage statistics are also used in other assemblers (Butler et al., 2008; Dayarian et al., 2010; Zerbino et al., 2009). An alternative approach relies on topological information: unitigs that have multiple conflicting neighbors (Li et al., 2010) can be inferred to represent repeats.
While the approaches described above work well in isolate genomes, they can lead to false positives in metagenomic datasets. Coverage-based methods can classify abundant organisms as repeats, preventing the assembly of exactly those segments of the community that should be easily assembled (Venter et al., 2004). Distinguishing between repeats within the same genome and conserved genomic segments shared by closely related organisms can be difficult. As seen in Figure 1, the local unitig graphs and coverage look identical in both cases. Below, we will describe new approaches for repeat detection that work well in metagenomic datasets.
Fig. 1.

(a) The unitig graph representation of a single unitig, 3, having double the coverage of the surrounding unitigs. Solid black arrows correspond to reads comprising a unitig. (b) One of the possible resolutions of the graph presented in (a). This example places unitig 3 in two locations along a single genome. (c) A second of the possible resolutions of the graph presented in (a). This example places unitig 3 at the same location in two genomes (highlighted in different colors).
Currently available scaffolders attempt to construct linear scaffolds, i.e. where unitigs can be placed in a linear, non-overlapping order. When multiple unitigs occupy the same genomic region, they are either collapsed into one or the scaffolds are broken apart. Collapsing unitigs assumes the differences are due to error (Zerbino and Birney, 2008). Breaking scaffolds assumes the ambiguity is due to repeats (Dayarian et al., 2010). In metagenomic assembly, such bubbles (multiple contigs occupying the same position in the assembly) are common due to polymorphisms between closely related strains, and fracturing the scaffolds at such positions leads to fragmented assemblies. Collapsing unitigs can lead to a ‘mosaic’ consensus sequence. If the variation occurs within genes, the consensus may contain frameshifts and even make it difficult to determine whether a gene exists. Previous attempts at untangling the genomic variation information from assembly data have relied on visualization techniques (Eppley et al., 2007a). While valuable insights have been obtained through such studies, these approaches are manually intensive and not scalable to large metagenomic datasets. In this article, we propose an approach that can preserve polymorphic bubbles within the assembly yet allows long-range scaffolds to be constructed.
2 OUR APPROACH
We propose that repeats and genomic variation can be distinguished from each other by examining the unitig graph. Repeats appear to ‘tangle’ the unitig graph, thereby masking the global structure of the genome. Genomic variants, on the other hand, lead to localized motifs in the graph. For example, assume that several strains of a same organisms are virtually identical with the exception of a region of variation (e.g. a locus of antigenic variation). The graph pattern corresponding to this situation in Figure 2a appears as a bubble in the unitig graph. We suggest that the global structure of the genome can be best recovered if the ambiguity due to genomic variation is maintained throughout the scaffolding process. Specifically, motifs due to genomic variation do not affect the long-range structure of the common backbone shared by related genomes. Instead of resolving the bubbles, we detect regions of variation and replace each of them with a single graph node, simplifying the graph without obscuring the structure. Through the iterative application of this process, interleaved with standard graph simplification procedures we can obtain scaffolds that capture a large fraction of the common genome structure of closely related organisms. For each variant, we output a main sequence along with alternatives corresponding to the haplotypes in the data. Fasulo and others (Fasulo et al., 2002) have previously presented an approach for detecting and representing variant bubbles during the assembly process, primarily targeting short-range variation that can be found within a single sequencing read. Our approach is more general and can tolerate larger scale variants (our approach detected variants with an average size of 5 606.2 ± 8 868.26 when scaffolding 75 bp reads). Used in concert with the algorithm described by Fasulo et al. (2002) our method will detect large-scale polymorphisms in addition to the short-range within-read variants.
Fig. 2.

(a) A variant motif detected on the Sim3 dataset. The motif corresponds to a ferrochelatase gene in E.coli. There are two alternate versions of the gene within the E.coli K12 (2338) and E.coli O157:H7 (2034) genomes. (b) A CLUSTAL W (Thompson et al., 1994) alignment of a subset of the fasta output from Bambus 2, with an edit region corresponding to (a).
Underlying the procedure above is the assumption that the ambiguity in the assembly graph is primarily caused by genomic variants, i.e. repeats have been detected and removed from the graph. We will describe two approaches for finding repeats in metagenomic samples. The first approach is based on the observation that repeat nodes appear to ‘tangle’ the graph structure—these nodes look like focal points in the graph, as in Figure 3. We detect such repeats using a measure of node centrality similar to the vertex-betweenness centrality measure used in social network analysis (Freeman, 1977, 1979). We also propose a variant of coverage-based repeat detection that tracks the change in coverage within-graph components instead of using a global coverage statistic. We will show that this localized coverage measure is less sensitive to coverage differences between organisms in the sample.
Fig. 3.

The figure shows a subset of a bacterial assembly where nodes are connected if they share paired-end reads. The shaded node, 119, is a repeat that occurs on many shortest paths.
3 METHODS
Our algorithms operate on a contig graph. A contig may represent a single unitig or an ungapped concatenation of multiple contigs. For each mate-pair connecting pairs of contigs, we generate a link l with length d(l) and orientation computed from the orientation and positions of the reads in the contigs. The SD σ(l) is provided as input to Bambus 2. Using the set of links between pairs of contigs, the orientation is set as the orientation of the majority of the links. Once an orientation is selected, we check whether the distance constraints implied by the links are consistent with each other. If not, we discard the smallest number of links that results in a consistent set S (the largest consistent set can be found in nlogn time using an algorithm for maximal clique finding in an interval graph). Each consistent set is output as an edge e with weight w(e)=|S|. The average length 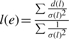 and SD
and SD 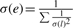 as suggested in Huson et al. (2001). Additional information, such as overlaps between adjacent contigs (contigs sharing common sequence), is also included when constructing the edges. The resulting graph is bidirected (Medvedev et al., 2007).
as suggested in Huson et al. (2001). Additional information, such as overlaps between adjacent contigs (contigs sharing common sequence), is also included when constructing the edges. The resulting graph is bidirected (Medvedev et al., 2007).
Scaffolding consists of three operations: orientation, positioning and simplification. Throughout the process, we prune the graph by removing contradictory edges and recording their reason for removal.
To avoid the ambiguity introduced by repeats, we start with a repeat detection step, then exclude all repeat contigs and incident edges from scaffolding. The (possibly multiple) placement of these nodes can be determined after the initial scaffolding is complete.
Centrality-based repeat detection: we calculate the all-pairs-shortest paths with each edge having weight w=1. For each node, v, we calculate the number of times it appears on a shortest path: Pv. Note that larger contigs are expected to have a higher degree because they contain more reads and, therefore, have a higher chance of being the end-point of a mate-pair link. To correct for this, we linearly scale Pv by the contig length. Such a length-dependent correction has been previously proposed in the context of estimation of gene abundance in metagenomic samples (Sharon et al., 2009). A node is declared repetitive if the scaled 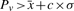 where c is a constant (usually set to 3),
where c is a constant (usually set to 3),  is the mean of all scaled Pv ∀v∈V and σ is the SD of all scaled Pv ∀v∈V.
is the mean of all scaled Pv ∀v∈V and σ is the SD of all scaled Pv ∀v∈V.
Local coverage statistic: for each connected component S and for each node v∈S, we compute the A-stat value (Myers et al., 2000). An abundant organism is less likely to appear repetitive in our approach as the connected component is more homogeneous. This operation is carried out after the repeat nodes identified by all-pairs-shortest paths have been removed.
Orientation: we must first convert the bidirected graph into a directed graph by choosing an orientation for each node in the graph. We call reverse edges any pairwise constraints that require the adjacent contigs to be in opposite orientations. It is impossible to assign a consistent order to nodes involved in a cycle with an odd number of reverse edges without discarding edges. We attempt to remove a minimum number of edges to allow a consistent orientation to be assigned. Finding such a minimum set is equivalent to the Maximal Bipartite Subgraph problem which is NP-hard (Garey and Johnson, 1979). We rely on a greedy heuristic proposed by Kececioglu and Myers (1995) that achieves a two-factor approximation. The algorithm runs in O(V+E) time.
Positioning: in addition to assigning an edge direction, we want to assign a position for each contig. There may be multiple edges assigning contradictory positions to a node. These imperfect data are the result of experimental errors and repeats (ambiguities in the placement of reads along a genome). We want to maximize the number of satisfied edges by placing nodes as close to the specified position as possible. This problem is similar to the Optimal Linear Arrangement problem which is also NP-hard (Garey and Johnson, 1979). We rely on the following greedy extension heuristic to linearly order the contigs: scaffolding starts by placing an arbitrary node at position 0. For each node without a position, compute an initial position based on all already-placed neighbors as a weighted average. Subsequent edges can reposition the node within a limit of 3σ(e) where σ(e) is the SD of the edge. The extension stops when the ratio of an edge weight w(e(u, v)) to the maximum weight edge incident on node u or v is below a threshold. Edges eliminated from the graph due to invalid orientation are not used in this step. The algorithm runs in O(V+E) time. This heuristic is sufficient once the graph is simplified as above and repeat contigs removed.
Simplification: a transitive reduction is applied to the contig graph and redundant edges are removed. Transitive edges [an edge e(u, v) such that there is a path p with a set of edges pe⊂E incident on nodes pv⊂V between u and v not including e(u, v)] are removed from acyclical components of the graph by performing a depth-first search from each node in topological order. Given the sequence lengths of contig in the graph l(v) ∀v∈V and a path p, we define the length of the path as l(p)=∑∀ contigs v∈pv l(v)+∑∀ edges e∈pe l(e). Define the SD of the path as σ(p)=∑∀ edges e∈pe σ(e). A transitive edge is removed when |l(e)−l(p)|≤σ(e)+σ(p). These edges can be removed without loss of information. Simple paths (all nodes have in- and out-degree equal to 1) are then collapsed: the nodes on the path are replaced with a single node representing the concatenation of the original nodes, and the intervening edges are removed from the graph. Finally, each simplified connected component in the graph gets reported as a scaffold.
Variant detection: once we have oriented and positioned the contigs and simplified the graph, we iteratively search for variation motifs. We search for subgraphs where multiple paths begin at a source node and collapse to one sink node within a certain number of hops. To allow for artifacts due to incomplete coverage, we allow subgraphs where paths terminate before reaching the sink.
Given graph G=(V,E) and motif set S⊂V
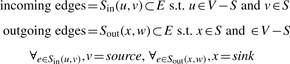 |
That is, the incoming edges may only be incident on the source node and the outgoing edges may only be incident on the sink node. Finally, to avoid false positives due to layouts that satisfy edge constraints but where nodes can be placed in a linear, non-overlapping order, we calculate the overlap ratio.
Given S⊂V, node v∈S, start coordinate of v, B(v) and end coordinate of v, E(v)
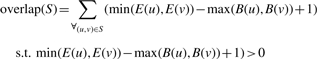 |
The overlap ratio is then 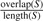 . Intuitively, it is the total number of bases covered by two or more nodes, divided by the total number of bases in the motif. Motifs whose overlap ratio exceeds a threshold are marked as a polymorphism. To make the problem tractable, only subgraphs with a diameter of 2 are detected in the current implementation of our algorithm. Each iteration of motif detection has a runtime of O(|V|×(Δ(G)3+3Δ(G))) where Δ(G) is the maximum degree of G. This algorithm has a worst-case runtime of O(|V|×(|E|3+3|E|)). However, in a contig graph it is likely that Δ(G)<<|E|. Every level of depth multiplies the runtime by a factor of Δ(G).
. Intuitively, it is the total number of bases covered by two or more nodes, divided by the total number of bases in the motif. Motifs whose overlap ratio exceeds a threshold are marked as a polymorphism. To make the problem tractable, only subgraphs with a diameter of 2 are detected in the current implementation of our algorithm. Each iteration of motif detection has a runtime of O(|V|×(Δ(G)3+3Δ(G))) where Δ(G) is the maximum degree of G. This algorithm has a worst-case runtime of O(|V|×(|E|3+3|E|)). However, in a contig graph it is likely that Δ(G)<<|E|. Every level of depth multiplies the runtime by a factor of Δ(G).
Output: Bambus 2 supports several output formats. Since we do not linearize scaffolds and maintain ambiguity due to variation in the graph, the native output is a graph [in Graphviz format Gansner and North (2000)]. Bambus 2 also finds the longest sequence reconstruction through each scaffold. That is, it will ignore variant motifs and generate a single self-consistent sequence for each scaffold. Additionally, Bambus 2 outputs each variation motif as a set of sequences. For each motif, S, we start from the source node, as defined above. For each child node c of source, we recursively compute the sequences starting at c. The longest sequence starting at source is the master sequence of the motif. The alternate sequences found in the graph are also output, including edit positions specifying where within the master sequence they belong. Figure 2b shows an example alignment of the fasta output for a variant region within E.coli.
Test data: we tested the algorithm using nine datasets. Brucella suis 1330 comprised 36 080 reads and available as NCBI Trace Archive Project ID 320. The reference includes: AE014291:AE014292 (2 107 792 bp, 1 207 381 bp). Three simulated datasets were generated using MetaSim (Richter et al., 2008) (Table 1). The acid mine drainage dataset, generated by Simmons et al. (2008); Tyson et al. (2004), consists of 179 770 reads and is available as NCBI Trace Archive Project ID 13696. The reference AMD dataset includes: Ferroplasma acidarmanus Type I, Ferroplasma sp. Type II, Leptospirillum sp. Group II 5-way CG, Leptospirillum sp. Group III and Thermoplasmatales archaeon Gpl and is available as CH003520:CH004435. The Twin Gut data were generated by Turnbaugh et al. (2008) and is available as SRA002775 (8.30M GS FLX fragments). The MetaHit datasets were generated by the MetaHit consortium (Qin et al., 2010) and are available as ERS006526, ERS006594 and ERS006494.
Table 1.
Simulated data
| Organism | Reference size | Identifier | ||||
|---|---|---|---|---|---|---|
| Psychromonas sp CNPT3 | 3 052 410 | AAPG00000000 | ||||
| Porphyromonas gingivalis W83 | 2 343 476 | AE015924 | ||||
| Escherichia coli K-12 MG1655 | 4 639 675 | U00096 | ||||
| Escherichia coli O157:H7 EDL933 | 5 528 445 | AE005174 | ||||
| Dataset | # Reads | Size of simulated paired-end libraries | P.sp CNPT3 | P.gingivalis W83 | E.coli K-12 | E.coli O157:H7 |
| Sim1 | 10 000 | 50% 5 kb, 50% 10 kb | 1.97X | 2.00X | 2.01X | 0.00X |
| Sim2 | 10 000 | 50% 5 kb, 50% 10 kb | 5.30X | 0.55X | 0.56X | 0.00X |
| Sim3 | 10 000 | 50% 5 kb, 50% 10 kb | 0.55X | 0.57X | 1.68X | 1.65X |
Four reference genomes were used to generate three simulated datasets. Organism: the reference used to generate simulated data. Reference size: the size (in base pair) of the reference. Identifier: the identifier of the reference in the NCBI Entrez database. # Reads: total number of reads simulated from the reference for a simulated dataset. The effective coverage for each reference is listed in each dataset.
4 RESULTS
In the following section, we demonstrate the performance of Bambus 2 by comparing it with two assemblers used in recent metagenomic projects [Celera Assembler (Myers et al., 2000) and SOAPdenovo (Li et al., 2010)]. We have not included a comparison with our previous scaffolder, Bambus (Pop et al., 2004), as it lacks the functionality necessary in a metagenomic setting. Also, we have omitted comparisons to Genovo (Laserson et al., 2011) and Meta-IDBA (Peng et al., 2011) as neither of these use mate-pair information during the assembly process.
4.1 Repeat detection
We benchmarked our algorithms for repeat detection using artificial and real datasets by comparing repeats identified by Bambus 2 with those identified by the Celera Assembler (Miller et al., 2008) with metagenomic settings (Rusch et al., 2007; Venter et al., 2004) (referred to as CA-met). The CA-met settings increase the tolerance for mismatches when building unitigs, providing longer range contiguity, but possibly leading to mis-assembly. The repeat detection from Celera Assembler relies on coverage, a common approach and procedures for tuning this assembler for both isolate and metagenomic assemblies have been documented (http://wgs-assembler.sf.net). Figure 4 shows the results.
Fig. 4.

Repeat detection comparison. Ideal repeat detection corresponds to the top-right corner of the graph, with 100% sensitivity and specificity. We vary the genome size estimate (a critical parameter in the procedure for detecting repeats) for CA, generating a curve for each dataset. The CA-met default is indicated by large shaded points. The Bambus 2 repeat detection is fully automated, generating a single point. As CA is designed for clonal organisms, only the default genome size estimate is used for B.suis. The gold standard is built from REPuter. All tests are run using the set of unitigs generated by CA-met. Sensitivity: 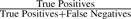 . Specificity:
. Specificity: 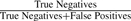 .
.
Ideally, the repeat detection should have both high sensitivity and specificity. Sensitivity reflects how many true repeats are detected. Detecting too few repeats can lead to assembly errors in scaffolding. Specificity reflects the false positives. Detecting too many repeats leads to a suboptimal assembly as these contigs do not fully participate in scaffolding. In the case of B.suis 1330, both methods have high sensitivity and specificity. Celera Assembler repeat detection was designed for clonal organisms. Since the B.suis dataset is clonal, CA can accurately detect repeats. In all other cases, Bambus 2 has a higher sensitivity and specificity than Celera Assembler. The default genome size estimates in CA are too sensitive, identifying too many repeats. While varying the genome size improves repeat detection, it is at the expense of sensitivity or specificity. On all datasets, this tuning, which is difficult when the true taxonomic distribution is unknown, still does not match Bambus 2's automated sensitivity and specificity result.
4.2 Scaffolding of simulated metagenomic datasets
We compared Bambus 2 to CA with default settings and CA-met. While other assemblers have been used in metagenomic studies [e.g. Phrap http://www.phrap.org/ and Newbler (Margulies et al., 2005)], as far as we are aware, they have not been extended to target metagenomic data. SOAPdenovo has also been used for metagenomic studies; however, no scaffolding results were reported (Qin et al., 2010).
We ran Bambus 2 to scaffold unitigs from CA-met and Minimus (Sommer et al., 2007). As seen in Figure 5, for all genomes, Bambus 2 outperforms CA. For all but one genome, Bambus 2 also outperforms CA-met. The only case where CA-met performs better than Bambus 2 is E.coli O157:H7 EDL933. The closely related E.coli strains are present at sufficient combined coverage for CA-met to obtain large scaffolds. However, the low-abundance genomes in the same sample are not assembled. In scaffolds over 2 kb, CA-met only includes 10.90 and 13.31% of the low−abundance genomes, versus 17.24 and 18.37% for Bambus 2. Additionally, CA-met constructs a ‘mosaic’ sequence of the two E.coli strains, masking variation and potentially introducing error (Supplementary Material). As we will show below, on the acid mine dataset, this ‘mosaic’ assembly leads CA-met makes more mistakes (chimeric scaffolds) than Bambus 2.
Fig. 5.

Assembly results for three simulated datasets. The y-axis represents the minimum number of scaffolds that add up to 1% of the genome size. Lower bars represent a better assembly. Bambus 2 produces large scaffolds for a wide range of coverage levels in our simulated datasets. Bambus 2 (CA-met) is Bambus 2 run using CA-met instead of using Minimus unitigs. We aligned the assembly (all contigs >2 kb) to the reference and counted coverage by reciprocal best matches over 95% identity. We use reciprocal best matches to avoid double counting Bambus 2 motifs that cover the same genomic region. We divide the number of scaffolds by the genome coverage and average the results, by genome, on all three simulated datasets to evaluate performance across varying coverage.
We examined all datasets for variation motifs detected by Bambus 2. A total of 16 motifs were found in the Sim1 dataset, and 6 motifs in the Sim2 dataset. Each of the motifs appear to be false positives (all the contigs comprising the motif originate from the same genome). The analysis of the sequence of the overlapping unitigs could be used to detect and correct such mistakes. Such analyses will be included in future versions of our software.
A total of 30 variation motifs were detected in the Sim3 dataset. The motifs detected include genes for ferrochelatase (Fig. 2a) as well as outer membrane proteins and integrase for prophage, which are known to vary across strains of E.coli (Perna et al., 2001) and more broadly, across other enterobacteria.
4.3 Scaffolding of the acid mine drainage metagenome
We tested Bambus 2 on an acid mine drainage (Tyson et al., 2004) metagenomic set. These data represent an ideal benchmark as they comprise a low number of organisms, and the genomic variation between related members of the community has been extensively studied. We generated unitigs using CA and scaffolded them with Bambus 2. The Bambus 2 assembly has fewer scaffolds in three of the five organisms present in this sample when compared with CA-met (Fig. 6). In two genomes, Leptospirillum sp Group III and Ferroplasma acidarmanus Type I, Bambus 2 halves the number of scaffolds while reconstructing a larger percentage of the references, as compared to CA-met. In one case, Ferroplasma sp Type II, CA-met produces fewer scaffolds than Bambus 2. However, we found that over 61% of the contigs in the Ferroplasma sp Type II CA-met assembly cannot be uniquely assigned to a single reference genome. We hypothesize that CA-met combined the assemblies of Ferroplasma acidarmanus Type I and Ferroplasma sp Type II, creating chimeric contigs and scaffolds.
Fig. 6.

Assembly results for the acid mine metagenomic dataset. The y-axis represents the minimum number of scaffolds that add up to 1% of the genome size. Lower bars represent a better assembly. Bambus 2 produces larger scaffolds that CA-met in three of the five genomes. We calculated assembly statistics as in Figure 5. In three genomes, both CA and Bambus 2 produced slightly >100% coverage. This is due to redundancy within the MUMmer alignments.
We validated our hypothesis by counting the fraction of contigs in chimeric scaffolds. Chimeric scaffolds either include a chimeric contig or contain contigs from different organisms (Supplementary Material). Bambus 2 had the lowest rate of chimeras, 5.66%, while CA-met had the highest at 23.07%. This is expected as CA-met was tuned to maximize scaffold size, possibly merging unrelated organisms. Bambus 2 built large scaffolds while making fewer mistakes.
The acid mine community used in our analysis is dominated by two genera: Leptospirillum bacteria and Ferroplasma archaea. A large extent of genomic variation, primarily due to recombination, was characterized in both these groups of organisms (Eppley et al., 2007b; Simmons et al., 2008; Tyson et al., 2004). Initial studies of this environment indicated that most genomic variation can be found in Ferroplasma sp Type II, with no predominant functional groups being associated with the variable regions (Tyson et al., 2004). Subsequent publications with additional sequencing (included in our dataset), also showed significant variation in Leptospirillum sp Group II ‘5-way CG’ (Simmons et al., 2008). Here we evaluated whether Bambus 2 is able to rediscover these results. We detected a total of 99 motifs, of which 66 represented alternate sequences (two contigs occupying the same positions) and 33 represent insertion/deletion of sequence. The majority of motifs could be assigned to regions from the Ferroplasma sp Type II, as expected. However, as a percentage of bases contained within variation motifs (the extent, rather than number of motifs), the most varied organisms appear to be Leptospirillum sp Group II ‘5-way CG’ and Leptospirillum sp Group III, followed by Ferroplasma sp Type II. The difference in the patterns of variation (frequent but small in Ferroplasma and less frequent but large in Leptospirillum) was also observed by Simmons et al. (2008) and could be explained by different biological mechanisms that drive the genomic variability. It was hypothesized (Eppley et al., 2007b) that recombination frequently occurs within Ferroplasma possibly due to the fact that that these organisms (as well as many other archaea) lack the mutS and mutL DNA repair systems. Conjugation or transduction, which produce large events (as they are dependent on the F-plasmid and phage size), was hypothesized to contribute to the genomic variation in Leptospirillum (Simmons et al., 2008).
We compare the genes within variation motifs to those identified in previous publications. We annotated the assembly by taking non-overlapping best BLASTX (Altschul et al., 1990) hits for each unitig and assigned a COG (Tatusov et al., 2000) functional category to each hit. We tabulated the counts of each COG category within the assembly and within the motifs. We then characterized the functional categories that are statistically enriched in motif regions (Supplementary Material). The functional category corresponding to ‘DNA replication, recombination and repair’ (category L) is significantly enriched (P=0.006, hypergeometric test). Also enriched (P=0.25, hypergeometric test) is one of the poorly characterized COG categories, ‘general function prediction’ (category R). Our results are consistent with previous analysis of the data (Simmons et al., 2008). One specific motif identified within Leptospirillum sp Group II ‘5-way CG’, corresponds to glycosyltransferase, a gene previously characterized as occurring within a mobile region of Leptospirillum sp Group II and Leptospirillum sp Group III (Goltsman et al., 2009). Thus, it is expected that this mobile element would mutate and recombine independently within the members of the Leptospirillum sp population, giving rise to the motif.
4.4 Scaffolding output of NGS assemblers
Finally, we tested Bambus 2 on four dataset composed of next-generation sequencing reads. The first dataset, comprising the gut microbiome of twins (Turnbaugh et al., 2008), was assembled using Newbler (Margulies et al., 2005) followed by Bambus 2. Our assembly combined all 18 individual samples from the original study. The assembly generated 3230 variation motifs. Since we lacked a reference, we could not map our assembly and tabulate statistics as with previous datasets. Instead, we evaluated the assembly contiguity. We sorted the scaffolds in decreasing order by size and counted the number and size of the smallest scaffold required to reach 5 Mb (Table 2). To assess scaffold correctness, we used BLAST (Altschul et al., 1990) to identify a dominant organism within the datset. The best hit was Bifidobacterium longum NCC2705 (AE014295). We mapped the assembled scaffolds to the reference using nucmer (Kurtz et al., 2004) and calculated the errors in scaffolds and the number of scaffolds to cover 1% of the reference. We also ran MetaGeneMark (Lukashin and Borodovsky, 1998) to identify open reading frames (ORFs) within the assemblies and include the results in Table 2. We annotated the assembly and evaluated COG functional category enrichment in motifs as before. The COG functional categories for ‘amino acid transport and metabolism’ (category E), ‘nucleotide transport and metabolism’ (category F), ‘carbohydrate transport and metabolism’ (category G), ‘DNA replication, recombination and repair’ (category L) and ‘cell envelope biogenesis, outer membrane’ (category M) were enriched, while categories for ‘cell motility and secretion’ (category N) and ‘unknown function’ (category S) were depleted in the variation motifs found by Bambus 2. Interestingly, the enriched functional categories were characterized as ‘core’ for the gut biome (categories universally found across all subjects) in Turnbaugh et al. (2008). Other categories classified by Turnbaugh et al. as core, such as ‘transcription’ (category K), were also found enriched in our motifs, but not significantly (Supplementary Material). Turnbaugh et al. noted that while no core microbiome exists at a taxonomic level, a core can be detected at a functional level. The over-abundance of these core genes in the detected motifs may explain this observation. We hypothesize that the core genes can occur in different genomic contexts due to lateral transfer, allowing a diverse set of organisms to survive within the human distal gut, and thereby explaining an enrichment of such genes within variation hotspots. These results would not be apparent from the analysis of the contig consensus sequences and demonstrate the importance of performing detailed analyses of the data underlying the assembly (i.e. the assembly graph) to characterize an environment.
Table 2.
Assembly results on test datasets
| Dataset | ASM | # Scaffolds | Mean | Max | # Scf | Len at 5 Mb | # ORFs / MB | # Scf in 1% | # Errors |
|---|---|---|---|---|---|---|---|---|---|
| GUT | Newbler | 11 012 | 4115.1 | 46 150 | 275 | 12 769 | 0.00217 | 19.18 | 7 |
| Bambus2 | 11 450 | 4778.9 | 80 512 | 134 | 25 370 | 0.00204 | 15.24 | 10 | |
| V1.CD-2 | SOAPdenovo | 5794 | 4889.0 | 84 000 | 230 | 14 207 | 0.00186 | 15.54 | 51 |
| Bambus2 | 4057 | 5680.6 | 237 167 | 166 | 18 210 | 0.00200 | 7.28 | 67 | |
| V1.UC-8 | SOAPdenovo | 15 029 | 5371.3 | 176 511 | 87 | 39 282 | 0.00178 | 1.34 | 13 |
| Bambus2 | 12 952 | 5954.0 | 257 939 | 58 | 55 905 | 0.00183 | 0.80 | 18 | |
| MH0012 | SOAPdenovo | 27 451 | 6470.1 | 356 312 | 30 | 115 466 | 0.00172 | 2.67 | 33 |
| Bambus2 | 23 994 | 5704.7 | 823 131 | 35 | 84 700 | 0.00180 | 0.99 | 43 | |
The contiguity results on four NGS datasets. #Scaffolds: the number of scaffolds > 2 kb. Mean: the average length of scaffolds. Max: the maximum length scaffold in the assembly. Scaffold at 5 Mb: we sort the scaffolds in decreasing order by length and count the number and size of the smallest scaffold required to reach 5 Mb. The #ORFs/MB measures the number of open reading frames (ORFs) identified by MetaGeneMark (Lukashin and Borodovsky, 1998) in the assembly. The counts are normalized by total sequence length in the assembly. The # Scf in 1% is reported as in Figures 5 and 6 using a reference identified by BLAST (Altschul et al., 1990); lower scores are better. The errors are reported by dnadiff from the MUMmer 3.20 package (Kurtz et al., 2004). The GUT dataset did not include paired-end information and we relied on the Newbler contig graph to perform scaffolding with Bambus 2. Therefore, the Newbler results are reported on contigs not scaffolds as no scaffolds were generated by Newbler on this dataset.
We selected three samples at random from the MetaHit consortium (V1.CD-2, V1.UC-8 and MH0012) (Qin et al., 2010). We reran SOAPdenovo to generate unitigs and scaffolds. We used Bambus 2 to scaffold the unitigs produced by SOAPdenovo (Li et al., 2010) and compare Bambus 2 scaffolds to those generated by SOAPdenovo (Table 2). One dataset (V1.CD-2), comprising over 51M Illumina reads was analyzed in ≈3.5 h with a peak RAM usage of 10.0 GB. The largest dataset (MH0012) comprising 186M reads was analyzed in ≈20 h.
In all cases, Bambus 2 produced more contiguous scaffolds than SOAPdenovo, in two cases more than doubling the largest scaffolds. We again identified a dominant organism within each dataset and map scaffolds to it. The best hits were Bacteroides coprophilus DSM 18228 (NZ_ACBW00000000), Methanobrevibacter smithii ATCC 35061 (NC_009515) and Akkermansia muciniphila ATCC BAA-835 (CP001071.1) for V1.CD-2, V1.UC-8 and MH0012, respectively. Bambus 2 produced more ORFs per MB of assembly. It also required fewer scaffolds to cover the reference while not introducing many errors.
We hypothesize that the improvement in contiguity is due to Bambus 2 overcoming genomic variation within the data, where we identified 2763 variation motifs. To evaluate our hypothesis, we aligned Bambus 2 motifs to SOAPdenovo scaffolds and counted motifs that span multiple scaffolds. Out of the 2763 motifs in the assemblies, 2554 mapped to multiple scaffolds, confirming that Bambus 2 motifs correspond to scaffold breaks in SOAPdenovo.
5 DISCUSSION
Bambus 2 is not a stand-alone assembler. Instead, it is a drop-in scaffolding module optimized for non-clonal data and is compatible with the output of many modern assemblers. Thus, Bambus 2 can be applied to virtually all existing sequencing technologies—it is sufficient to start with an assembler that is best suited for that type of data. We have shown that it can easily be applied to the output of Celera Assembler, Newbler, SOAPdenovo and Minimus, and have demonstrated its performance in Sanger, 454 and Illumina data. Bambus 2 includes an executive script (named goBambus2) that will automatically process input data in a variety of formats and run the pipeline, making it easy to use.
The current version of our code does not make use of sequence information when performing graph simplification. We plan to incorporate such information in the future, allowing Bambus 2 to merge contigs, when appropriate. In addition, using sequence information can allow Bambus 2 to avoid false positives in detecting variation motifs. We also plan to distribute a visualization tool to allow users to interact with the variants and the assembly graph.
The repeat detection procedures used in Bambus 2 are sensitive without sacrificing specificity, and could also be applied to the assembly of single genomes, in particular in single-cell projects where depth-of-coverage artifacts are common. The scaffolds generated by Bambus 2 cover a large percentage of the genomes in the samples, while largely avoiding misjoins. The fasta output of variants motifs facilitates analysis of the full diversity in an environment. Furthermore, the ability to highlight regions of variation has proven useful in detecting biologically meaningful patterns that match previously published results.
Accurately assembling metagenomic datasets automatically is challenging with current assemblers, and often requires manual tuning of parameters and post-processing. Bambus 2 represents a first step toward automated metagenomic assembly, and is able to obtain long-range contiguity in metagenomic datasets while also characterizing regions of variation.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Kate Koren, Jason Miller and Granger Sutton for suggestions and discussion on the manuscript and algorithms. We thank Dan Sommer and Bo Liu for helping to download data and annotate assemblies.
Funding: National Institutes of Health (R01-HG-004885 to M.P.); National Science Foundation (IIS-0812111 to M.P.).
Conflict of Interest: none declared.
REFERENCES
- Altschul S, et al. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Arumugam M, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler J, et al. ALLPATHS: De novo assembly of whole-genome shotgun microreads. Genome Res. 2008;18:810–820. doi: 10.1101/gr.7337908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayarian A, et al. Sopra: scaffolding algorithm for paired reads via statistical optimization. BMC Bioinformatics. 2010;11:345. doi: 10.1186/1471-2105-11-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eppley J, et al. Strainer: software for analysis of population variation in community genomic datasets. BMC Bioinformatics. 2007a;8:398. doi: 10.1186/1471-2105-8-398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eppley JM, et al. Genetic exchange across a species boundary in the archaeal genus ferroplasma. Genetics. 2007b;177:407–416. doi: 10.1534/genetics.107.072892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasulo D, et al. Efficiently detecting polymorphisms during the fragment assembly process. Bioinformatics. 2002;18:294–302. doi: 10.1093/bioinformatics/18.suppl_1.s294. [DOI] [PubMed] [Google Scholar]
- Freeman L. A set of measures of centrality based on betweenness. Sociometry. 1977;40:35–41. [Google Scholar]
- Freeman L. Centrality in social networks conceptual clarification. Soc. Netw. 1979;1:215–239. [Google Scholar]
- Gansner ER, North SC. An open graph visualization system and its applications to software engineering. Softw. Pract. Exp. 2000;30:1203–1233. [Google Scholar]
- Gao S, et al. Opera: reconstructing optimal genomic scaffolds with high-throughput paired-end sequences. Lect. Notes Comput. Sci. 2011;6577:437–451. doi: 10.1089/cmb.2011.0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garey M, Johnson D. Computers and Intractability: a Guide to NP-Completeness. San Francisco, CA: WH Freemanand Company; 1979. [Google Scholar]
- Goltsman DS, et al. Community genomic and proteomic analyses of chemoautotrophic iron-oxidizing “Leptospirillum rubarum” (Group II) and “Leptospirillum ferrodiazotrophum” (Group III) bacteria in acid mine drainage biofilms. Appl. Environ. Microbiol. 2009;75:4599–4615. doi: 10.1128/AEM.02943-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess M, et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science. 2011;331:463–467. doi: 10.1126/science.1200387. [DOI] [PubMed] [Google Scholar]
- Huson D, et al. Proceedings of the Fifth Annual International Conference on Computational Biology, RECOMB'01. New York, NY, USA: Association for Computing Machinery; 2001. The greedy path-merging algorithm for sequence assembly; pp. 157–163. [Google Scholar]
- Kececioglu J, Myers E. Combinatorial algorithms for DNA sequence assembly. Algorithmica. 1995;13:7–51. [Google Scholar]
- Kingsford C, et al. Assembly complexity of prokaryotic genomes using short reads. BMC Bioinformatics. 2010;11:21. doi: 10.1186/1471-2105-11-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig JE, et al. Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl Acad. Sci. USA. 2011;108(Suppl. 1):4578–4585. doi: 10.1073/pnas.1000081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laserson J, et al. Genovo: de novo assembly for metagenomes. J. Comput. Biol. 2011;18:429–443. doi: 10.1089/cmb.2010.0244. [DOI] [PubMed] [Google Scholar]
- Li R, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20:265–272. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashin AV, Borodovsky M. Genemark.hmm: new solutions for gene finding. Nucleic Acids Res. 1998;26:1107–1115. doi: 10.1093/nar/26.4.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies M, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavromatis K, et al. Use of simulated data sets to evaluate the fidelity of metagenomic processing methods. Nat. Methods. 2007;4:495–500. doi: 10.1038/nmeth1043. [DOI] [PubMed] [Google Scholar]
- Medvedev P, et al. Computability of models for sequence assembly. In: Giancarlo R, Hannenhalli S, editors. Algorithms in Bioinformatics. Vol. 4645. Berlin/Heidelberg: Springer; 2007. pp. 289–301. of Lecture Notes in Computer Science. [Google Scholar]
- Miller JR, et al. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics. 2008;24:2818–2824. doi: 10.1093/bioinformatics/btn548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers EW, et al. A whole-genome assembly of Drosophila. Science. 2000;287:2196–2204. doi: 10.1126/science.287.5461.2196. [DOI] [PubMed] [Google Scholar]
- Nagarajan N, Pop M. Parametric complexity of sequence assembly: theory and applications to next generation sequencing. J. Comput. Biol. 2009;16:897–908. doi: 10.1089/cmb.2009.0005. [DOI] [PubMed] [Google Scholar]
- Peng Y, et al. Meta-idba: a de novo assembler for metagenomic data. Bioinformatics. 2011;27:i94–i101. doi: 10.1093/bioinformatics/btr216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perna N, et al. Genome sequence of enterohaemorrhagic Escherichia coli O157: H7. Nature. 2001;409:529–533. doi: 10.1038/35054089. [DOI] [PubMed] [Google Scholar]
- Pop M, et al. Hierarchical scaffolding with Bambus. Genome Res. 2004;14:149–159. doi: 10.1101/gr.1536204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter DC, et al. MetaSim: a sequencing simulator for genomics and metagenomics. PLoS One. 2008;3:e3373. doi: 10.1371/journal.pone.0003373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusch DB, et al. The Sorcerer II global ocean sampling expedition: Northwest atlantic through eastern tropical pacific. PLoS Biol. 2007;5:e77. doi: 10.1371/journal.pbio.0050077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon I, et al. A statistical framework for the functional analysis of metagenomes. Res. Comput. Mol. Biol. 2009;5541:496–511. [Google Scholar]
- Simmons SL, et al. Population genomic analysis of strain variation in Leptospirillum Group II bacteria involved in acid mine drainage formation. PLoS Biol. 2008;6:e177. doi: 10.1371/journal.pbio.0060177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer D, et al. Minimus: a fast, lightweight genome assembler. BMC Bioinformatics. 2007;8:64. doi: 10.1186/1471-2105-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, et al. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, et al. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh P, et al. A core gut microbiome in obese and lean twins. Nature. 2008;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson G, et al. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 2004;428:37–43. doi: 10.1038/nature02340. [DOI] [PubMed] [Google Scholar]
- Venter J, et al. Environmental genome shotgun sequencing of the sargasso sea. Science. 2004;304:66–74. doi: 10.1126/science.1093857. [DOI] [PubMed] [Google Scholar]
- Vignuzzi M, et al. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature. 2005;439:344–348. doi: 10.1038/nature04388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinson JP, et al. Assembly of polymorphic genomes: Algorithms and application to Ciona savignyi. Genome Res. 2005;15:1127–1135. doi: 10.1101/gr.3722605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yooseph S, et al. The Sorcerer II global ocean sampling expedition: expanding the universe of protein families. PLoS Biol. 2007;5:e16. doi: 10.1371/journal.pbio.0050016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, et al. Pebble and rock band: Heuristic resolution of repeats and scaffolding in the velvet short-read de Novo assembler. PLoS One. 2009;4:e8407. doi: 10.1371/journal.pone.0008407. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.