

Abstract
Earlier electron microscopic data had shown that a hallmark of the vascular remodeling in pulmonary arterial hypertension (PAH) in man and experimental models includes enlarged vacuolated endothelial and smooth muscle cells with increased endoplasmic reticulum and Golgi stacks in pulmonary arterial lesions. In cell culture and in vivo experiments in the monocrotaline model, we observed disruption of Golgi function and intracellular trafficking with trapping of diverse vesicle tethers, SNAREs and SNAPs in the Golgi membranes of enlarged pulmonary arterial endothelial cells (PAECs) and pulmonary arterial smooth muscle cells (PASMCs). Consequences included the loss of cell surface caveolin-1, hyperactivation of STAT3, mislocalization of eNOS with reduced cell surface/caveolar NO and hypo-S-nitrosylation of trafficking-relevant proteins. Similar Golgi tether, SNARE and SNAP dysfunctions were also observed in hypoxic PAECs in culture and in PAECs subjected to NO scavenging. Strikingly, a hypo-NO state promoted PAEC mitosis and cell proliferation. Golgi dysfunction was also observed in pulmonary vascular cells in idiopathic PAH (IPAH) in terms of a marked cytoplasmic dispersal and increased cellular content of the Golgi tethers, giantin and p115, in cells in the proliferative, obliterative and plexiform lesions in IPAH. The question of whether there might be a causal relationship between trafficking dysfunction and vasculopathies of PAH was approached by genetic means using HIV-nef, a protein that disrupts endocytic and trans-Golgi trafficking. Macaques infected with a chimeric simian immunodeficiency virus (SIV) containing the HIV-nef gene (SHIV-nef), but not the non-chimeric SIV virus containing the endogenous SIV-nef gene, displayed pulmonary arterial vasculopathies similar to those in human IPAH. Only macaques infected with chimeric SHIV-nef showed pulmonary vascular lesions containing cells with dramatic cytoplasmic dispersal and increase in giantin and p115. Specifically, it was the HIV-nef–positive cells that showed increased giantin. Elucidating how each of these changes fits into the multifactorial context of hypoxia, reduced NO bioavailability, mutations in BMPR II, modulation of disease penetrance and gender effects in disease occurrence in the pathogenesis of PAH is part of the road ahead.
Keywords: Pulmonary vascular remodeling, intracellular organelles, Golgi apparatus
INTRODUCTION
To quote the first sentence of the recent comprehensive review by Stenmark and colleagues[1] on animal models for pulmonary hypertension, “Pulmonary hypertension is not a disease per se but rather a pathophysiological parameter defined by a mean pulmonary arterial pressure exceeding the upper limits of normal, i.e. ≥25 mmHg at rest”. Intrinsic to this understanding is that this chronic disease will perforce have many steps in its pathogenesis that follow one or, perhaps, more than one “initiating” event(s), likely different for different etiologies, which cascade through multiple pathways flowing both in series and parallel, culminating in the pathophysiological changes at the organ, tissue, cellular and subcellular levels evident as the eventual proliferative, obliterative and plexiform pulmonary arterial lesions characteristic of pulmonary arterial hypertension (PAH) [Figure 1]. The focus of this review is on those aspects of the cell biology in PAH that relate to vascular remodeling.[2]
Figure 1.
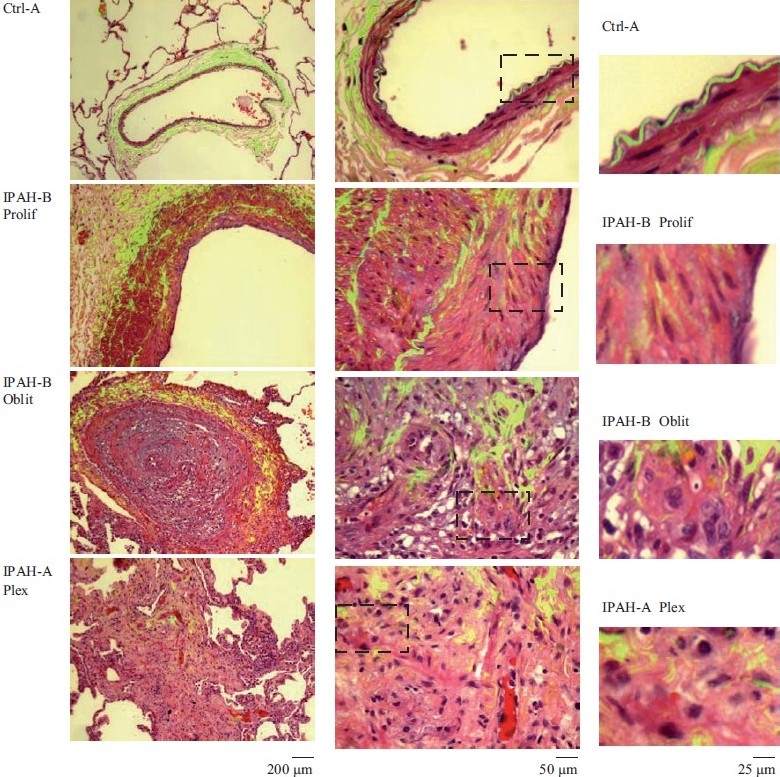
Representative histopathologic changes observed in idiopathic pulmonary hypertension. Sections of human lungs (Ctrl-A, IPAH-A and IPAH-B) were stained using H&E and imaged using a ×40 objective in visible light. Elastin autofl uoresence was simultaneously imaged in green and the visible light and autofluoresence images merged. Representative images showing neointimal proliferation (Prolif), obliterative (Oblit) and plexiform (Plex) lesions are illustrated. Side sets on the right show higher magnifi cation views of the boxed areas within panels in the middle column. (Adapted from ref. 2.)
We were attracted to the question of dysfunctional intracellular trafficking in PAH by observations of an inverse relationship between the levels of the plasma membrane raft/caveolar protein caveolin-1 (cav-1) and development of PAH in the rat/monocrotaline model.[3] The observations by Zhao and colleagues that cav-1-/- mice spontaneously developed pulmonary hypertension and dilated cardiomyopathy,[4,5] and reports that cav-1 and cav-2 were reduced in the cells in plexiform lesions in patients with severe PAH[6] , heightened interest in this inverse relationship. Our initial focus in PAH was in terms of the structure and function of plasma membrane rafts and caveolae and the trafficking of vasorelevant proteins to such specialized subcellular regions on the cell surface and effects on the transmission of ligand-activated cell surface signals to the cell interior [e.g. the hyperactivation of the IL-6/STAT3 and IL-6/ERK pathways inversely with loss of cav-1 from plasma membrane rafts in pulmonary arterial endothelial cells (PAECs)].[3] In recent years, this focus has expanded to a consideration of broader dysfunctions in anterograde and retrograde vesicular trafficking in the development of PAH.[7,8] Numerous studies have elucidated the molecular and vesicular machineries involved in the trafficking of vasorelevant growth factor and cytokine receptors (as examples, the trafficking of BMPR I and BMPR II, VEGFR, IL-6R and gp130, etc.) from the endoplasmic reticulum (ER) through the Golgi apparatus (abbreviated to “Golgi”) and thence to the plasma membrane or via the alternative pathways that “bypass” the Golgi [Figure 2].[7–10] The secretion of cytokines and growth factors by different cell types is also intricately regulated by distinct vesicular trafficking pathways and molecules in different types of cells.[10] Moreover, numerous studies have elucidated the obligatory involvement of membrane-associated pathways (clathrin- or caveolin-mediated endocytic pathways) in the inward transcription-targeted signaling initiated by growth factors, cytokines and ligands (e.g. signaling by transforming growth factor beta (TGF-β), bone morphogenetic proteins (BMPs), interleukin-6 (IL-6), epidermal growth factor (EGF), platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), Notch-3 and Wnt.[11–16] With several of these receptors and ligands implicated in the pathogenesis of PAH,[17–20] it becomes increasingly important to consider the potential involvement of dysfunctional membrane- and protein-trafficking pathways in the pathogenesis of this disease. The observations that some mutant BMPR II species, such as those observed in patients with familial PAH, failed to traffic normally from the ER to the Golgi and thence to the plasma membrane[20] highlight the importance of investigating the role of protein trafficking dysfunctions in the pathogenesis of PAH.
Figure 2.
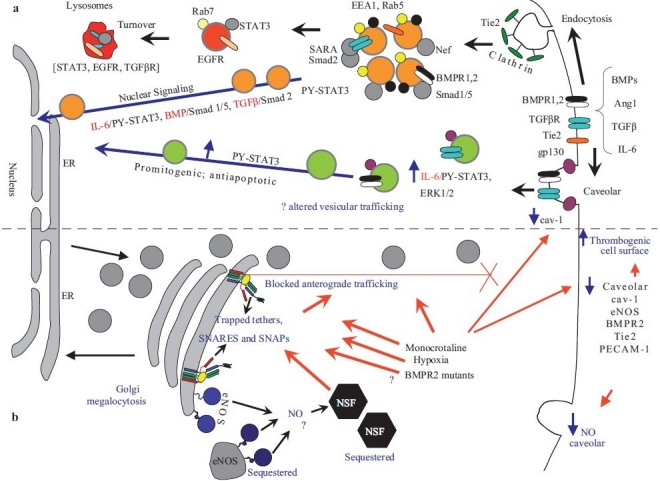
Dysfunctional intracellular trafficking in the pathobiology of pulmonary arterial hypertension. (a) Productive transcriptional signaling from the plasma membrane to the nucleus along the BMP/Smad1/5, TGFβ/Smad2 and IL-6/PY-STAT-3 signaling pathways is membrane associated. IL-6/STAT3 and ERK1/2 signaling is inversely related to loss of caveolar/raft cav-1. (b) Golgi blockade mechanisms in PAH. MCTP and hypoxia lead to a trapping of vesicle tethers, SNAREs and SNAPs in the Golgi of affected pulmonary arterial endothelial cells. This leads to a block in anterograde trafficking of vasorelevant cargo proteins such as cav-1 and eNOS and reduced caveolar NO production. The intracellularly sequestered eNOS produces NO which may potentially S-nitrosylate cysteine-rich proteins like NSF, further inhibiting traffi cking. Golgi-trapped dominant negative BMPR 2 mutants may also potentially block trafficking of cargo proteins to the plasma membrane. (Adapted from ref. 8.)
SPATIAL CELL BIOLOGY–AT THE SUBCELLULAR LEVEL
A constant refrain in our perspective on the involvement of various molecules and mediators of interest in the pathogenesis of PAH is the question of where exactly inside the cell is that particular molecule located.[7,8] Is it in the correct place inside the cell or is its location aberrant? As a consequence of aberrant location, is the function aberrant? As one example, when investigators report the increase in eNOS in PAECs in, for example, the monocrotaline/rat model of PAH,[21] we wondered exactly where within the cell was the eNOS located. Was it at the cell surface and available for function or was it trapped in a non-functional intracellular compartment? As another example, do mutant BMPR II species[20] have dominant negative effects on the intracellular trafficking of other molecules, including eNOS, other receptors and membrane-associated signaling pathways?
INSIGHTS FROM PRIOR LITERATURE
In 1914, Cajal reported finding a compact juxtanuclear “apparato reticular de Golgi” in endothelial cells and the fragmentation and pulverization of this Golgi apparatus in cells (neuronal cells) subjected to hypoxia.[22] The pulmonary hypertension literature over the last four decades contains numerous pieces of data indicative of defects in intracellular trafficking in the cellular elements in pulmonary arterial lesions.[7,8] Noteworthy among these are histologic and electron microscopic (EM) studies reporting “plump” and “enlarged” endothelial and smooth muscle cells with increased vacuolation, Golgi stacks and ER in hypoxic and primary PAH.[23–30] Particularly evocative are the EM studies reporting extensive rough ER with dilated cisternae in a cell within a plexiform lesion in idiopathic PAH,[26] and a cell showing distended globular and granular structures.[28] Meyrick and Reid[25] reported “hypertrophied smooth muscle cells (with) a significant increase in relative areal proportions of Golgi apparatus and rough sarcoplasmic reticulum in the tunica media of the hilar artery of rats with hypoxia-induced PAH” and further commented that such rats have “thickened endothelial cells (with) a more extensive Golgi apparatus than normal and many have swollen cisternae of rough endoplasmic reticulum”. Increased accumulations of Weibel-Palade bodies (which are vesicular structures involved in the exocytosis of Factor VIII) were reported in endothelial cells in hypoxic rats with PAH[29] and in human primary PAH.[27] Mette and colleagues[30] reported marked accumulations of vesicular cytoplasmic structures in cells in pulmonary vascular lesions of patients with PAH with HIV infection. Taken together, these prior observations point to dysfunctions in intracellular membrane trafficking in cells located within lesions in PAH.
A major line of research extending back to the observations of Harris et al. in 1944,[31,32] Bull in 1955[33] and the EM studies of Klienerman and Merkow in 1966[34] and of Afzelius and Schoental in 1967[35] is also particularly insightful. These and subsequent investigators[36–39] studied the effects of pyrrolizidene alkaloids on various cell types and tissues. One of these alkaloids, monocrotaline (MCT), is the basis for a widely used, and widely debated, experimental model of PAH in rats, pigs and dogs.[1,36–39] Harris et al.[31,32] and Bull[33] reported that pyrrolizidene alkaloid treated liver cells (which themselves metabolically activate the alkaloid) continued to grow in size but not divide. This phenotype was termed “megalocytosis” by Bull in 1955.[33] PAECs and alveolar type II cells in lungs of rats exposed to MCT that developed PAH showed increased vacuolation, increased ER and extensive Golgi complexes.[34,37–40] Afzelius and Schoental[35] pointed out that “one of the most striking features of the enlarged cells is the lack of normal distribution of organelles”. Specifically, Afzelius and Schoental[35] linked the “large size of the Golgi complexes” to defects in “sorting out of organelles into separate regions” as well as a defect in entry of cells into mitosis, yielding “growing, but non-dividing cells”.
In 1991, Reindel and Roth[40] extended the above insightful observations to changes in bovine pulmonary artery endothelial and smooth muscle cells in culture exposed to the bioactive monocrotaline pyrrole (MCTP). Ultrastructurally, the hypertrophic MCTP-treated bovine pulmonary artery endothelial cells (BPAECs) had enlarged cell profiles with enlarged nuclei and “prominent” Golgi apparatus and endoplasmic reticulum.[40] In the last 20 years, these investigators as well as the research groups of Segall and Wilson pioneered extensive cell culture studies of the MCTP-induced megalocytosis in pulmonary vascular cells.[39,41,42] A landmark observation by the Segall and Wilson groups has been the identification of specific Cys-containing and Cys-free proteins that are covalently derivatized by MCTP when human PAECs were exposed by radioisotope labeled MCTP – this list included proteins that help fold newly synthesized proteins in the lumen of the ER (such as the protein disulfide isomerases, ERp57 and PDI) as well as cell surface proteins such as galectin-1.[41,42] The above data in the prior literature taken together point to dysfunctions in intracellular membrane trafficking in cells exposed to bioactive pyrrolizidene alkaloids.
Following the discovery of the relationship between mutations in BMPR II and idiopathic PAH (IPAH) (both the familial and sporadic types) beginning approximately 10 years ago,[43] it was realized that several of the relevant mutant BMPR II species failed to traffic correctly to the plasma membrane.[20,44,45] Subsequent detailed studies have identified PAH disease-related BMPR II mutants that remain sequestered in the ER, sequestered in the Golgi or even those that traffic to the plasma membrane and yet exhibit defective signaling.[20,44,45] These data raise the question of potential dominant negative effects of such BMPR II mutants on the intracellular trafficking of other vasorelevant molecules. Mice harboring such mutant BMPR II genes have proven informative in exploring the pathogenesis of PAH.[1,46,47] However, the incomplete penetrance of disease parameters in even the BMPR II knockout model[46] suggests that additional environmental and genetic factors influence PAH pathogenesis.[1,20]
The observations by Zhao and colleagues in 2002 that cav-1-/- mice spontaneously developed pulmonary hypertension and dilated cardiomyopathy[4,5] highlight the ability of defects in trafficking scaffolding proteins to initiate a cascade of changes including pulmonary hypercellularity, interstitial fibrosis, thickening of the alveolar septa and reduced exercise tolerance.[48–50]
In pioneering observations Flores and colleagues[51] reported in 2006 that macaques infected with a chimeric simian immunodeficiency virus (SIV) in which the endogenous SIV-nef had been replaced by the HIV-nef gene (dubbed SHIV-nef virus) developed pulmonary arterial lesions similar to those seen in IPAH. These lesions included complex plexiform-like lesions characterized by medial hypertrophy and luminal obliteration. Such lesions included cells that were nef–, Factor VIII–, and/or smooth muscle-specific actin-positive. Such lesions were not observed in macaques infected with the non-chimeric SIV virus. In parallel, Flores and colleagues[51] reported that HIV-nef-positive cells were also observed in pulmonary vascular lesions in HIV-positive patients but not in patients with IPAH. The fact that HIV-nef is a protein that is well known to interfere with and modulate endocytic and Golgi trafficking pathways, and has been best investigated in terms of its effects on intracellular trafficking pathways involved in major histocompatibility complex (MHC) antigen presentation,[9,52–55] again point to dysfunctional protein trafficking as, at the very least, part of the overall process leading to pulmonary vascular lesions.
The above brief overview of the prior PAH literature highlights previously published data that clearly pointed to the occurrence of protein trafficking dysfunctions in PAH. The seminal hypothesis of Afzelius and Schoental[35] linking Golgi dysfunction to organellar defects on the one hand and to entry into mitosis and effects on cell proliferation on the other hand represents an important link in discussions of how trafficking defects can underlie effects on cell size and cell proliferation. Organelles such as the Golgi apparatus stand at the interface between trafficking and cell proliferation.
DISRUPTION OF ENDOTHELIAL CELL CAV-1/RAFT SCAFFOLDING IN PAH
In 2002, we observed that normal IL-6 signaling in different cell types to the cell interior was dependent on the integrity of cholesterol-rich plasma membrane raft microdomains.[12,56] In 2004, we reported that in the MCT/rat model although PAH and right ventricular hypertrophy developed by 2 weeks after MCT, a reduction in cav-1 levels in the lung was apparent within 48 hrs, declining to 30% of control levels by 2 weeks, accompanied by an increase in the activation of the transcription factor, PY--STAT3, which also began within 48 hrs.[3] Immunofluorescence studies showed a loss of cav-1 in PAECs within 48 hrs after MCT but an increase in PY-STAT3. At the single-cell level in pulmonary arteries of MCT-treated rats, the particular PAECs that showed reduced cav-1 were the ones that showed increased PY-STAT3 and nuclear immunostaining for proliferating cell nuclear antigen (PCNA). Cell fractionation studies showed that there was a loss of cav-1 from detergent-resistant membrane raft fraction concomitant with hyperactivation of PY-STAT3. Additionally, PAECs treated with MCTP in culture developed megalocytosis associated with hypo-oligomerization and reduction of cav-1, hyperactivation of PY-STAT3 and ERK1/2, and stimulation of DNA synthesis. These data suggested the occurrence of defects in the trafficking of cav-1 to plasma membrane rafts in MCTP-treated cells with a consequent promitogenic cascade of events.[3]
Lisanti and colleagues[50] confirmed and extended the above observations in the rat/MCT model and showed that the decline in cav-1 and cav-2 and development of PAH as well as the activation of PY-STAT3 and upregulation of cyclins D1 and D3 could be blocked by a cav-1-mimetic peptide (AP-Cav) administered 30 min after MCT administration. The loss of cav-1 from cells in plexiform lesions in severe IPAH,[6] in rats with PAH following experimental myocardial infarction,[49] those administered SU5419[6] and the ability of the cholesterol reducing agent, simvastatin, to reduce the severity of PAH in the MCT/rat model[57,58] further supported interest in the mechanisms underlying the trafficking and loss of cav-1 in PAECs in PAH.
In studies that recapitulated the earlier work of Riendel and Roth and of the Segall and Wilson research groups on the effects of MCTP on pulmonary vascular cells in culture,[39,40] we observed that pulmonary vascular cells exposed to MCTP displayed megalocytosis within 24–48 hrs of exposure with the trapping of cav-1 in the enlarged Golgi apparatus. In these studies, the Golgi apparatus was observed using immunostaining for a Golgi tether/scaffolding protein called GM130 [Figure 3].[3,59] Subsequent studies showed that this phenotype in PAECs was characterized by a cascade of changes including increased DNA synthesis but reduced entry into mitosis, increased PY-STAT3, an incomplete unfolded protein response, reduced cdc2 but increased p21, and various alterations in cyclins D1, D3, E, A and B1.[59–62] It was noteworthy that eNOS was also lost from the plasma membrane and now increasingly sequestered in the Golgi and in intracellular vesicular structures.[62–64] The increased sequestration of cav-1 in the Golgi membranes was apparent within 6–8 hrs after MCTP exposure, that is, in advance of any other phenotypic alteration. Similar alterations were also observed in PAECs exposed to hypoxia.[62] These observations led to the hypothesis that defective trafficking of proteins through the Golgi apparatus might represent an event in how MCTP and hypoxia affected pulmonary vascular cells and that this trafficking disruption might also be of relevance in the human disease (Golgi blockade hypothesis).[59]
Figure 3.
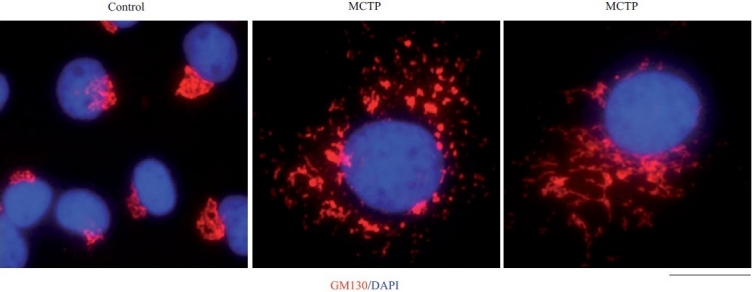
Golgi enlargement and fragmentation in primary bovine PAECs in culture exposed to MCTP for 4 days. BPAEC cultures in a 6-well plate were exposed to MCTP and megalocytosis was allowed to develop for 4 days.[3,7,8] The cultures were then fixed and immunostained for the Golgi tether, GM130, and for nuclei using 4’,6-diamidino-2-phenylindole (DAPI). Scale bar=4 μm.
BMPR II has been reported to be located in lipid rafts, including caveolae, in human and rat pulmonary arterial endothelium.[65] Additionally, cav-1 has been shown to regulate the caveolar localization and transcriptional activation function of BMPR II in mouse aortic smooth muscle cells.[66] BMPR II interacted with cav-1 and downregulation of cav-1 using an siRNA approach decreased BMP-dependent Smad phosphorylation and gene regulation. Reduction in cav-1 or a dominant-negative mutant of cav-1 reduced BMPR II plasma membrane localization. A reduction in cav-1 also reduced the association between BMPR II and BMPR I.[66] In MCTP-treated PAECs, BMPR II was increasingly trapped in the Golgi membranes together with increased trapping of cav-1 and eNOS,[62] and such cells displayed altered functional BMP/Smad signaling.[67,68]
However, several puzzling questions about the relationship between cav-1 and development of PAH prompt a more detailed look at the subcellular/intracellular events that might be at play. On the one hand, overexpression of wild-type (wt) cav-1 in PAECs inhibited IL-6/STAT3 transcriptional signaling,[15] and thus, the inverse relationship between cav-1 levels and PY-STAT3 levels in PAECs in rats exposed to MCT or in the cav-1 knockout mouse appears reasonable. In the latter case, an interpretation would be that disruption of cav-1/rafts per se produced the hyperactivation of IL-6/PY-STAT3 because cav-1 in rafts is inhibitory to such signaling. On the other hand, the Antennapedia-Cav-1 membrane-permeable small peptide used by Jasmin et al.[50] that inhibited MCT-induced PAH and also inhibited the loss of cav-1 (and cav-2) and the increase in PY-STAT3 corresponds to the scaffolding domain of cav-1 and should have also disrupted cav-1 rafts and had the opposite effects – if the effects were solely due to events at the level of the plasma membrane caveolae. It is known from our prior work that raft disruption by other means (such as using the cholesterol binding compounds, filipin III or β-methylcyclodextrin) inhibited IL-6/STAT3 signaling, even in cells low in cav-1 (in such cells, “plasma membrane rafts” are formed by other proteins such as flotillin-1 or flotillin-2).[12,56] One possible way to resolve these seemingly contradictory observations is to extend the discussion to intracellular trafficking pathways. The cav--1/caveosome retrograde trafficking has been shown to inhibit TGF-β/Smad transcriptional signaling by shunting the activated Smad molecules toward lysosomal degradation, while clathrin/endocytic trafficking leads to transcriptionally productive signaling.[11] Similarly, the majority of activated PY-STAT3 in the cytoplasm is associated with the early endosome compartment[13,69] and overexpression of cav-1 increasingly targets STAT3 toward the lysosomal compartment.[15] The possibility of cav-1 levels affecting intracellular retrograde signaling pathways has not been explored in the context of PAH. To add to the puzzle are more recent observations of Zhou et al.[5] showing that lungs in the cav-1-/-mouse display persistent eNOS activation and that the PAH observed can be reduced by a superoxide scavenger (MnTMPyP) or a NOS inhibitor (L-NAME). However, it was not evaluated where exactly within the endothelial and/or bronchial epithelial cells was the eNOS located. Additionally, Patel et al.[70] reported increased cav-1 in PASMCs in patients with IPAH and interpreted their cell culture experiments to indicate that increased cav-1 and caveolae “contribute to IPAH-PASMC pathophysiology”. Moreover, Patel et al.[70] suggest that “disruption of caveolae in PASMC may provide a novel therapeutic approach to attenuate disease manifestations of IPAH”, which would be the opposite of the inference of an inverse relationship between cav-1 and PAH from the studies summarized above. Perhaps, the many unanswered puzzles at hand relate to cell type, disease heterogeneity, lesion heterogeneity and the multiple different levels at which cav-1 can affect intracellular trafficking pathways and functions of vasoactive molecules (such as eNOS and BMPR II). There is a clear need for a more detailed understanding of the subcellular events that connect cav-1–mediated trafficking and regulatory pathways to the development of PAH.
The above considerations led us to evaluate various molecules and cellular organelles involved in both Golgi to plasma membrane (anterograde; exocytosis) and plasma membrane to cell interior (retrograde; endocytosis) trafficking in pulmonary vascular cells, both in pulmonary vascular cells in culture under various experimental conditions and in cells in pulmonary vascular disease lesions in IPAH and in the MCT/rat and SHIV-nef/macaque models.
BRIEF OVERVIEW OF VESICULAR TRAFFICKING MACHINERIES AND MOLECULES
Intracellular protein trafficking involves the selective scission of vesicles from membranous organelles, followed by the selective and targeted capture of such vesicles by other membrane organelles and membrane fusion.[7–10,62,64,71,72] This process requires molecules on the surface of vesicles and on acceptor membranes that recognize each other with specificity (the tethers and SNAREs (soluble N-ethylmaleimide-sensitive factor attachment protein receptor)], molecules that are involved in the recruitment of an ATPase to the SNARE complexes [the SNAPs (soluble N-ethylmaleimide-sensitive factor attachment protein) and NSF (N-ethylmaleimide-sensitive factor)] and an additional hierarchy of molecules belonging to the complexin and Sec-Munc (SM) family of proteins that provide an additional layer of regulation [Figures 4 and 5]. Several reviews provide considerable details about these molecules and mechanisms and show that this is an exciting new area of considerable ongoing research in cell biology today.[8,10,71,72]
Figure 4.

Tethers, SNAREs, SNAPs, NSF and additional molecules involved in vesicular trafficking
Figure 5.
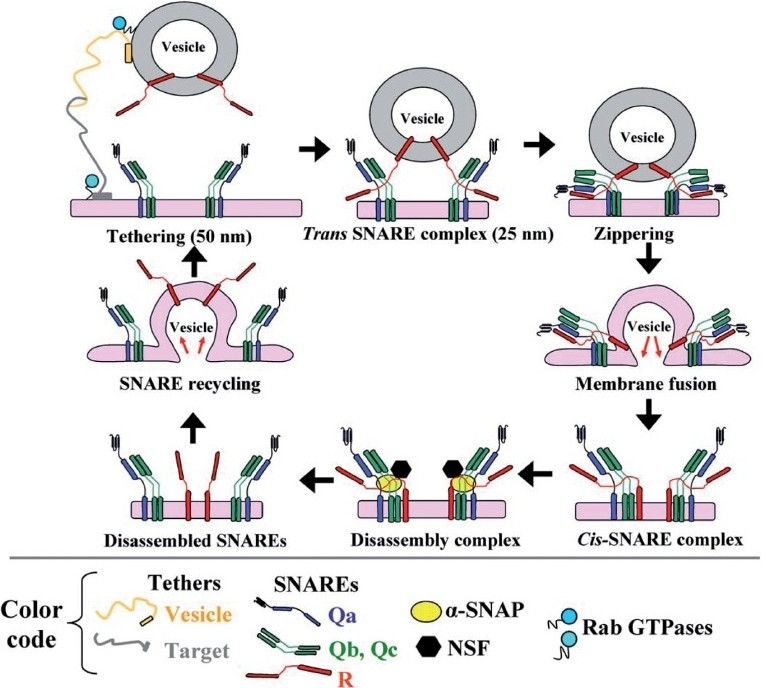
The SNARE cycle in membrane fusion. Initial interaction between a vesicle and its target membrane is mediated by cognate tethers. Membrane fusion is then mediated by the formation of a quaternary-α-helical trans-SNARE complex consisting of one v- (or R -) SNARE on the vesicle and two or three t- (or Q-) SNAREs on the target membrane. After membrane fusion, the cis-SNARE complex is disassembled by the ATPase NSF which is recruited to the cis-SNARE complex from the cytosol by α-SNAP. (Adapted from ref. 8.)
Figure 4 is a brief compilation of the molecules that are part of the discussion relating to pathogenesis of PAH. “Tethers” represent cell surface molecules on respective vesicle and target membrane surfaces that search for and mediate binding of cognately recognized molecules at long distance (up to 50 nm) [Figure 5]. Each organelle and vesicle type contains several such tethers. In the Golgi, these tethers include giantin, p115 and Golgi matrix 130 kDa (GM130). In addition to tethering, these molecules also mediate more specific functions in the respective organelle (such as structural scaffolding, phosphorylation-mediated organelle fragmentation and apoptosis). Tether interactions draw the vesicle and target membrane surface closer to each other, and at distances of 25 nm or less, groups of proteins called SNAREs come together as part of a four-helix complex called a trans-SNARE complex [Figure 5]. One of the helices is contributed by the SNARE on the vesicle membrane, while the other three are contributed by two or three SNAREs on the target membrane. There are 38 different SNAREs in human cells and these mediate specific interactions with cognate molecules [Figures 4 and 5]. Thus, the combinations of specific SNAREs provide the addressing mechanisms (like Zip Codes) for vesicular trafficking. The SNAREs are classified as Q- and R-SNARES depending upon their underlying amino acid motifs (Q-SNAREs are typically t- or target-SNAREs, while R-SNAREs are typically v- or vesicle SNAREs).
The trans-SNARE complex drives the membrane fusion reaction [Figure 5]. However, this membrane fusion event is regulated by the family of complexin and Sec-Munc proteins.[71,72] Subsequent to membrane fusion, the now cis-SNARE complex recruits a soluble cytosolic protein called a SNAP (α-, β- or γ-SNAP) (not to be confused with SNAP proteins identified by numerical designations such as SNAP23, etc.; the latter are SNARE proteins). Of the SNAP proteins, α-SNAP is the most ubiquitous (present in PAECs and PASMCs) and this protein, in turn, recruits the cytosolic ATPase called NSF to the cis-SNARE complex. NSF, in an energy dependent manner, leads to the disassembly of the four-helix complex into its component SNAREs, preparing this machinery for another round of membrane fusion. Thus, in PAECs and PASMCs, α-SNAP and NSF participate in virtually all vesicular trafficking. Moreover, it is noteworthy that the ATPase activity of NSF is inhibited by NO through S-nitrosylation at multiple Cys residues.[73] Finally, a range of additional GTPases (the dynamin family and the Rab family of GTPases) are involved in the process of scission of the newly forming vesicle for organellar membranes with different membrane trafficking pathways requiring different GTPases in a selective manner.[9–11]
Dysfunctions of Golgi tethers, SNAREs, α-SNAP and NSF in pulmonary vascular cells exposed to MCTP, hypoxia, NO scavenging and senescence
PAECs exposed to MCTP in cell culture had shown increased trapping of cav-1 in Golgi membranes as early as 6 hrs after MCTP.[62] In this experimental system, a combination of immunofluorescence and cell-fractionation studies revealed the considerable accumulation of diverse Golgi trafficking mediator proteins in the enlarged/circumnuclear Golgi apparatus by 1–2 days after exposure to MCTP or the NO scavenger, c-PTIO [Figures 3 and 6]. These proteins included GM130, p115, giantin, golgin 84, syntaxin-4, -6, Vti1a, Vti1b, GS15, GS27, GS28, SNAP23 and α-SNAP. Strikingly, NSF, the ATPase required in all vesicular trafficking, was increasingly sequestered in non-Golgi vesicular elements and was depleted from Golgi membranes.[64] The sequestration of a large collection of trafficking-mediator molecules in the Golgi was accompanied by intracellular accumulation of eNOS and reduced caveolar NO production as assayed using live-cell DAF2-DA imaging.[62–64] The reduced levels of caveolar NO in MCTP-treated cells were consistent with the observation of reduced S-nitrosylation of NSF, eNOS and cav-1 in such cells.[63,64] Thus, although total eNOS levels were either unchanged (on a total cell protein basis) after MCTP or were increased, this eNOS was mislocalized to the incorrect intracellular compartment and was thus hypofunctional. These effects were evident in cultures of both bovine and human PAECs [Figures 3 and 6].[74,75]
Figure 6.
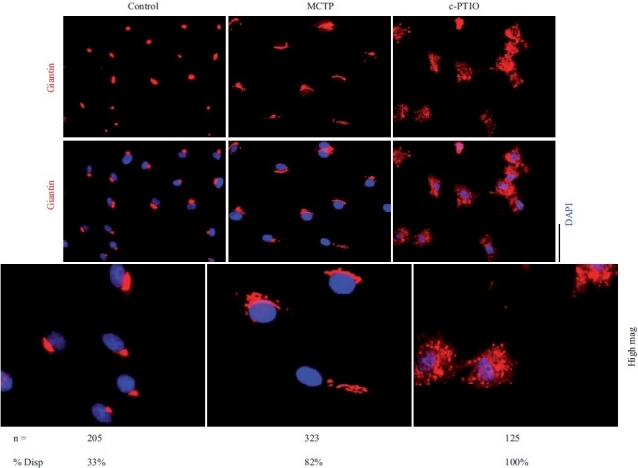
Golgi fragmentation and dispersal in human PAECs after exposure to MCTP or the NO scavenger, c-PTIO. Primary HPAEC cultures in 6-well plates were exposed to MCTP once or to c-PTIO (100 μM) continuously for 48 hrs. The integrity of the Golgi was assayed by immunotagging for the Golgi tether, giantin, together with DAPI staining for demarcating nuclei. NIH Image J software using Otsu segmentation analyses were used to determine Golgi structures with fragment number greater than 1[75] and are enumerated as % cells with dispersed Golgi. Scale bar=20 μm
An enlarged Golgi, as evidenced by increased circumnuclear accumulation of GM130 and giantin, was observed in PAECs in rat lungs within 4 days after administration of MCT to rats.[62] Thus, this Golgi alteration was observed at a time when there was no evidence of PAH which takes 10–14 days to develop.[3,62] Because the changes in the Golgi preceded development of PAH, the data suggested that such changes might lie in the pathway of causality leading to the disease and were not a consequence of increased pulmonary arterial pressure per se.
PAECs exposed to hypoxia (1.5% v/v oxygen in room air) for 4 days also showed a similar phenotype.[63] The cells were enlarged and contained enlarged circumnuclear Golgi with increased trapping of GM130, p115, syntaxin 6, GS28, together with cav-1 and eNOS in Golgi membranes. In such cells, the eNOS was mislocalized away from the plasma membrane. Senescent PAECs also exhibited a similar phenotype with enlarged cells with increased GM130, giantin, p115, GS28, syntaxin 6, Vti1a in the enlarged Golgi with marked intracellular mislocalization of eNOS and cav-1.[63]
Various investigators had previously suggested that at least some instances of PAH might include reduced bioavailability of NO.[75] We observed that scavenging NO from cultures of bovine PAECs or human PAECs and PASMCs led to marked Golgi fragmentation [Figure 6].[74,75] However, although MCTP exposure reduced entry of PAECs into mitosis, exposure of PAECs to the NO scavenger, c-PTIO, markedly increased the entry of endothelial cells into mitosis.[74] Thus, a hypo-NO state was promitogenic, at least for PAECs in culture.
Inhibition of functional endocytosis and an unusual secretory phenotype
In functional ligand uptake assays, we observed that PAECs exposed to MCTP showed intact uptake and sequestration of C5-ceramide to the Golgi membranes but a marked inhibition of the endocytic uptake of LDL (clathrin-mediated endocytosis), transferrin (clathrin-mediated endocytosis) and cholera toxin (caveolar endocytosis).[74] In comparison, NO scavenging using c-PTIO inhibited the uptake of all four tracers.[74] Both MCTP and c-PTIO reduced the cell surface concentrations of low density lipoprotein receptor (LDLR), TfnR, BMPR II, Tie-2 and PECAM-1, albeit to differing extents. Neither MCTP nor c-PTIO affected the uptake and subcellular localization of LysoTracker, ERTracker or MitoTracker. Thus, there was clear selectivity in the effects of these agents on intracellular trafficking.
However, both MCTP and c-PTIO generated an unusual secretory phenotype in PAECs.[74] After an initial inhibition of the secretion of exogenously expressed soluble HRP over the first day following exposure to either MCTP or c-PTIO,[62,74] there was a marked increase in secretion over and above that seen in untreated PAECs.[74] This marked increase in secretion was sustained for the duration of the 5-day assay despite development of clear megalocytosis. This unusual secretory phenotype recalls (1) the senescence-associated secretory phenotype reported recently by Campisi and colleagues[77,78] in which senescent breast cancer cells secrete growth factors (including IL-6 and IL-8) which then recruit and enhance the function of infiltrating immune cells leading to increased tumorigenicity with increasing age and (2) the thesis, often stated, that the endothelial cells in pulmonary vascular lesions release cytokines and growth factors that then lead to the migration of smooth muscle cells into previously non-muscular arteries or even the conversion of arterial pericytes into smooth muscle cells.[3,17,18] The spectrum of cytokines and growth factors secreted by, for example, say, human PAECs in response to a hypo-NO state[74–76] would be important to elucidate as part of delineating a “PAH-associated persistent secretory phenotype” of the affected endothelial cells.
Dysfunctions of Golgi tethers, SNAREs and SNAPs in IPAH
The histological and EM data in IPAH reviewed in the section “Insights from Prior Literature” already included evidence of alterations in the structure of the Golgi apparatus and of various membrane organelles such as the endoplasmic reticulum in cells of IPAH lesions. Given this prior information, we asked whether we could extend this to identifying dysfunctions of specific Golgi tethers, SNAREs and α-SNAP. A technical limitation in carrying out these analyses was the availability, at the moment, of only archived formalin-fixed, paraffin-embedded tissue from patients with IPAH. For now, we have been limited to evaluating only those tethers, SNAREs and α-SNAP for which the respective antibodies can react with tissue antigens still immunoreactive despite the fixation, embedding and long-term storage.[2,75]
Sections of lung tissue corresponding to the IPAH patients and lesions shown in part in Figure 1 were subjected to immunofluorescence imaging for the Golgi tethers, giantin and p115.[2] Compared to pulmonary vascular cells in controls without PAH, there was an increase in the amounts of these Golgi tethers on a per cell basis in lung tissue from patients with IPAH, accompanied by marked cytoplasmic dispersal of giantin- and p115-bearing vesicular elements in the vascular cells in the proliferative, obliterative and plexiform lesions[2] [Figures 7 and 8]. High-resolution subcellular 3D imaging of immunotagged giantin confirmed marked Golgi fragmentation and increased cell size of individual cells in pulmonary arterial lesions in IPAH compared to compact Golgi in controls.[75] Immunofluorescence tagging followed by high-resolution 3D imaging of subcellular structures also revealed increased levels of the SNARE Vti1a and α-SNAP cells in IPAH lesions in dispersed cytoplasmic vesicular elements.[75]
Figure 7.
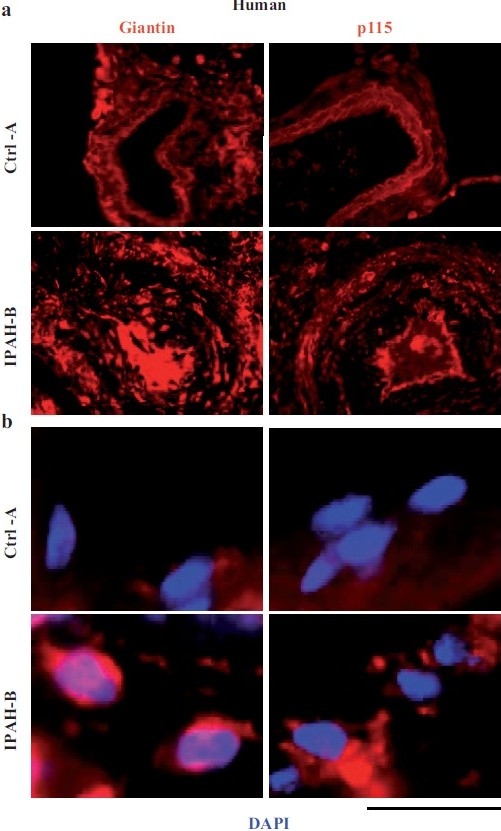
Increased accumulation of the Golgi matrix proteins/tethers, giantin and p115, in cellular elements in pulmonary arterial vasculopathies in human IPAH. (a) Representative images of respective vasculopathies probed for giantin or p115 compared to representative controls. Scale bar=85 μm. (b) Representative higher magnifi cation images of giantin and p115 immunostaining from analyses as in Figure 7a. Scale bar=10 μm. (Adapted from ref. 2.)
Figure 8.
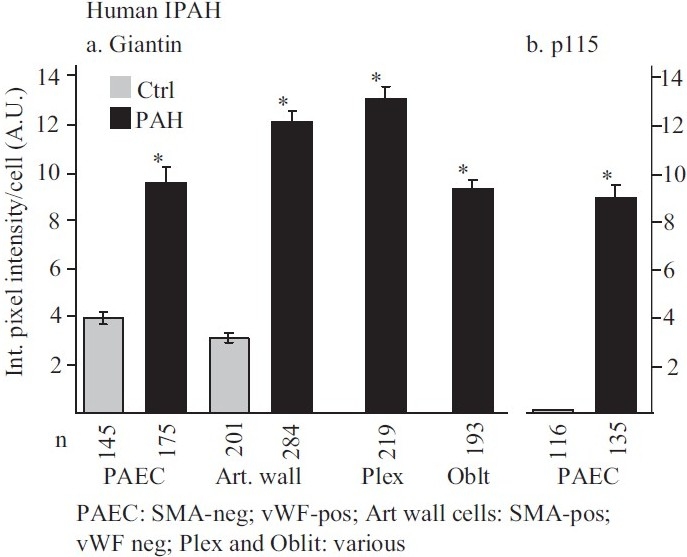
Summary of the quantitative immunomorphometry data for the Golgi tethers, giantin and p115, in cellular elements in pulmonary arterial vasculopathies in IPAH. Integrated pixel intensity/cell was computed using Image J software by outlining individual cells within immunofluorescence images corresponding to luminal endothelium (PAEC), cells in arterial walls, in plexiform lesions and in obliterative lesions and was expressed in arbitrary units (A.U.). Images for quantitation were derived from sections in the set of the human control and IPAH patients as in ref. 2. All the per cell data (n as indicated in the figure) were pooled into either control or IPAH and evaluated using Student's two-tailed t-test. Asterisks indicate P<0.0001 for groups compared with the corresponding control groups; cells in plexiform lesions and in the obliterative lesions were compared with the control PAEC group. In Panel b, there was little detectable p115 signal in the control PAECs evaluated. (Adapted from ref. 2.)
Dysfunctions of Golgi tethers, SNAREs and SNAPs in the SHIV-nef/macaque model
The SHIV-nef–infected macaque model has proven informative in helping address the question whether Golgi and trafficking dysfunctions might lie in the chain of causality of pulmonary vasculopathies. Flores and colleagues[51] had previously shown that macaques infected with the chimeric SHIV-nef virus, but not those infected with the non-chimeric SIV virus, developed pulmonary vasculopathies similar to those seen in IPAH. Figure 9 shows the pulmonary vascular histopathology observed in SHIV-nef–infected macaques to that observed in IPAH in the proliferative, obliterative and plexiform lesions in a manner that is directly comparable to the histopathology shown in Figure 1 for IPAH.[2] Since HIV-nef had been previously shown to interfere with endocytic and Golgi trafficking, the data implied that these vasculopathies might be initiated by such trafficking defects.[9,52–55] The question remained whether cells in pulmonary vascular lesions in SHIV-nef–infected macaques did indeed display Golgi defects. This was addressed by quantitative immunofluorescence assays as well as high-resolution 3D imaging of subcellular organelles in cells in SHIV-nef–induced pulmonary vasculopathies compared to those in SIV-infected or uninfected macaques.[2,75] Only macaques infected with the chimeric SHIV-nef virus showed pulmonary vascular lesions containing cells with dramatic increase in and cytoplasmic dispersal of the Golgi tethers, giantin and p115 [Figures 10 and 11]. High-resolution 3D subcellular imaging confirmed the extensive fragmentation of the Golgi in such cells. Moreover, it was specifically only those cells that contained endocytic nef that showed enlarged dispersed vesicular Golgi elements [Figure 12].[2,75] In contrast, giantin and p115 immunostaining of pulmonary vascular cells in SIV-infected or uninfected macaques showed a discrete compact Golgi.
Figure 9.
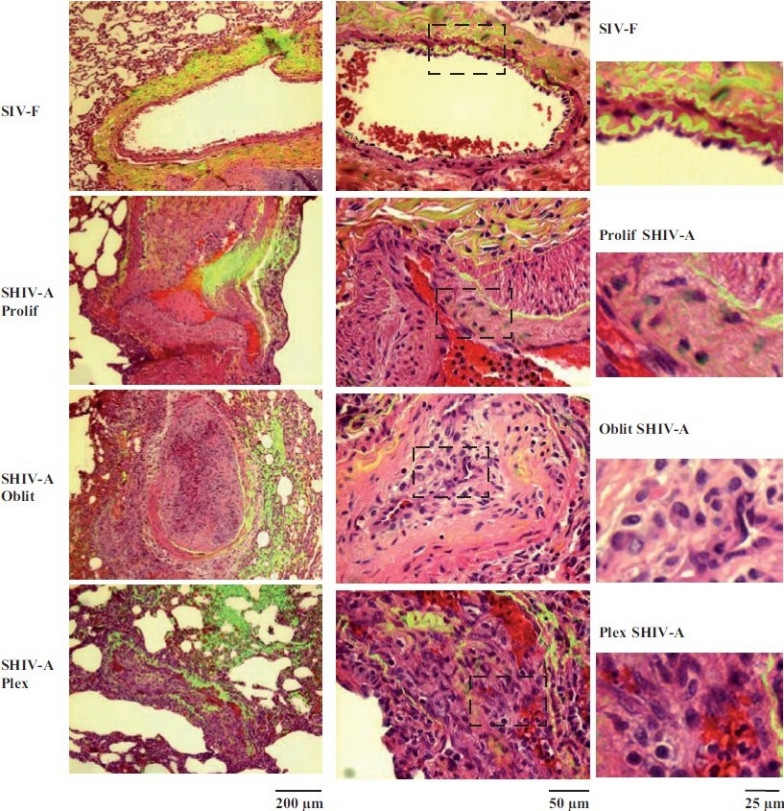
Representative histopathologic changes observed in pulmonary vascular lesions in SHIV-nef–infected macaques. Sections of lungs from macaquesinfected with the chimeric SHIV-nef virus (SHIV-A) or a non-chimeric SIV virus (SIV-F) were stained using H&E and imaged using a ×40 objective in visiblelight. Elastin autofl uoresence was simultaneously imaged in green and the visible light and autofl uoresence images merged. Representative images showing neointimal proliferation (Prolif), obliterative (Oblit) and plexiform (Plex) lesions are illustrated. Side sets on the right show higher magnifi cation views of the boxed areas within each panel in the middle column. (Adapted from ref. 2.)
Figure 10.
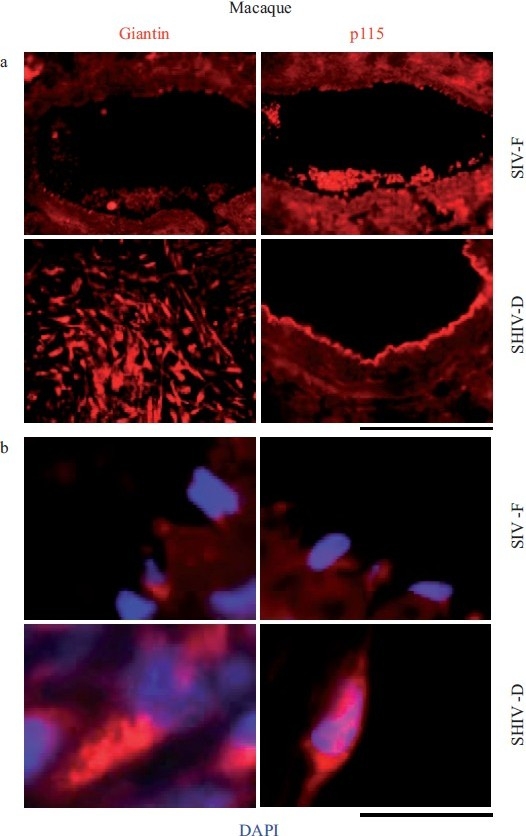
Increased accumulation of the Golgi matrix proteins/tethers, giantin and p115, in cellular elements in pulmonary arterial vasculopathies in the chimeric SHIV-infected macaque model. (a) Representative images of respective vasculopathies probed for giantin or p115 compared to representative controls. Scale bar=85 μm. (b) Representative higher magnification images of giantin and p115 immunostaining from analyses as in Figure 10a. Scale bar=10 μm. The SIV-F, p115 segment in the upper right is the same as in Figure 9, SIV-F at the top of the middle column. (Adapted from ref. 2.)
Figure 11.
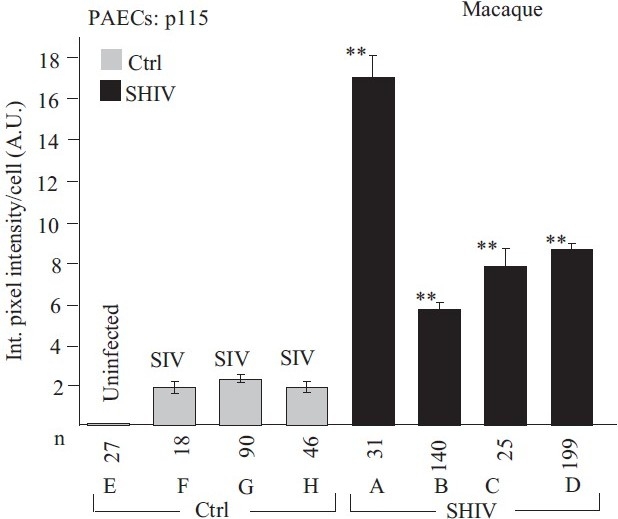
Quantitative immunomorphometry data for the Golgi tether, p115, in PAECs in SHIV-nef–infected macaques. Integrated pixel intensity/cell was computed using Image J software by outlining individual cells within immunofl uorescence images corresponding to luminal endothelium (PAEC) in sections from each of the control/SIV- and SHIV-infected macaques (n=4 in each group) and was expressed in arbitrary units (A.U.) (mean±SE; n is number of cells quantitated). PAECs in group Ctrl-E had little or no detectable p115. Post-hoc between-group comparisons were carried out using the Tukey–Kramer Multiple Comparison test with an alpha setting of 0.05 (NCSS 2007). Double asterisks indicate that the particular group was different from all control/SIV groups. (Adapted from ref. 2.)
Figure 12.
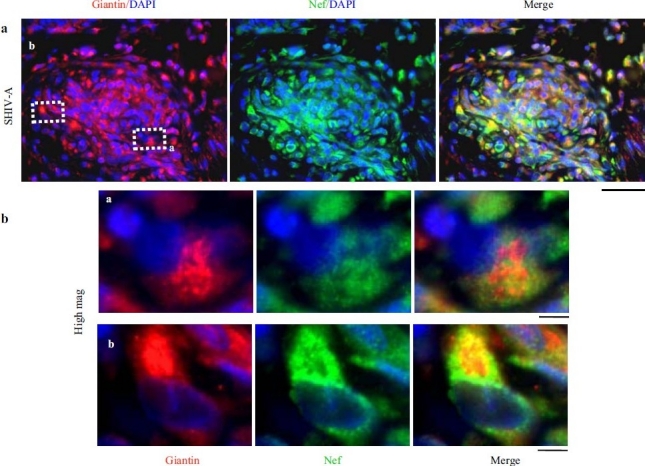
HIV-nef–positive vascular cell elements have increased giantin. (a) Lung sections from a SHIV-nef–infected macaque with increased giantin in the obliterative vascular lesion probed for HIV-nef and giantin. Scale bar=25 μm. (b) Higher magnifi cation images of the insets depicted in Figure 12a. Scale bar=4μm. Respective 3D intracellular immunoimaging is in ref. 75. (Adapted from ref. 2.)
Additionally, both Vti1a and α-SNAP were increased in per cell amounts and localized to cytoplasmic vesicular structures in cells in the pulmonary vascular lesions in SHIV-nef–infected macaques.[75] These data, especially the more recent high-resolution 3D imaging studies,[75] provide evidence in support of an underlying trafficking defect in nef-containing cells in this model of pulmonary vasculopathy. Flores and colleagues are currently engaged in the isolation of HIV virus from AIDS patients with PAH or without PAH and carrying out genetic sequence analyses of the respective HIV-nef genes in order to help identify mutations that predispose toward PAH.[79]
It has been recently reported that exposure of porcine pulmonary artery rings and human PAECs in culture to HIV-nef protein for 24 hrs led to decreased vasorelaxation, decreased eNOS expression, decreased eNOS antigen by immunohistochemistry and reduced NO bioavailability.[80] It is an exciting possibility that reduced NO bioavailability subsequent to endosomal uptake of HIV-nef might mechanistically contribute to Golgi fragmentation in a manner similar to that observed after NO scavenging[64,74–76] and it can be tested in future studies using the subcellular 3D imaging approach.
Defective trafficking of mutants of BMPR II
The pioneering studies of Morrell and colleagues[20,43,45] and of Nishihara and colleagues[44] have drawn attention to the observations that several of the mutants of BMPR II found in patients with PAH show defective trafficking in their traverse from the ER to the Golgi and thence to the plasma membrane. Using green fluorescent protein (GFP) -tagged constructs of wt and mutant BMPR II species, Rudarakanchana et al.[45] and Nishihara et al.[44] showed the failure of several such mutants to traffic to the plasma membrane. BMPR II proteins containing mutations in the conserved Cys residues in the ligand binding remained increasingly trapped in the ER and the Golgi apparatus, while those with mutations in the kinase domain appeared to traffic to the plasma membrane but displayed reduced BMP/Smad signaling. However, all mutants investigated showed a gain in function involving upregulation of p38MAPK-dependent proproliferative pathways.[20,81] Chemical chaperones (thapsigargin, glycerol or sodium 4-phenylbutyrate) increased the trafficking of the C118W BMPR II mutant to the cell surface, accompanied by enhanced BMP/Smad signaling.[82] However, several parts of the puzzle of how mutations in BMPR II eventually lead to PAH disease manifestations include (1) the incomplete and delayed penetrance,[17–20] (2) the gender dependence,[17–20,83] (3) the postulated roles of additional unknown genetic and/or environmental factors,[17–20] and (4) that in some cases of PAH, there are reduced levels of BMPR II and reduced BMP/Smad signaling even though there are no mutations in BMPR II.[84]
The consequences of mutations in BMPR II on the trafficking of other vasorelevant proteins (such as eNOS) and receptors (such as gp130 or IL-6R or VEGFR) in trans have not been adequately investigated. Moreover, since Smad signaling is obligatorily dependent on membrane-associated endocytic pathways for transcriptional signaling,[11,13,69] it is important to consider the dependence of this inward signaling on the integrity of the underlying vesicular transport and fusion machineries. Perhaps, defects in the retrograde membrane-associated trafficking pathways might explain defects in BMP/Smad signaling in IPAH despite a wt BMPR II. The reduced expression of eNOS antigen in arterial endothelium in lungs of BMPR II mutant transgenic mice subjected to hypoxia[85] suggests a multifactorial pathophysiology. As this review makes clear, reduced NO bioavailability, hypoxia and mutations in BMPR II all affect intracellular trafficking pathways.
Estradiol and trans-Golgi trafficking
There is a gender bias in the development of IPAH with the female to male ratio recently quoted as 1.64–3.88:1.[83] Familial PAH due to mutations in BMPR II also appears to have an earlier onset in women.[17–20,82] However, it has been long known that in the case of the MCT/rat model, the situation is the opposite – estradiol-17β and related compounds inhibit the development of PAH.[86,87] In this context, it is worth noting the studies of Greenfield et al.[88] who investigated the mechanisms by which estrogen therapy reduced the risk of Alzheimer's disease in post-menopausal women, β-amyloid burden in animal models of Alzheimer's disease, and secretion of β-amyloid peptides by neuronal cells. These investigators observed that estradiol-17β stimulates trans-Golgi network (TGN) biogenesis including increased TGN phospholipids levels, recruitment of soluble Rab 11 to the TGN and regulating traffic within the late secretory pathway.[88] Earlier, Hendrix et al.[89] reported the effects of estrogens and progestins and their antagonists in modulating the intracellular trafficking of integral membrane proteins. We have recently discovered two specific estrogen-binding proteins in the Golgi apparatus of human PAECs and PASMCs that await further characterization(Lee JE, Sehgal PB, unpublished data). There is thus a growing nexus of data pointing to the ability of estrogens in regulating intracellular trafficking, in particular, through the Golgi. The ability of estrogens to regulate intracellular trafficking remains unexplored in the context of PAH.
A LOOK AHEAD
We have been struck by the presence of almost all of our current ideas already extant in the PAH and related literature extending back over the last four decades. The EM data of Heath and colleagues,[26–28] and others clearly pointed to defects in intracellular trafficking in cells in pulmonary vascular lesions in IPAH. From our perspective, we have added a vocabulary long established in cell biology in terms of the molecules and machineries that mediate and regulate intracellular trafficking and targeted vesicular transport to consideration of role(s) in the pathogenesis PAH. In doing so, our focus remains on the subcellular mechanisms at the level of cytoplasmic organelles and trafficking, which might contribute to the net consequence of vascular remodeling observed in PAH. We clearly envisage such contributions to be at multiple levels in terms of anterograde and retrograde trafficking and in the regulation of cell motility and cell mitosis. Consequences of such dysfunctions would be widespread changes in the cell surface texture and landscape of the affected cells, leading to global changes in cell surface receptors, cell surface ligands, signaling pathways, the unusual secretory phenotype, prothrombogenicity and lumen closure [Figure 2]. A separate major line of research with a focus on subcellular organellar biology not addressed in the present review involves the role of mitochondria in pulmonary vascular remodeling.[90] Elucidating how each of these changes fits into the multifactorial context of hypoxia, reduced NO bioavailability, mutations in BMPR II, modulation of disease penetrance and gender effects in disease occurrence in the pathogenesis of PAH is part of the road ahead.
Footnotes
Source of Support: NIH R01 HL-087176 and F31 HL-107013
Conflict of Interest: None
REFERENCES
- 1.Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: The hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1013–32. doi: 10.1152/ajplung.00217.2009. [DOI] [PubMed] [Google Scholar]
- 2.Sehgal PB, Mukhopadhyay S, Patel K, Xu F, Almodóvar S, Tuder RM, et al. Golgi dysfunction is a common feature in idiopathic human pulmonary hypertension and vascular lesions in SHIV-nef-infected macaques. Am J Physiol Lung Cell Mol Physiol. 2009;297:L729–37. doi: 10.1152/ajplung.00087.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mathew R, Huang J, Shah M, Patel K, Gewitz M, Sehgal PB. Disruption of endothelial-cell caveolin-1alpha/raft scaffolding during development of monocrotaline-induced pulmonary hypertension. Circulation. 2004;110:1499–506. doi: 10.1161/01.CIR.0000141576.39579.23. [DOI] [PubMed] [Google Scholar]
- 4.Zhao YY, Lui Y, Stan RV, Fan L, Gu Y, Dalton N, et al. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc Natl Acad Sci U S A. 2002;99:11375–80. doi: 10.1073/pnas.172360799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao YY, Zhao YD, Mirza MK, Huang JH, Potula HH, Vogel SM, et al. Persistent eNOS activation secondary to caveolin-1 deficiency induces pulmonary hypertension in mice and humans through PKG nitration. J Clin Invest. 2009;119:2009–18. doi: 10.1172/JCI33338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Achcar RO, Demura Y, Rai PR, Taraseviciene-Stewart L, Kasper M, Voelkel NF, et al. Loss of caveolin and heme oxygenase expression in severe pulmonary hypertension. Chest. 2006;129:696–705. doi: 10.1378/chest.129.3.696. [DOI] [PubMed] [Google Scholar]
- 7.Sehgal PB, Mukhopadhyay S. Pulmonary arterial hypertension: A disease of tethers, SNAREs, and SNAPs. Am J Physiol Heart Circ Physiol. 2007;293:H77–85. doi: 10.1152/ajpheart.01386.2006. [DOI] [PubMed] [Google Scholar]
- 8.Sehgal PB, Mukhopadhyay S. Dysfunctional intracellular trafficking in the pathobiology of pulmonary arterial hypertension. Am J Respir Cell Mol Biol. 2007;37:31–7. doi: 10.1165/rcmb.2007-0066TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell. 2004;116:153–66. doi: 10.1016/s0092-8674(03)01079-1. [DOI] [PubMed] [Google Scholar]
- 10.Jahn R, Scheller RH. SNAREs–engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–43. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- 11.Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL. Distinct endocytic pathways regulate TGF-beta receptor signaling and turnover. Nat Cell Biol. 2003;5:410–21. doi: 10.1038/ncb975. [DOI] [PubMed] [Google Scholar]
- 12.Sehgal PB, Guo GG, Shah M, Kumar V, Patel K. Cytokine signaling: STATs in plasma membrane rafts. J Biol Chem. 2002;277:12067–74. doi: 10.1074/jbc.M200018200. [DOI] [PubMed] [Google Scholar]
- 13.Shah M, Patel K, Mukhopadhyay S, Xu F, Guo G, Sehgal PB. Membrane-associated STAT3 and PY-STAT3 in the cytoplasm. J Biol Chem. 2006;281:7302–8. doi: 10.1074/jbc.M508527200. [DOI] [PubMed] [Google Scholar]
- 14.Xu F, Mukhopadhyay S, Sehgal PB. Live cell imaging of interleukin-6-induced targeting of the “transcription factor” STAT3 to sequestering endosomes in the cytoplasm. Am J Physiol Cell Physiol. 2007;293:C1374–82. doi: 10.1152/ajpcell.00220.2007. [DOI] [PubMed] [Google Scholar]
- 15.Mukhopadhyay S, Shah M, Xu F, Patel K, Tuder RM, Sehgal PB. Cytoplasmic provenance of STAT3 and PY-STAT3 in the endolysosomal compartments in pulmonary arterial endothelial and smooth muscle cells: Implication in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;294:L449–68. doi: 10.1152/ajplung.00377.2007. [DOI] [PubMed] [Google Scholar]
- 16.Le Borgne R. Regulation of Notch signaling by endocytosis and endosomal sorting. Curr Opin Cell Biol. 2006;18:213–22. doi: 10.1016/j.ceb.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 17.Tuder RM, Marecki JC, Richter A, Fijlkowska I, Flores S. Pathology of pulmonary hypertension. Clin Chest Med. 2007;28:23–42. doi: 10.1016/j.ccm.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rabinovitch M. Molecular pathogenesis of pulmonary hypertension. J Clin Invest. 2008;118:2372–9. doi: 10.1172/JCI33452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Runo JR, Loyd JE. Primary pulmonary hypertension. Lancet. 2003;361:1533–44. doi: 10.1016/S0140-6736(03)13167-4. [DOI] [PubMed] [Google Scholar]
- 20.Morrell NW. Role of bone morphogenetic protein receptors in the development of pulmonary hypertension. Adv Exp Med Biol. 2010;661:251–64. doi: 10.1007/978-1-60761-500-2_16. [DOI] [PubMed] [Google Scholar]
- 21.Resta TC, Gonzales RJ, Dail WG, Sanders TC, Walker BR. Selective upregulation of arterial endothelial nitric oxide synthase in pulmonary hypertension. Am J Physiol Heart Circ Physiol. 1997;41:H806–13. doi: 10.1152/ajpheart.1997.272.2.H806. [DOI] [PubMed] [Google Scholar]
- 22.Cajal SR. Algunas variaciones fisilógicas y patalógicas del aparato reticular de Golgi. Trab Del Lab De Inv Biol Madrid. 1914;12:127–227. [Google Scholar]
- 23.Jaenke RS, Alexander AF. Fine structural alterations of bovine peripheral pulmonary arteries in hypoxia-induced hypertension. Am J Pathol. 1973;73:377–98. [PMC free article] [PubMed] [Google Scholar]
- 24.Meyrick B, Reid L. The effect of continued hypoxia on rat pulmonary arterial circulation. An ultrastructural study. Lab Invest. 1978;38:188–200. [PubMed] [Google Scholar]
- 25.Meyrick B, Reid L. Hypoxia-induced structural changes in the media and adventitia of the rat hilar pulmonary artery and their regression. Am J Pathol. 1980;100:151–78. [PMC free article] [PubMed] [Google Scholar]
- 26.Smith P, Heath D. Electron microscopy of the plexiform lesion. Thorax. 1979;34:177–86. doi: 10.1136/thx.34.2.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heath D, Smith P, Gosney J, Mulcahy D, Fox K, Yacoub M, et al. The pathology of the early and late stages of primary pulmonary hypertension. Br Heart J. 1987;58:204–13. doi: 10.1136/hrt.58.3.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith P, Heath D, Yacoub M, Madden B, Caslin A, Gosney J. The ultrastructure of plexogenic pulmonary arteriopathy. J Pathol. 1990;160:111–21. doi: 10.1002/path.1711600204. [DOI] [PubMed] [Google Scholar]
- 29.King AP, Smith P, Heath D. Ultrastructure of rat pulmonary arterioles after neonatal exposure to hypoxia and subsequent relief and treatment with monocrotaline. J Pathol. 1995;177:71–81. doi: 10.1002/path.1711770112. [DOI] [PubMed] [Google Scholar]
- 30.Mette SA, Palevsky HI, Pietra GG, Williams TM, Bruder E, Prestipino AJ, et al. Primary pulmonary hypertension in association with human immunodeficiency virus infection.A possible viral etiology for some forms of hypertensive pulmonary arteriopathy. Am Rev Respir Dis. 1992;145:1196–200. doi: 10.1164/ajrccm/145.5.1196. [DOI] [PubMed] [Google Scholar]
- 31.Harns PN, Anderson RC, Chen KK. The action of senecionine, integerrimine, jacobine, longilobine and spartiodine, especially on the liver. J Pharmacol Exp Ther. 1942;69:69–77. [Google Scholar]
- 32.Harns PN, Anderson RC, Chen KK. The action of monocrotaline and retronecine. J Pharmacol Exp Ther. 1942;75:78–82. [Google Scholar]
- 33.Bull LB. The histological evidence of liver damage from pyrrolizidene alkaloids: Megalocytosis of the liver cells and inclusion globules. Aust Vet J. 1955;31:33–40. [Google Scholar]
- 34.Merkow L, Kleinerman J. An electron microscopic study of pulmonary vasculitis induced by monocrotaline. Lab Invest. 1966;15:547–64. [PubMed] [Google Scholar]
- 35.Afzelius BA, Schoental R. The ultrastructure of the enlarged hepatocyes induced in rats with a single oral dose of retrorsine, a pyrrolozidine (Senecio) alkaloid. J Ultrastruct Res. 1967;20:328–45. doi: 10.1016/s0022-5320(67)80103-5. [DOI] [PubMed] [Google Scholar]
- 36.Todorovich-Hunter L, Johnson DJ, Ranger P, Keeley FW, Rabinovitch M. Altered elastin and collagen synthesis associated with progressive pulmonary hypertension induced by monocrotaline. A biochemical and ultrastructural study. Lab Invest. 1988;58:184–95. [PubMed] [Google Scholar]
- 37.Rosenberg HC, Rabinovitch M. Endothelial injury and vascular reactivity in monocrotaline pulmonary hypertension. Am J Physiol. 1988;255:H1484–91. doi: 10.1152/ajpheart.1988.255.6.H1484. [DOI] [PubMed] [Google Scholar]
- 38.Sugita T, Stenmark KR, Wagner WW, Jr, Henson PM, Henson JE, Hyers TM, et al. Abnormal alveolar cells in monocrotaline induced pulmonary hypertension. Exp Lung Res. 1983;5:201–15. doi: 10.3109/01902148309061515. [DOI] [PubMed] [Google Scholar]
- 39.Wilson DW, Segall HJ. Changes in type II cell populations in monocrotaline pneumotoxicity. Am J Pathol. 1990;136:1293–1299. [PMC free article] [PubMed] [Google Scholar]
- 40.Reindel JF, Roth RA. The effects of moncrotaline pyrrole on cultured bovine pulmonary artery endothelial smooth muscle cells. Am J Pathol. 1991;138:707–19. [PMC free article] [PubMed] [Google Scholar]
- 41.Lamé MW, Jones AD, Wilson DW, Dunston SK, Segall HJ. Protein targets of monocrotaline pyrrole in pulmonary artery endothelial cells. J Biol Chem. 2000;275:29091–9. doi: 10.1074/jbc.M001372200. [DOI] [PubMed] [Google Scholar]
- 42.Lamé MW, Jones AD, Wilson DW, Segall HJ. Monocrotaline pyrrole targets proteins with and without cysteine residues in the cytosol and membranes of human pulmonary artery endothelial cells. Proteomics. 2005;5:4398–413. doi: 10.1002/pmic.200402022. [DOI] [PubMed] [Google Scholar]
- 43.Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, 3rd, Loyd JE, et al. Heterozygous germline mutations in BMPR2, encoding a TGF-â receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26:81–4. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 44.Nishihara A, Watabe T, Imamura T, Miyazono K. Functional heterogeneity of bone morphogenetic protein receptor-II mutants found in patients with primary pulmonary hypertension. Mol Biol Cell. 2002;13:3055–63. doi: 10.1091/mbc.E02-02-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rudarakanchana N, Flanagan JA, Chen H, Upton PD, Machado R, Patel D, et al. Functional analysis of bone morphogenetic protein type II receptor mutations underlying primary pulmonary hypertension. Hum Mol Genet. 2002;11:1517–25. doi: 10.1093/hmg/11.13.1517. [DOI] [PubMed] [Google Scholar]
- 46.Hong KH, Lee YJ, Lee E, Park SO, Han C, Beppu H, et al. Genetic ablation of the BMPR2 gene in pulmonary endothelium is sufficient to predispose to pulmonary arterial hypertension. Circulation. 2008;118:722–30. doi: 10.1161/CIRCULATIONAHA.107.736801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.West J. Cross talk between Smad, MAPK, and actin in the etiology of pulmonary arterial hypertension. Adv Exp Med Biol. 2010;661:265–78. doi: 10.1007/978-1-60761-500-2_17. [DOI] [PubMed] [Google Scholar]
- 48.Razani B, Wang XB, Engelman JA, Battista M, Lagaud G, Zhang XL, et al. Caveolin-2-deficient mice show evidence of severe pulmonary dysfunction without the disruption of caveolae. Mol Cell Biol. 2002;22:2329–44. doi: 10.1128/MCB.22.7.2329-2344.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jasmin JF, Mercier I, Hnasko R, Cheung MW, Tanowitz HB, Dupuis J, et al. Lung remodeling and pulmonary hypertension after myocardial infarction: Pathogenic role of reduced caveolin expression. Cardiovasc Res. 2004;63:747–55. doi: 10.1016/j.cardiores.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 50.Jasmin JF, Mercier I, Dupuis J, Tanowitz HB, Lisante MP. Short-term administration of a cell-permeable caveolin-1 peptide prevents the development of monocrotaline-induced pulmonary hypertension and right ventricular hypertrophy. Circulation. 2006;114:912–20. doi: 10.1161/CIRCULATIONAHA.106.634709. [DOI] [PubMed] [Google Scholar]
- 51.Marecki JC, Cool CD, Parr JE, Beckley VE, Luciw PA, Tarantal AF, et al. HIV-1 Nef is associated with complex pulmonary vascular lesions in SHIV-nef-infected macaques. Am J Respir Crit Care Med. 2006;174:437–45. doi: 10.1164/rccm.200601-005OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chaudhri R, Lindwasser OW, Smith WJ, Hurley JH, Bonifacino JS. CD4 downregulation by HIV-1 Nef is dependent on clathrin and involves a direct interaction of Nef with AP2 clathrin adaptor. J Virol. 2007;81:3877–90. doi: 10.1128/JVI.02725-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Collette Y, Aroid S, Picard C, Janvier K, Benichou S, Benarous R, et al. HIV-2 and SIV nef proteins target different src family SH3 domains than does HIV-1 Nef because of a triple amino acid substitution. J Biol Chem. 2000;275:4171–6. doi: 10.1074/jbc.275.6.4171. [DOI] [PubMed] [Google Scholar]
- 54.Hiyoshi M, Suzu S, Yoshidomi Y, Hassan R, Harada H, Sakashita N, et al. Interaction between Hck and HIV-1 Nef negatively regulates cell surface expression of M-CSF receptor. Blood. 2008;111:243–50. doi: 10.1182/blood-2007-04-086017. [DOI] [PubMed] [Google Scholar]
- 55.Roeth JF, Williams M, Kasper MR, Filzen TM, Collins KL. HIV-1 Nef disrupts MHC-1 trafficking by recruiting AP-1 to the MHC-1 cytoplasmic tail. J Cell Biol. 2004;167:903–13. doi: 10.1083/jcb.200407031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shah M, Patel K, Fried VA, Sehgal PB. Interactions of STAT3 with caveolin-1 and heat shock protein 90 in plasma membrane raft and cytosolic complexes. Preservation of cytokine signaling during fever. J Biol Chem. 2002;277:45662–9. doi: 10.1074/jbc.M205935200. [DOI] [PubMed] [Google Scholar]
- 57.Nishimura T, Faul JL, Berry GJ, Vaszar LT, Qui D, Pearl RG, et al. Simvastatin attenuates smooth muscle neointimal proliferation and pulmonary hypertension in rats. Am J Respir Crit Care Med. 2002;166:1403–8. doi: 10.1164/rccm.200203-268OC. [DOI] [PubMed] [Google Scholar]
- 58.Nishimura T, Vaszar LT, Faul JL, Zhao G, Berry GJ, Shi L, et al. Simvastatin rescues rats from fatal pulmonary hypertension by inducing apoptosis of neointimal smooth muscle cells. Circulation. 2003;108:1640–5. doi: 10.1161/01.CIR.0000087592.47401.37. [DOI] [PubMed] [Google Scholar]
- 59.Shah M, Patel K, Sehgal PB. Monocrotaline pyrrole-induced endothelial cell megalocytosis involves a Golgi blockade mechanism. Am J Physiol Cell Physiol. 2005;288:C850–62. doi: 10.1152/ajpcell.00327.2004. [DOI] [PubMed] [Google Scholar]
- 60.Mukhopadhyay S, Sehgal PB. Discordant regulatory changes in monocrotaline ¬induced megalocytosis of lung arterial endothelial and alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1216–26. doi: 10.1152/ajplung.00535.2005. [DOI] [PubMed] [Google Scholar]
- 61.Mukhopadhyay S, Shah M, Patel K, Sehgal PB. Monocrotaline pyrrole-induced megalocytosis of lung and breast epithelial cells: Disruption of plasma membrane and Golgi dynamics and an enhanced unfolded protein response. Toxicol Appl Pharmacol. 2006;211:209–20. doi: 10.1016/j.taap.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 62.Sehgal PB, Mukhopadhyay S, Xu F, Patel K, Shah M. Dysfunction of Golgi tethers, SNAREs, and SNAPs in monocrotaline-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;290:L1526–42. doi: 10.1152/ajplung.00463.2006. [DOI] [PubMed] [Google Scholar]
- 63.Mukhopadhyay S, Xu F, Sehgal PB. Aberrant cytoplasmic sequestration of eNOS in endothelial cells after monocrotaline, hypoxia, and senescence: Live-cell caveolar and cyotplasmic NO imaging. Am J Physiol Heart Circ Physiol. 2007;292:H1373–89. doi: 10.1152/ajpheart.00990.2006. [DOI] [PubMed] [Google Scholar]
- 64.Mukhopadhyay S, Lee J, Sehgal PB. Depletion of the ATPase NSF from Golgi membranes with hypo-S-nitrosylation of vasorelevant proteins in endothelial cells exposed to monocrotaline pyrrole. Am J Physiol Heart Circ Physiol. 2008;295:H1943–55. doi: 10.1152/ajpheart.00642.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ramos M, Lame MW, Segall HJ, Wilson DW. The BMP type II receptor is located in lipid rafts, including caveolae, of pulmonary endothelium in vivo and in vitro. Vasc Pharmacol. 2006;44:50–9. doi: 10.1016/j.vph.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 66.Wertz JW, Bauer PM. Caveolin-1 regulates BMPR II localization and signaling in vascular smooth muscle cells. Biochem Biophys Res Commun. 2008;375:557–61. doi: 10.1016/j.bbrc.2008.08.066. [DOI] [PubMed] [Google Scholar]
- 67.Ramos M, Lame MW, Segall HJ, Wilson DW. Monocrotaline pyrrole induces Smad nuclear accumulation and altered signaling expression in human pulmonary arterial endothelial cells. Vasc Pharmacol. 2007;46:439–48. doi: 10.1016/j.vph.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ramos MF, Lame MW, Segall HJ, Wilson DW. Smad signaling in the rat model of monocrotaline pulmonary hypertension. Toxicol Pathol. 2008;36:311–20. doi: 10.1177/0192623307311402. [DOI] [PubMed] [Google Scholar]
- 69.Sehgal PB. Paradigm shifts in the cell biology of STAT signaling. Semin Cell Dev Biol. 2008;19:329–40. doi: 10.1016/j.semcdb.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Patel HH, Zhang S, Murray F, Suda RY, Head BP, Yokoyama U, et al. Increased smooth muscle cell expression of caveolin-1 and caveolae contribute to the pathophysiology of idiopathic pulmonary arterial hypertension. FASEB J. 2007;21:2970–9. doi: 10.1096/fj.07-8424com. [DOI] [PubMed] [Google Scholar]
- 71.Maximov A, Tang J, Yang X, Pang ZP, Sudhof TC. Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science. 2009;323:516–21. doi: 10.1126/science.1166505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Giraudo CG, Garcia-Diaz A, Eng WS, Chen Y, Hendrickson WA, Melia TJ, et al. Alternative zippering as an on-off switch for SNARE-mediated fusion. Science. 2009;323:512–6. doi: 10.1126/science.1166500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Matsushita K, Morrell CN, Cambein B, Yang SX, Yamakuchi M, Bao C, et al. Nitric oxide regulates exocytosis by S-nitrosylation of N-ethlymaleimide sensitive factor. Cell. 2003;115:139–50. doi: 10.1016/s0092-8674(03)00803-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee J, Reich R, Xu F, Sehgal PB. Golgi, trafficking, and mitotis dysfunctions in pulmonary arterial endothelial cells exposed to monocrotaline pyrrole and NO scavenging. Am J Physiol Lung Cell Mol Physiol. 2009;297:715–28. doi: 10.1152/ajplung.00086.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee JE, Patel K, Almodovar S, Tuder RM, Flores SC, Dehgal PB. Dependence of Golgi apparatus on nitiric oxide in vascular cells; implications in pulmonary artirial hypertension. Am J Physiol Heart Circ Physiol. 2011 doi: 10.1152/ajpheart.00767.2010. [In Press] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zuckerbraun BS, Shiva S, Ifedigbo E, Mathier MA, Mollen KP, Rao J, et al. Nitrite potently inhibits hypoxic and inflammatory pulmonary arterial hypertension and smooth muscle proliferation via xanthine oxidoreductase-dependent nitric oxide generation. Circulation. 2010;121:98–109. doi: 10.1161/CIRCULATIONAHA.109.891077. [DOI] [PubMed] [Google Scholar]
- 77.Orialo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J. Cell surface-bound IL 1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci U S A. 2009;106:17031–6. doi: 10.1073/pnas.0905299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rodier F, Coppé JP, Patil CK, Hoeijmakers WA, Muñoz DP, Raza SR, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–9. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Almodovar S, Cicalini S, Petrosillo N, Flores SC. Pulmonary hypertension associated with HIV infection: Pulmonary vascular disease: The global perspective. Chest. 2010;137:6S–12S. doi: 10.1378/chest.09-3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Duffy P, Wang X, Lin PH, Yao Q, Chen C. HIV Nef protein causes endothelial dysfunction in porcine pulmonary arteries and human pulmonary artery endothelial cells. J Surg Res. 2009;156:257–64. doi: 10.1016/j.jss.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yang X, Long Lu, Southwood M, Rudarakanchana N, Upton PD, Jeffery TK, et al. Dysfunctional Smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ Res. 2005;96:1053–63. doi: 10.1161/01.RES.0000166926.54293.68. [DOI] [PubMed] [Google Scholar]
- 82.Sobolewski A, Rudarakanchana N, Upton PD, Yang J, Crilley TK, Trembath RC, et al. Failure of bone morphogenetic protein receptor trafficking in pulmonary arterial hypertension: Potential for rescue. Hum Mol Genet. 2008;17:3180–90. doi: 10.1093/hmg/ddn214. [DOI] [PubMed] [Google Scholar]
- 83.Sakao S, Tanabe N, Tatsumi K. The estrogen paradox in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2010;299:L435–8. doi: 10.1152/ajplung.00057.2010. [DOI] [PubMed] [Google Scholar]
- 84.Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC, et al. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. 2002;105:1672–8. doi: 10.1161/01.cir.0000012754.72951.3d. [DOI] [PubMed] [Google Scholar]
- 85.Frank DB, Lowery J, Anderson L, Brink M, Reese J, de Caestecker M. Increased susceptibility to hypoxic pulmonary hypertension in Bmpr2 mutant mice is associated with endothelial dysfunction in the pulmonary vasculature. Am J Physiol Lung Cell Mol Physiol. 2008;294:L98–109. doi: 10.1152/ajplung.00034.2007. [DOI] [PubMed] [Google Scholar]
- 86.Farhat MY, Chen MF, Bhatti T, Iqbal A, Cathapermal S, Ramwell PW. Protection by oestradiol against the development of cardiovascular changes associated with monocrotaline pulmonary hypertension in rats. Br J Pharmacol. 1993;110:719–23. doi: 10.1111/j.1476-5381.1993.tb13871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tofovic SP, Zhang X, Jackson EK, Dacic S, Petrusevska G. 2-methoxyestradiol mediates the protective effects of estradiol in monocrotaline-induced pulmonary hypertension. Vascul Pharmacol. 2006;45:358–67. doi: 10.1016/j.vph.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 88.Greenfield JP, Leung LW, Cai D, Kaasik K, Gross RS, Rodriguez-Boulan E, et al. Estrogen lowers Alzheimer beta-amyloid generation by stimulating trans-Golgi network vesicle biogenesis. J Biol Chem. 2002;277:12128–36. doi: 10.1074/jbc.M110009200. [DOI] [PubMed] [Google Scholar]
- 89.Hendrix EM, Myatt L, Sellers S, Russell PT, Larsen WJ. Steroid hormone regulation of rat myometrial gap junction formation: Effects on cx43 levels and trafficking. Biol Reprod. 1995;52:547–60. doi: 10.1095/biolreprod52.3.547. [DOI] [PubMed] [Google Scholar]
- 90.Dromparis P, Sutendra G, Michelakis ED. The role of mitochondria in pulmonary vascular remodeling. J Mol Med. 2010;88:1003–10. doi: 10.1007/s00109-010-0670-x. [DOI] [PubMed] [Google Scholar]