

CLINICAL CLASSIFICATION
The clinical classification of pulmonary hypertension (PH) has gone through a series of changes since the first classification was proposed in 1973 at the World Health Organization international conference on primary pulmonary hypertension (PPH) in Geneva, Switzerland.[1,2] The initial classification designated only two categories, primary pulmonary hypertension (PPH) and secondary PH, depending on the presence or absence of identifiable causes or risk factors.
Twenty-five years later, a second World Symposium on Pulmonary Arterial Hypertension (PAH) was held in 1998 in Evian, France. Based on the research that had ensued in studying the pulmonary circulation since 1973, by 1998 significant advances had been made in our understanding of PH. Further, the first drug for PPH was approved during this time, i.e., in 1995. Thus, because the aim of a clinical classification is to individualize different categories sharing similarities in pathobiology, clinical presentation, and therapeutic approaches, the “Evian Classification” was based on defining categories of PH that shared similar histopathology and clinical characteristics. The Evian Classification expanded the prior 1973 classification from 2 groups to 5 major groups with Group 1 PH being the most studied, i.e., PAH.[3] In 2003, during the third World Symposium on PH held in Venice, Italy, the clinical classification was slightly modified,[4] with further modification most recently during the fourth World Symposium held in Dana Point, California in 2008[5] (Table 1). Although not the basis for the modifications, these modifications also permitted clinical investigators to conduct randomized controlled drug trials in patients with a shared underlying pathobiology (resulting in our currently having 8 drugs approved for the treatment of adult subjects with PAH, i.e., Group 1 PH).
Table 1.
Dana Point clinical classification of pulmonary hypertension (2008)

The classifications were developed, and revised, based on the current understanding of PH, i.e., its etiology, pathobiology, clinical course and response to therapeutic interventions. The majority of the work in this field has focused on PAH, i.e., Group 1 PH. And although these classifications were never specifically limited to adult subjects, utilizing them for all pediatric subjects can be less than ideal. Reasons for this include unique aspects of childhood PH, not the least of which is that PH can start in utero resulting in growth abnormalities that can persist into adulthood. Further, with improvements in our ability to effectively take care of premature infants, children are now living longer with many children not infrequently having various forms of pulmonary vascular disease that did not exist several decades ago (and thus are not included in the most recent Dana Point Classification). Nevertheless, the Dana Point Classification has been, and continues to be, invaluable in our advancing the PH field. It has also been invaluable for adolescent patients and has facilitated obtaining therapy for younger children.
However, the Dana Point Classification has limitations for classifying pulmonary vascular disease in childhood. Thus, at the 2011 Pulmonary Vascular Research Institute (PVRI) meeting in Panama, a new classification for Pediatric Pulmonary Hypertensive Vascular Disease was developed by a group of investigators focused on pulmonary hypertension in childhood.[6,7] The aim was not to replace the Dana Point Classification of 2008 but rather to augment it for specific pediatric disorders. The proposed classification, presented in this issue of Pulmonary Circulation, may be called the “Panama Classification, sponsored by the Pulmonary Vascular Research Institute,” or, “the Panama Classification (2011).” This classification now allows those interested in pediatric pulmonary hypertensive vascular disease to more critically identify specific diseases and disorders that we hope will improve our ability to move the field forward. In many aspects, pediatric PH is now where PH in the adult was 2 decades ago; including our lagging behind in developing evidence-based treatment guidelines for children with PH with increased pulmonary vascular resistance (PVR). The current consensus-based treatment recommendations for children with PAH are based on extrapolation from the evidence-based adult PAH treatment guidelines. And although due to the similarities between children and adults there is no reason to believe that drugs approved to treat PAH in adults will not be efficacious in pediatric PAH, determining optimal dosing and long-term safety, including potential effects on growth and development, cannot merely be extrapolated from adult data. Children are not “small adults.”
There are also important aspects of neonatal and childhood pulmonary hypertensive vascular disease that are not included in the 2008 Dana Point Classification. These include: (1) fetal origins of pulmonary vascular disease; (2) the potential importance of developmental mechanisms in both childhood and adult onset disease; (3) an inconsistent approach to neonatal pulmonary vascular disease; (4) the importance of perinatal maladaptation, maldevelopment and pulmonary hypoplasia in pulmonary vascular disease; (5) variability in the heterogeneity of factors that contribute to pulmonary vascular disease in children compared with adults; and (6) adult survivors of pediatric pulmonary vascular disease (e.g., children with pulmonary vascular disease who in the past never survived long enough to reach adulthood are now surviving into adulthood). Additionally, there are risk factors that may be significant in children that have not been considered significant in adult PH (Table 2).
Table 2.
Updated risk factors and associated conditions for PAH

The Dana Point Classification Supplements that focus on the specifics of the congenital heart defects are useful but remain less than ideal for use in patients of all ages. For example, a 1-cm defect may be large in an infant but small in an adult (Table 3). Nevertheless, defining the congenital heart defects as accurately as possible is useful. I find the Groups A, B, C and D in patients with congenital systemic to pulmonary shunts very informative and recommend that they be included in the pediatric classification (Table 4).
Table 3.
Anatomic-pathophysiologic classification of congenital systemic-to-pulmonary shunts associated with pulmonary arterial hypertension

Table 4.
Clinical classification of congenital systemic-to-pulmonary shunts associated with pulmonary arterial hypertension

Classifications are useful in medicine because they provide a framework for diagnosis and management, and encourage epidemiological insight. They should also include categories for undiscovered diseases and undiscovered mechanisms of known disease complexes but this is more difficult. As the authors of this new proposed pediatric classification point out, the aims are to: Improve diagnostic strategies, promote clinical investigation in pathobiology, pathophysiology and outcomes, provide guidance for human disease modeling in laboratory and animal studies, and serve as an educational resource. However, it is important to note that this new classification was not designed solely to serve as a therapeutic guide.
This “Panama Classification (2011)” has been termed “pulmonary hypertensive vascular disease” as opposed to PH to exclude children with PH who do not have increased PVR. A significant number of children have PH without increased PVR, e.g., children with large left to right systemic to pulmonary shunts, in whom the treatment should be closure of the shunts and not treatment for PAH (which by definition requires an increase in PVR in addition to PH). Unfortunately, the term PAH does not immediately equate to PH with increased PVR to the pediatric cardiology community; it never has, and I doubt it ever will regardless of education attempts at conveying this.
This pediatric classification also includes children with an increase in PVR in whom the mean pulmonary artery pressure (PAPm) may be less than 25 mmHg but the children are symptomatic from the increased PVR, e.g. children undergoing staged repair for a single ventricle. And as surgical procedures improve, many of these children will survive into adulthood with symptomatic pulmonary vascular disease due to increased PVR without a mean PAP≥25 mmHg. The proposal for the definition of pediatric pulmonary hypertensive vascular disease is: an increased pulmonary vascular resistance index (PVRI), i.e. ≥3 Wood units times m2 whether or not the PAPm is ≥25 mmHg.
No classifications are perfect; their value is if and only if they are used, thereby permitting colleagues in different specialties around the world to communicate with one another using the same language. To balance being all-inclusive with adequate simplicity is the challenge. At first review, this new proposal appears overwhelming; however, the 10 divisions are quite straightforward when viewed individually. There will always be overlap as there is in the Dana Point Classification, since patients have genetic, epigenetic, environmental and unknown factors triggering the development of pulmonary vascular disease. In the Dana Point Classification, a patient may be considered to have Group 1 PH, i.e., PAH, as his primary diagnosis, but he may also have COPD, left ventricular diastolic dysfunction, etc., that may be contributing in some way to the patient's PH. Multi-factorial conditions come into play in subjects of all ages. However, in addition to multi-factorial conditions in the pediatric population, pathological insults on the growing lung and pulmonary developmental abnormalities can be quite significant. Additionally, children with chromosomal or genetic syndromes that may not have previously survived to adulthood are surviving longer and not infrequently these syndromes appear to contribute to the development of pulmonary vascular disease. And whether some of these factors will affect the response to various therapies remains unclear but certainly is possible and will require careful study.
Utilizing this new pediatric clinical classification will undoubtedly result in modifications; if not, then the classification will not have been successful, as one of its goals is to advance research and further our understanding of pulmonary vascular disease in subjects of all ages.
FUNCTIONAL CLASSIFICATION
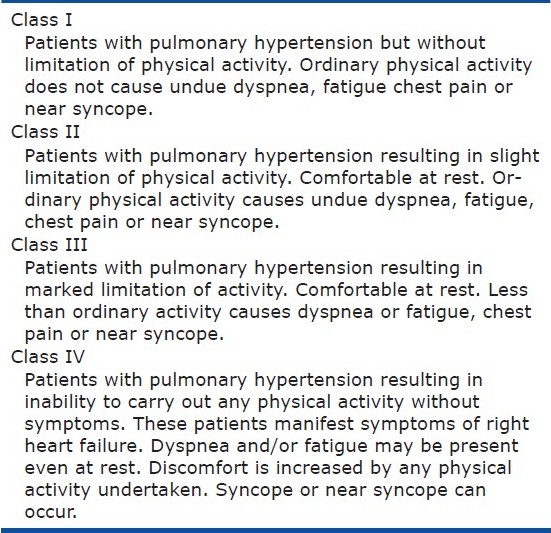
What about how we assess patients‘ functional capacity? Can we use the same functional classification for children as we do for adults? And what about children of differing ages, e.g. infants, toddlers, school-age children, adolescents? As one of our goals in treating patients is to improve their overall quality of life (in addition to increasing survival), how should we assess children? For adult PH, we modified the NYHA functional classification specifically for PH (Table 5). And although clinicians may vary from one to another in how they classify patients, for a given patient, classification performed by the same clinician appears quite useful in assessing functional capacity. But when we look at this classification for children, there are significant difficulties in using it. The members of the 2011 PVRI pediatric task force therefore also proposed a new functional classification for children with PH. The adult classification was modified based on differences in age, maturity, and motor and language development. Further, how a parent perceives their child is doing is also important and thus assessments from the children (adapted for various aged children) and from their parents were developed. Similar to the pediatric clinical classification, this pediatric functional capacity classification may appear quite complicated when first looking at it, but that is because of the significant changes that occur normally from infancy through adolescence and the inability to have a “one size fits all” functional capacity tool. Thus when you look at the classification specifically for the age child you want to assess, this proposed classification should permit us to accurately assess how a child is coping with his illness and functionally in his family, school and interacting with his peers.
Table 5.
Modified NYHA functional classification for PH
I commend these investigators for their dedication to advancing our understanding of pulmonary vascular disease in childhood. As we move forward in the field of PH and pulmonary vascular disease in children and in adults, an increased collaboration between investigators committed to this disease will improve outcomes for all aged patients; life is a continuum.
REFERENCES
- 1.Hatano S, Strasser T. Geneva: WHO; 1973. Oct 15-17, Primary pulmonary hypertension. Report on a WHO meeting. 1975. [Google Scholar]
- 2.Wagenvoort CA, Wagenvoort N. Primary pulmonary hypertension: A pathologic study of the lung vessels in 156 clinically diagnosed cases. Circulation. 1970;42:1163–84. [Google Scholar]
- 3.Fishman AP. Clinical classification of pulmonary hypertension. Clin Chest Med. 2001;22:385–91. doi: 10.1016/s0272-5231(05)70278-1. vii. [DOI] [PubMed] [Google Scholar]
- 4.Simonneau G, Galie N, Rubin LJ, Langleben D, Seeger W, Domenighetti G, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2004;43:S5–S12. doi: 10.1016/j.jacc.2004.02.037. [DOI] [PubMed] [Google Scholar]
- 5.Simonneau G, Robbins IM, Beghetti M, Channick RN, Denton CP, Elliott GE, et al. Updated Clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S43–54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 6.Del Cerro MJ, Abman S, Diaz G, Freudenthal AH, Freudenthal F, Harikrishnan S, et al. A consensus approach to the classification of pediatric pulmonary hypertensive vascular disease: Report from the PVRI Pediatric Taskforce, Panama 2011. Pulm Circ. 2011;2:286–98. doi: 10.4103/2045-8932.83456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lammers AE, Adatia I, del Cerro MJ, Diaz G, Freudenthal AH, Freudenthal F, et al. Functional classification of pulmonary hypertension in children: Report from the PVRI pediatric taskforce, Panama 2011. Pulm Circ. 2011;2:280–5. doi: 10.4103/2045-8932.83445. [DOI] [PMC free article] [PubMed] [Google Scholar]