

Abstract
The histaminergic tuberomamillary nucleus (TMN) controls arousal and attention, and the firing of TMN neurons is state-dependent, active during waking, silent during sleep. Thyrotropin-releasing hormone (TRH) promotes arousal and combats sleepiness associated with narcolepsy. Single-cell reverse-transcription-PCR demonstrated variable expression of the two known TRH receptors in the majority of TMN neurons. TRH increased the firing rate of most (ca 70%) TMN neurons. This excitation was abolished by the TRH receptor antagonist chlordiazepoxide (CDZ; 50 μm). In the presence of tetrodotoxin (TTX), TRH depolarized TMN neurons without obvious change of their input resistance. This effect reversed at the potential typical for nonselective cation channels. The potassium channel blockers barium and cesium did not influence the TRH-induced depolarization. TRH effects were antagonized by inhibitors of the Na+/Ca2+ exchanger, KB-R7943 and benzamil. The frequency of GABAergic spontaneous IPSCs was either increased (TTX-insensitive) or decreased [TTX-sensitive spontaneous IPSCs (sIPSCs)] by TRH, indicating a heterogeneous modulation of GABAergic inputs by TRH. Facilitation but not depression of sIPSC frequency by TRH was missing in the presence of the κ-opioid receptor antagonist nor-binaltorphimine. Montirelin (TRH analog, 1 mg/kg, i.p.) induced waking in wild-type mice but not in histidine decarboxylase knock-out mice lacking histamine. Inhibition of histamine synthesis by (S)-α-fluoromethylhistidine blocked the arousal effect of montirelin in wild-type mice. We conclude that direct receptor-mediated excitation of rodent TMN neurons by TRH demands activation of nonselective cation channels as well as electrogenic Na+/Ca2+ exchange. Our findings indicate a key role of the brain histamine system in TRH-induced arousal.
Introduction
The tripeptide thyrotropin-releasing hormone (TRH) was the first identified hypothalamic releasing factor. It is mainly produced in neurons of the paraventricular nucleus and triggers thyroid-stimulating hormone release from the adenohypophysis. Beyond neuroendocrine function, two TRH receptors, TRH and the TRH-degrading enzyme, are expressed in many brain regions, where they can modulate neuronal activity, suggesting a role of TRH as a neurotransmitter or neuromodulator (Yarbrough, 1979; Gershengorn and Osman, 1996; Heuer et al., 2000). Clinical and experimental reports demonstrated a role of TRH in the modulation of locomotion, cognition, mood, and sleep. Biologically stable TRH analogues such as CG3703 (montirelin) and TA0910 increase wakefulness and decrease sleep time in narcoleptic canines (Nishino et al., 1997; Riehl et al., 2000). TRH analogues are also known as antiepileptics in animal seizure models (Nillni and Sevarino, 1999) and in clinical use (Kubek and Garg, 2002). Although the antiepileptic function of TRH can be explained by its excitatory action on hippocampal interneurons (Atzori and Nistri, 1996; Deng et al., 2006), cellular mechanisms underlying the promotion of arousal are not fully elucidated. Broberger and McCormick (2005) demonstrated a depolarization of perigeniculate and thalamocortical cells in the lateral geniculate nucleus by TRH and a transformation of perigeniculate neurons from bursting to the tonic, single-spike mode of action potential generation. These actions shift the behavioral state from sleep to waking. However, the wake-promoting potential of TRH involves other structures and neurons, too. The posterior hypothalamus with the histaminergic and orexinergic (hypocretinergic) neurons has a crucial function for the waking state, and TRH provides excitatory drive in this location (Hara et al., 2007; Sergeeva et al., 2007). The histaminergic tuberomamillary nucleus (TMN) plays a prominent role in sleep-waking regulation (Haas and Panula, 2003; Haas et al., 2008). In freely moving animals, histaminergic neurons discharge tonically and specifically during waking (Steininger et al., 1999; Vanni-Mercier et al., 2003; Takahashi et al., 2006). Enhancing histaminergic transmission promotes wakefulness (Lin et al., 1988; Monti et al., 1991). Finally, abolition of histamine synthesis in knock-out mice affects the cortical electroencephalogram (EEG) during all sleep–wake states and causes behavioral deficits, indicating a key role in the maintenance of an awake state, notably in the presence of behavioral challenges (Parmentier et al., 2002). Some histaminergic neurons show TRH immunoreactivity (Airaksinen et al., 1992) and express TRH receptors (Gotoh et al., 2007), whereas release of TRH from neurosecretory nerve endings in the mediobasal hypothalamus is stimulated by histamine through H2 receptors (Charli et al., 1978). The histaminergic system has been made responsible to some extent for the effects of TRH on the regulation of feeding: TRH-induced suppression of feeding after food deprivation was missing in histamine receptor-1 (H1) knock-out mice and in histamine-depleted rats (Gotoh et al., 2007).
The aim of the present study was the elucidation of molecular and electrophysiological actions of TRH on histaminergic neurons and of the role of histamine in TRH-induced arousal. We describe the expression of TRH receptors and demonstrate, that similar to the orexin- and serotonin-mediated excitation (Eriksson et al., 2001a,b), TRH-receptor mediated excitation of TMN neurons demands activation of a Na+/Ca2+ exchanger. Parts of this work have been presented in abstract form (Sergeeva et al., 2007).
Materials and Methods
Slice preparation.
Coronal brain slices from the posterior hypothalamus with a thickness of ∼450 μm (for conventional slice recording) or 350 μm [for recording under differential interference contrast (DIC) microscopy] containing the TMN were prepared from 21- to 28-d-old male Wistar rats and 5- to 10-week-old mice (129/Sv strain). Animal experiments were conducted according to German law and the local guidelines (Bezirksregierung Duesseldorf). All efforts were made to minimize the number of animals and their suffering. The animals were quickly decapitated and the brains transferred to ice-cold modified artificial CSF (ACSF), saturated with carbogen (95% O2/5% CO2), in which NaCl had been replaced by 207 mm sucrose. In this solution, slices were cut with a vibroslicer (Campden Instruments) and placed into ACSF containing the following (in mm): 124 NaCl, 3.7 KCl, 2.0 CaCl2, 1.3 MgSO4, 1.24 NaH2PO4, 25.6 NaHCO3, 10 d-glucose, 0.01% phenol red, bubbled with carbogen (pH 7.4) for at least 1 h at room temperature and then transferred to the recording chamber at 32°C, where they were constantly perfused with the same ACSF at a flow rate of 1–2 ml/min.
Slice electrophysiology.
Extracellular recordings were obtained using glass microelectrodes filled with ACSF (resistance, 4–8 MΩ). According to Ericson et al. (1987), the TMN is subdivided into three subgroups: a diffuse part (neurons are scattered within the lateral hypothalamic area) and two compact (nucleus-like) parts: the ventral TMN (neurons situated at the ventral surface of the brain) and the medial TMN (dense neuronal groups on each side of the mammillary recess of the third ventricle). Neurons were recorded in the ventral, most dense part of TMN, identified under a dissecting microscope. Signals were recorded using an Axoclamp 2B amplifier and a Digidata 1200 interface board (Axon Instruments), filtered between 0.5–10 kHz, sampled at 20 kHz, and analyzed with pClamp8 software (Axon Instruments). The frequency of extracellular action potentials was determined online in bins of 15 s duration. Intracellular recordings from TMN neurons were obtained using sharp glass microelectrodes filled with 3 m KCl (if not mentioned otherwise) with resistances of 80–110 MΩ. Biocytin (Sigma; 1%) was added to the electrode solution. We used the following electrophysiological criteria to identify TMN neurons. They exhibit a regular, spontaneous firing rate (typically 2–6 Hz) and no burst firing at a resting membrane potential of approximately −50 mV, a broad action potential with a Ca2+ shoulder, and a long after-hyperpolarization. Finally, an inward current is activated during a large hyperpolarizing step, and a transient outward K+ current is activated after removing the hyperpolarization. For the voltage-ramp experiments, K+ acetate 4 m intracellular solution was used and CdCl2 (10 μm), d-AP5 (50 μm), CNQX (20 μm), tetrodotoxin (TTX; 1 μm), and bicuculline methiodide (10 μm) were added to the bath solution. Neurons were filled with biocytin at the end of experiments by 200-ms-long anodal (depolarizing) current pulses (frequency 1 Hz) for at least 20 min. Slices were fixed after recording overnight in 4% paraformaldehyde (prepared in 0.1 m PBS, pH 7.4) and cryoprotected in PBS with 20% sucrose, than cryosectioned at 40 μm thickness and mounted on gelatin-coated slides, dried, and stained according to the immunofluorescence staining protocol. The sections were first washed in PBS with 0.25% Triton X-100 (PBS-T) for 5 min and then preincubated with 2% normal goat serum in PBS-T for 30 min at room temperature. This solution was also used to dilute primary guinea pig polyclonal antibody to histidine decarboxylase (HDC; Acris) to 1:600. This antibody was applied to the sections for 12–16 h at 4°C. After washing, sections were incubated with Alexa Fluor 488-labeled goat-anti-guinea pig IgG (1:500; Invitrogen) to reveal HDC immunoreactivity and Texas Red-streptavidin (1:200; Invitrogen) to stain biocytin-filled neurons, for 90 min at room temperature.
Whole-cell patch-clamp recordings of GABAergic spontaneous IPSCs (sIPSCs) were made from ventral TMN neurons in coronal rat brain slices. Cells were visually identified and approached with the help of infrared DIC (IR-DIC). Voltage-clamp (at −70 mV) recordings of sIPSCs were done either at room temperature (22–24°C) or at 32 ± 0.5°C, with a flow rate of 2–2.5 ml/min in the presence of AMPA and NMDA receptor blockers: d-AP5 (100 μm) and DNQX (10 μm), using an EPC9 patch-clamp amplifier (Heka Elektronik). The patch pipette solution contained the following (in mm): 135 KCl, 1 CaCl2, 2 MgCl2, 1 EGTA, 10 HEPES, 2 Na2ATP, and 0.5 Na2GTP (pH 7.2 adjusted with KOH). Histaminergic neurons were identified by presence of the inwardly rectifying current activated by hyperpolarization (Ih) and the transient outward current (IA) (Haas and Reiner, 1988).
Spontaneous IPSCs were analyzed with MiniAnalysis 4.2 (Synaptosoft): Peak amplitude, the 10–90% rise time, τdec (exponential decay time constant in a 100 ms window from the time of peak) and frequency of sIPSCs were calculated. All events were visually inspected before analysis to exclude obvious artifacts. Cumulative interevent intervals, kinetics (τdec), and amplitudes were compared between control (before TRH application) versus a period of recording with TRH using the Kolmogorov–Smirnov two-sample test in every cell. Each of three testing periods lasted 180–360 s. The nonparametrical Wilcoxon test was used for comparison between groups. The significance level was set at p < 0.05.
Single-cell reverse-transcription-PCR.
Whole-cell patch-clamp recordings from dissociated hypothalamic neurons and single-cell reverse-transcription (RT)-PCR procedures were performed as described previously (Sergeeva et al., 2002). Briefly, acutely isolated neurons were prepared from the brains of 22- to 28-d-old male Wistar rats (n = 6) or 21- to 60-d-old male129/Sv mice (n = 4). Transverse slices containing the TMN region were cut and incubated for 1 h in a solution containing the following (mm): 125 NaCl, 3.7 KCl, 1.0 CaCl2, 1.0 MgCl2, 1.3 NaH2PO4, 23 NaHCO3, 10 d-glucose, 0.01% phenol, bubbled with carbogen, pH 7.4. TMN was dissected from the slice and incubated with papain in crude form (0.3–0.5 mg/ml) for 30 min at 37°C. After rinsing, the tissue was placed in a small volume of recording solution with the following composition (in mm): 150 NaCl, 3.7 KCl, 2.0 CaCl2, 2.0 MgCl2, 10 HEPES, pH adjusted to 7.4 with NaOH. Cells were separated by gentle pipetting and placed in the recording chamber, where selected cells were digitally photographed on an inverted microscope. Whole-cell patch-clamp recordings in voltage-clamp mode were used to determine the electrophysiological properties and viability of the neurons, which responded with a sodium current to depolarizing voltage steps. After recording, the cytoplasm of the cell was sucked into the electrode in a stream of sterile control solution. The content of the electrode (8 μl) was expelled into an Eppendorf tube, containing 7 μl of a mixture prepared according to the protocol of the “first strand cDNA synthesis kit” (GE Healthcare). After incubation for 1 h at 37°C, for RT, this reaction was stopped by freezing at −20°C. Care was taken to ensure that the PCR signal arose from the single-cell mRNA. Three negative controls were taken in every experiment (Sergeeva et al., 2002).
Cell identification was performed by HDC–cDNA amplification. For the first amplification round, primer HDC up (5′-GAT GAT GGA GCC C(A/T)G TGA ATA-3′) was used with HDC lo (5′-CTG GTC AGA GGC ATA GGC AAC A-3′) in rats and with mHDC lo (5′-TCA GAG GTG TAG GCA ACG A-3′) in mice. For the second round of amplification in rats, HDC up 2 primer (5′-AGT CCT CTG CAA GAC GCC TC-3′) was taken in combination with HDC lo primer, generating PCR products of 457 bp size. Mouse HDC was amplified with the HDC up primer in combination with HDC lo 2 primer: 5′-GAT GCT GTC CCA GCT GTC G-3′ (expected size of amplimer 193 bp). In mice and rats, cDNAs encoding for the TRH receptors were amplified in the first amplification round with degenerate primers Dg up [5′-TGGCTGC(AG)GG-(AG)CT(GC)CCCAA-3′] and Dg lo [5′-TGGTG(AG)CCTG-CTTCCTGGA-3′]. For the TRH receptor 1 (TRHR1)-specific amplification, primer R1 lo [5′-TGGCTCTGGAAAA(CT)GTGCA(GC)AG-3′] was used in combination with Dg up (amplimer size, 201 bp), and for the TRHR2-specific amplification, R2 up (5′-TGAGAGCACAGACCGTGTGCACTG-3′) and R2 lo [5′-TC(CA)CCAGCAAGGGT(GC)C(AG)ATGAA-3′] primers were used (amplimer size, 219 bp). Randomly selected PCR products obtained after two amplification rounds were purified in water and sequenced. The obtained sequences corresponded to the known one for the rat or mouse (GenBank, accession number): mouse TRHR2 receptor (BC117988), mouse TRHR1 (BC128269), rat TRHR1(M90308), and rat TRHR2 (AB015645).
Thin-walled PCR tubes contained a mixture of first strand cDNA template (1–1.5 μl), 10× PCR buffer, 10 pm each of sense and antisense primer, 200 μm of each deoxyNTP (dNTP) and 2.5 units Taq polymerase. The final reaction volume was adjusted to 10 μl, with nuclease-free water (Promega). The magnesium concentration was 3 mm in all PCRs. The Taq enzyme, PCR buffer, Mg2+ solution, and four dNTPs were all purchased from Qiagen. All oligonucleotides were synthesized by MWG-Biotech. Amplification was performed on a thermal cycler (Mastercycler). A two round amplification strategy was used in each protocol. In each round, 35 cycles of the following thermal programs were used: denaturation at 94°C for 48 s, annealing at 53°C for 48 s, and extension at 72°C for 1 min. For the second amplification round, 1 μl of the product of the first PCR was used as a template. Products were visualized by staining with ethidium bromide and analyzed by electrophoresis in 2% agarose gels.
Real-time RT-PCR analysis of TRH receptor expression in HDC knock-out and wild-type mice.
Total cellular mRNA was isolated from posterior hypothalamic slices using an mRNA isolation kit (GE Healthcare) from 6- to 11-week-old knock-out (KO; n = 4) and wild-type (WT; n = 4) mice according to the manufacturer's protocol. Total mRNA was eluted from the matrix with 200 μl of RNase-free water. For the reverse-transcription, 8 μl of eluted mRNA was added to 7 μl of reagents mixture prepared according to the protocol of the “first strand cDNA synthesis kit” (GE Healthcare). After incubation for 1 h at 37°C, the reverse-transcription reaction was stopped by freezing at −20°C. The reverse-transcription reactions were not normalized to contain the equivalent amounts of total mRNA. The PCR was performed in a PE Biosystems GeneAmp 5700 sequence detection system using the SYBR green master mix kit. Each reaction contained 2.5 μl of the 10× SYBR green buffer, 200 nm dATP, dGTP, and dCTP, 400 nm dUTP, 2 mm MgCl2, 0.25 units of uracil N-glycosylase, 0.625 units of Amplitaq Gold DNA polymerase, 10 pm forward and reverse primers, 5 μl of 1:4 diluted cDNA, and water to 25 μl. All reactions were normalized on β-actin expression, which was amplified with the primers β-actin up (5′-CGT GAA AAG ATG ACC CAG ATC ATG TT-3′; β-actin lo (5′-GCT CAT TGC CGA TAG TGA TGA CCT G-3′.
The reactions were performed in optical tubes capped with MicroAmp optical caps. The reactions were incubated at 50°C for 2 min to activate uracil N′-glycosylase and then for 10 min at 95°C to inactivate the uracil N′-glycosylase and activate the Amplitaq Gold polymerase followed by 40 cycles of 15 s at 95°C, 1 min at 60°C. The PCRs were subjected to a heat dissociation protocol (PE Biosystems; 5700 software). After the final cycle of the PCR, the reactions were heat denaturated over a 35°C temperature gradient at 0.03°C/s from 60 to 95°C. Each PCR product showed a single peak in the denaturation curves. Standard curves for real-time PCR protocols with all primer-pairs obtained with sequential dilutions of one cDNA sample (till 1:128) were found optimal (linear regression coefficients were >0.95). Semiquantitative analysis of TRH receptor expression relative to the β-actin endogenous control was performed according to the “2−ΔΔCt” (ΔFold) method as described previously (Sergeeva et al., 2003a). The nonparametrical Mann–Whitney U test was used for the comparison between averages (6–9 data points for each animal).
Surgery, polygraphic recordings in the mouse, and analysis of sleep–wake parameters.
All experiments followed European Economic Community (86/609/EEC) directives. Histidine decarboxylase knock-out mice were offspring of the mouse strain generated by Ohtsu et al. (2001) and kept on 129Sv genomic background and genotyped by PCR (Parmentier et al., 2002). At the age of 12 weeks and with a body weight of 30 ± 2 g, mice used for EEG and sleep–wake studies were chronically implanted under deep isoflurane anesthesia (2%; 200 ml), with six cortical electrodes (gold-plated tinned copper wire, Ø = 0.4 mm; Filotex) and three neck muscle electrodes (fluorocarbon-coated gold-plated stainless steel wire, Ø = 0.03 mm; Cooner Wire) to record the EEG and electromyogram (EMG) and to monitor the sleep–wake cycle. Finally, the electrode assembly was anchored and fixed to the skull with Super-Bond (Sun Medical) and dental cement. This implantation allows stable and long-lasting polygraphic recordings (Parmentier et al., 2002).
After surgery, the animals were housed individually in barrels placed in an insulated sound-proof recording room maintained at an ambient temperature of 22 ± 1°C and on a 12 h light/dark cycle (lights on at 7:00 A.M.), standard food and water being available ad libitum. After a 7-d-recovery period, mice were habituated to the recording cable for 7 d before polygraphic recordings were started. Cortical EEG (contralateral frontoparietal leads) and EMG signals were amplified, digitized with a resolution of 256 and 128 Hz, respectively, and computed on a Cambridge Electronics Design instrument (CED 1401 Plus). Using a Spike2 script and with the assistance of spectral analysis using the fast Fourier transform, polygraphic records were visually scored by 30 s epochs for wakefulness, slow-wave sleep (SWS), and paradoxical sleep (PS), according to previously described criteria validated for mice (Valatx and Bugat, 1974; Parmentier et al., 2002).
Animals were subjected to sleep–wake recordings after administration of either a vehicle (0.9% NaCl) or montirelin (TRH analog, known as NS3, CG3703) at 1 or 3 mg/kg. Drugs were dissolved in the vehicle, fresh before each administration, and were administered intraperitoneally. All administrations were performed at 10:00 A.M., i.e., during the sleepy period. The order of administration was randomized. An inhibitor of histidine decarboxylase, (S)-α-fluoromethylhistidine, was injected at 7:00 A.M., 3 h before the montirelin injection. Polygraphic recordings were made immediately after administration and maintained during 24 h. Two administrations were separated by a period of 7 d (washout). Each animal served as its own control, and each administration was repeated two times. Statistical analysis was performed with Dunnett's t test and ANOVA for repeated measures. Significance level was set at p < 0.05 or 0.01. Data are presented as mean ± SEM.
Drugs and statistical analysis.
Drugs used in the present study were as follows: R-α-methyl-histamine, gabazine (SR-95531), KB-R7943, nor-binaltorphimine dihydrochloride, d-AP5, and DNQX from Biotrend; (S)-α-fluoromethylhistidine from (Merck Sharp & Dohme); Chlordiazepoxide, benzamil hydrochloride hydrate, tetrodotoxin, and TRH were obtained from Sigma/RBI. Montirelin (CG3703) was a gift from Gruenenthal. Drugs were diluted and stored as recommended. Neurons were recorded for at least 15 min to obtain a stable baseline before perfusion of drugs for 5–10 min in the recording chamber. Statistical analysis was performed with the nonparametrical Mann–Whitney U test. Significance level was set at p < 0.05. Data are presented as mean ± SEM.
Results
Extracellular firing rate of TMN neurons is increased by TRH
Several criteria were used to identify TMN neurons in the present study. The recordings were performed in the ventral part of the TMN, a region where histaminergic neurons are encountered almost exclusively when recording intracellularly with sharp electrodes (Eriksson et al., 2001a). For extracellular recording, ventral TMN neurons were selected on the basis of their location, regular firing in the range of 1–8 Hz [most typically 2–4 Hz and a broad triphasic action potential (2–4 ms)] (Sergeeva et al., 2006). In addition, pharmacological identification was used as the most reliable criterion: inhibition by the H3-receptor agonist R-α-methyl-histamine.
In mouse slices, 6 of 8 TMN neurons (75%) responded to TRH (1.5 μm) with an enhancement of firing rate to190 ± 61% of the control level. In rat slices, 10 of 14 cells (71%) were excited by TRH (2 or 10 μm; there was no difference in the response amplitude between these two concentrations) to 262 ± 66% of control (Fig. 1A–C). The effect was followed by a fast desensitization in the presence of TRH. A second TRH response was not observed in the same neuron within 1 h (n = 4; data not shown).
Figure 1.

TRH enhances firing rate of TMN neurons. A, B, Averaged ratemeter recordings of responses to bath applied TRH in mouse (A; 1.5 μm TRH, n = 6) and rat (B; 2–10 μm TRH, n = 10) TMN neurons. Four nonresponding cells are not included. C, Extracellular action potential recordings in rat TMN neuron: regular firing, excitation by TRH 2 μm, and recovery after 15 min. Averaged extracellular triphasic action potentials (80–150 APs for each), reduction of amplitude during TRH indicating depolarization. D, Chlordiazepoxide, a TRH receptor antagonist (CDZ; open bar indicates period of bath application), reduces the TRH response in rat TMN neurons at 10 μm (n = 4) and abolishes it at 50 μm (n = 6). E, TRH applied in the presence of benzamil-enhanced firing rate in a significantly smaller fraction of TMN neurons to a significantly smaller extent (n = 10) compared with the control group (n = 14; top trace).
The TRH effect is antagonized by chlordiazepoxide
CDZ (10 μm), a benzodiazepine also known as an antagonist at TRH receptors, attenuated the effect of TRH in rat TMN neurons; the enhancement of firing reached 215 ± 51% (n = 14) in control and 144 ± 35% in the presence of CDZ (p = 0.19; Mann–Whitney U test; n = 4). At 50 μm CDZ, a concentration used also in previous studies for the blockade of TRH receptors (Deng et al., 2006), the TRH effect was abolished (n = 6; p = 0.0005; Mann–Whitney U test) (Fig. 1D). Interestingly, CDZ (50 μm) significantly reduced the firing rate of TMN neurons by 20 ± 5% (n = 6; p = 0.065) on its own. In acutely isolated mouse TMN neurons, CDZ potentiated GABA-evoked whole-cell currents with an EC50 of 0.7–1 μm and maximally at 10 μm (no further potentiation at 50 μm). Therefore, the observed inhibition of firing cannot be attributed to the modulatory action of CDZ at GABAA receptors. Moreover, GABAA receptors do not exhibit a tonic influence on TMN firing in slices, as bicuculline (50 μm; n = 4) and gabazine (up to 100 μm; n = 6) did not change the firing rate of TMN neurons. This indicates either an endogenous tone of TRH or the presence of constitutively active TRH receptors (Heinflink et al., 1995; Jinsi-Parimoo and Gershengorn, 1997).
Inhibiton of Na+/Ca2+ exchange by benzamil blocks the TRH effect
In the presence of benzamil (20 μm), TRH increased the firing rate in two TMN neurons recorded in rat slices and did not affect firing frequency in eight neurons (Fig. 1E). The difference to the occurrence of the excitatory TRH effect under control conditions (10 neurons of 14 tested) was significant (p = 0.04; Fisher's exact probability test). On the average, in the presence of benzamil, TRH increased the firing frequency to 134.1 ± 29.4% of control (n = 10), to a significantly lower level (p = 0.036) compared with the control condition (215 ± 51% of control; n = 14).
Transcriptional analysis of TRH receptor expression
Mouse and rat slices prepared in the same way as for electrophysiological recordings were used for the acute isolation of histaminergic TMN neurons. After whole-cell voltage-clamp recordings of voltage-dependent sodium currents, TMN neurons were subjected to single-cell RT-PCR (Sergeeva et al., 2003b). Among 26 mouse TMN neurons (positive for the histamine-producing enzyme HDC), nine cells expressed TRH R2 (35%), six cells TRH R1 (23%), five cells (19%) contained mRNAs for both receptor types, and six cells (23%) were TRHR-negative (Fig. 2). Among 19 rat TMN neurons, four cells were negative for TRH receptors (21%), eight cells (42%) expressed TRH R1, one cell (5%) only TRH R2, and six cells both receptors (32%). The TRH R2 was less frequently detected in rats than in mice: 37% versus 54%, respectively, whereas the occurrence of TRH R1 transcripts was opposite: 74% of neurons in rats versus 42% in mice were found to be TRH R1-positive.
Figure 2.

Expression of TRH receptors in rat and mouse TMN neurons. A, B, Single-cell RT-PCR analysis of TRH receptor expression in five mouse (A) and five rat (B) TMN neurons (phase-contrast light microscope photographs of corresponding cells are given). Pc, Positive control: posterior hypothalamus; nc, negative control; M-DNA size marker: 100 bp ladder (500 bp, most intense line). C, Summary of single-cell RT-PCR analysis done in 26 mouse and 19 rat TMN neurons positive for the histamine synthesizing enzyme HDC.
Thus, the transcriptional analysis of TRH receptor expression revealed that TRH R-negative cells represent the same small population among histaminergic neurons (ca 20%) as the TMN neurons not responding to TRH (21–29%) in electrophysiological experiments (see above).
Intracellular recordings in rat slices
Stable recordings were obtained from 32 neurons with electrophysiological characteristics of TMN neurons (Haas and Reiner, 1988). Recorded in current-clamp mode, they exhibited spontaneous firing at 3.8 ± 0.2 Hz (n = 29) and a typical Ih sag of 78 ± 5%, measured as the percentage reduction from the peak at the end of an 0.5 s, 400 pA current injection. Under tetrodotoxin, their resting membrane potential was −52.5 ± 0.8 mV (n = 25). Bath application of TRH (1.5 μm) rapidly depolarized the TMN neurons and increased their firing rate; this effect recovered completely 20–40 min after TRH withdrawal (Fig. 3A). During a washout period of 1 h, repeated application of TRH caused neither depolarization nor an increase in firing rate. In the presence of tetrodotoxin, which prevents firing and causes synaptic isolation, a depolarization by 15.3 ± 2.4 mV was detected in four cells (out of six tested) (Fig. 3B). No obvious change in the input resistance was observed. The following experiments were designed to elucidate the mechanism of the depolarization by TRH (summarized in Fig. 3C). Only the cells which responded to TRH were used (21 of 28). First, the effect of cations that can block potassium conductances were tested. The effect of TRH was not inhibited by 500 μm BaCl2 (n = 5) (Fig. 3C) or by 3 mm CsCl (n = 3). The selective blocker of the Na+/Ca2+ exchanger (NCX), KB-R7943 (Iwamoto et al., 1996) at 80 μm, strongly suppressed the depolarization (p < 0.01), but a residual 2–6 mV depolarization remained in five responding cells. Voltage–current curves obtained before and during 1.5 μm TRH intersected at −4.3 ± 6.3 mV (n = 4) (Fig. 3D), a potential close to the predicted one for a mixed cationic conductance. Electrophysiological identification of TMN neurons was confirmed post hoc by the colocalization of biocytin (delivered to the cell through the recording electrode) with HDC immunoreactivity (Fig. 4A).
Figure 3.

Intracellular sharp-electrode recordings from rat TMN neurons. A, Depolarization and increased firing in the presence of TRH. After washout inhibition by an H3-receptor agonist (R-α-methylhistamine). B, During 1 μm TTX, TRH depolarizes the neuron, indicating a postsynaptic site of action. During maximal depolarization and manual clamping to the resting potential, no obvious change in input resistance is seen. C, Depolarization by TRH is not significantly affected by 500 μm BaCl2 and 3 mm CsCl but markedly reduced by 80 μm KB-R7943. **p < 0.01, Student's two-tailed t test. Right, Average depolarizations (mean ± SE) under each condition in 1 μm TTX, for responding cells; numbers of cells are near bars. D, The voltage–current plots recorded in the same neuron before and during TRH treatment. The curves intersect at −4.3 ± 6.3 mV, close to the reversal potential predicted for a mixed cationic conductance.
Figure 4.
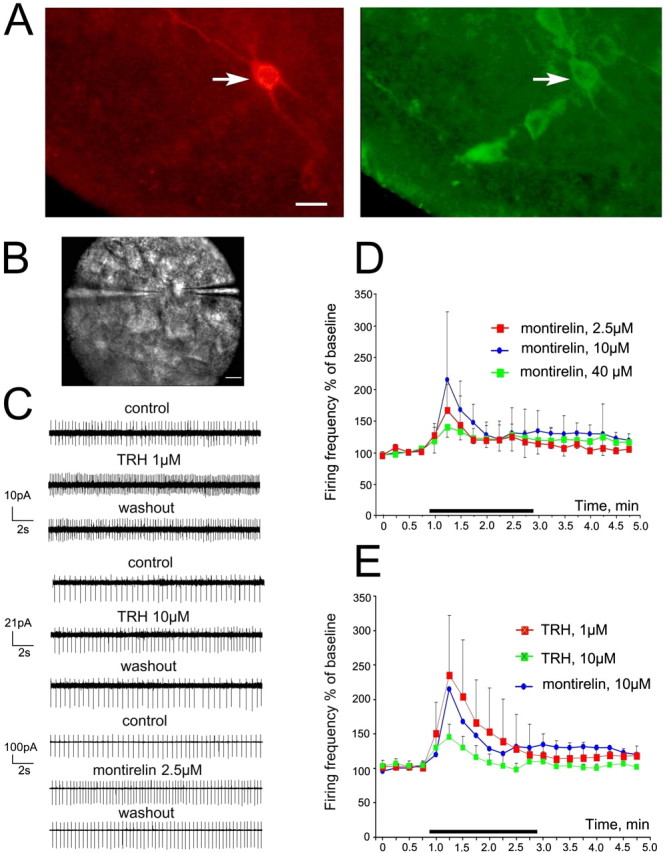
Ventrolateral TMN neurons respond to TRH and montirelin in a dose-dependent manner. A, TMN neuron stained with both biocytin (red) and HDC (green). Scale bar, 25 μm. B, Photograph of TMN neuron in slice approached with a recording patch pipette (right) and puff pipette filled with TRH (left); scale bar, 20 μm. C, Examples of TMN neuronal firing in control and in the presence of TRH or montirelin. D, Time course diagram illustrating change in averaged (±SE) firing frequency of TMN neurons in the presence of different concentrations of montirelin: n = 5, 4, and 4 cells tested for montirelin 2.5, 10, and 40 μm, respectively. E, Averaged time course diagrams for TRH 1 μm (n = 5) and 10 μm (n = 6).
Although we tried to use maximal concentrations of TRH known to be within 0.5–10 μm (Deng et al., 2006) in our initial sharp electrode recordings, 10 μm of TRH caused suppression of firing and irreversible depolarization in some of TMN neurons. Therefore, we selected for all aforementioned experiments a TRH concentration of 1.5 μm. Construction of complete dose–response curve for TRH was not possible for the following reasons: first, TRH could be applied only once to a single slice, and different cells recorded from different slices showed considerable variation in the TRH response magnitude; second, the number of responding cells was <50% with concentrations of TRH ≤1 μm. The TRH-degrading enzyme is strongly expressed in the hypothalamus (Heuer et al., 2000), making the real concentration at the recording site (within 450-μm-thick slice) after bath delivery of TRH unpredictable in each single case. Therefore, we performed in the following experiments local application of TRH receptor agonists (through a patch pipette with a pressure pulse) to the TMN neurons recorded under visual control (IR-DIC microscopy) in voltage-clamp (holding potential 0 mV) cell-attached mode (Fig. 4B–E). Cells were visually identified within the compact ventral TMN part and were inhibited by R-α-methyl-histamine (200 nm). With local application, 1 μm TRH enhanced the firing frequency of TMN neurons to the same extent (to 236 ± 39% of control; n = 5) (Fig. 4E) as 2–10 μm TRH with bath perfusion (to 262 ± 66% of control; n = 10; p = 0.58) (Fig. 1). The response to local application of 10 μm was significantly smaller than that to 1 μm (34%; p = 0.002) (Fig. 4C,E), in accordance with the bell-shaped dose–response curve obtained by Deng et al. (2006), where 0.5 and 1 μm evoked maximal responses, whereas responses to 10 μm TRH reached only ∼30% of the maximum. Montirelin at 10 μm mimicked the increase in firing rate caused by TRH 1 μm (Fig. 4D); however, because of the large heterogeneity of the montirelin response amplitude, the difference between three tested concentrations (2.5, 10, and 40 μm) was not significant.
Frequency of sIPSCs is differently modulated by TRH in different cells
In our initial experiments, sIPSCs were recorded from TMN neurons, identified by the presence of Ih and IA currents (Fig. 5A) in rat coronal slices at room temperature. Surprisingly, they were either significantly increased (n = 2) or decreased (n = 5) in frequency after bath application of TRH (2.5 μm) for 5 min. The amplitude was not changed significantly under TRH, indicating a presynaptic effect. After TRH washout (5–10 min), the GABAA receptor antagonist gabazine (10 μm) abolished sIPSCs in all cells (n = 7) (Fig. 5B) in accordance with our previous studies (Sergeeva et al., 2002; Eriksson et al., 2004), showing that spontaneous synaptic currents recorded from somata of TMN neurons are exclusively carried through the postsynaptic GABAA receptors. No postsynaptic inward currents in response to TRH were seen in these experiments, indicating a high probability that Na+/Ca2+ exchange is involved in TRH-induced excitation, as this transport is functionally inactive at room temperature in TMN neurons (Eriksson et al., 2001b).
Figure 5.

Whole-cell voltage-clamp sIPSC recordings from TMN neurons in rat thin slices. A, TMN neuron approached with a patch pipette (scale bar, 20 μm) and identified with the stimulation protocol shown at right. Hyperpolarization-activated inward current (Ih) becomes obvious after voltage jumps from −50 to −90 mV and to more negative values. Maximal outward IA current is seen after return to the holding membrane potential from −120 mV. B, TRH (2.5 μm)-induced depression of sIPSC frequency (at room temperature). The GABAA receptor antagonist gabazine (10 μm) blocks spontaneous synaptic activity. C, AMPA and NMDA receptor antagonists (DNQX and d-AP5, respectively) reduce frequency of sIPSCs without affecting their kinetics or amplitude.
As several factors could contribute to the heterogeneous responses to TRH of GABAergic cells, we performed all following experiments in the presence of AMPA/NMDA receptor blockers and at 32°C. A neurochemical study revealed recently that TMN neurons are under tonic NMDA-receptor-mediated inhibition (Faucard et al., 2006), as NMDA receptor antagonists increase TMN histamine production and its release in different brain regions. As inhibitory NMDA receptors do not exist, these findings are explained by the tonic activation of inhibitory GABAergic cells projecting to the TMN through these receptors.
In the presence of AMPA/NMDA receptor antagonists, the sIPSC frequency was significantly reduced in all investigated neurons (n = 13), from 1.07 ± 0.25 Hz to 0.57 ± 0.12 Hz (p < 0.0001), whereas amplitudes (45 ± 5.8 pA in control vs 38 ± 4.1 pA in DNQX/d-AP5) and decay kinetics (14.2 ± 0.9 ms vs 14 ± 0.72 ms) were not significantly different (Fig. 5C), indicating a presynaptic site of action. The extent of frequency and amplitude modulation of sIPSCs by AMPA/NMDA receptor antagonists did not correlate with the, again, diverse effects of TRH: in six neurons, TRH caused increases (group 1) in sIPSC frequency from 0.85 ± 0.27 Hz to 1.37 ± 0.49 Hz; in four cells (group 2), decreases from 0.61 ± 0.27 Hz to 0.43 ± 0.18 Hz; and in three cells (group 3), the frequency was not significantly changed (0.87 ± 0.29 Hz). There was no difference in the control sIPSC frequencies between the three groups. The reduction of sIPSC frequencies obtained before TRH application with DNQX/d-AP5 were to 62 ± 6.2% (n = 6), 55.3 ± 18.7% (n = 4), and 72.9 ± 11.6% (n = 3) of control in the first, second, and third cellular group, respectively (difference between groups was not significant). Postsynaptic inward currents (24.9 ± 3.38 pA) during TRH-perfusion were seen in 11 of 17 cells (65%); however, these responses were found in neurons belonging to all three groups (Fig. 6A), indicating that postsynaptic and presynaptic effects of TRH are not coherent, at least in a slice preparation in vitro, creating large scale neuronal heterogeneity on the output of the histaminergic system.
Figure 6.

TRH induces inward current and modulates frequency of sIPSCs in TMN neurons recorded in whole-cell voltage-clamp at 32°C. A, TRH-mediated inward current in TMN neuron where frequency of sIPSCs was increased. Summary diagram at right shows three neuronal groups: no change, decrease, and increase in sIPSC frequency under TRH (control 100%). Dark gray bars show percentage frequency changes in neurons, where no direct postsynaptic currents in response to TRH were recorded. Light gray bars show percentage frequency changes in cells where TRH-evoked currents measured; averages of these currents are given as black bars at the right side of the box. B, Neuron displaying an increased sIPSC frequency in response to TRH and summary of frequency changes in all cells belonging to the same group at right. co, Control; wa, washout. C, Neuron displaying decreased sIPSC frequency under TRH. As no difference in sIPSC occurrence (suppression or enhancement) by TRH was obtained with Fisher's exact probability test (p = 0.36) between neurons recorded at room or more physiological temperature, all recordings were pooled. Cells with control sIPSC frequency <0.1 Hz were not included in summary diagrams.
TTX (1 μm) abolished INa+ in TMN neurons (evoked by a +40 mV, 200 ms depolarizing step from the holding potential of −50 mV) and significantly reduced the frequency of spontaneous IPSCs in all recorded cells from 1.28 to 0.62 Hz (n = 12). In the presence of TTX, a reduction of frequency of mIPSCs by TRH was never observed, and in the majority of recorded neurons (n = 8), the frequency did not change. However, in four cells, TRH significantly increased the frequency of mIPSCs (Kolmogorov–Smirnov test) from 0.55 ± 0.13 Hz to 0.86 ± 0.23 Hz (Fig. 7A), indicating that TRH enhanced action-potential independent release of GABA at some synapses either directly through presynaptic TRH receptors or indirectly, through interaction with neuronal postsynaptic or glial receptors triggering the release of some yet unknown transmitter. The occurrence of facilitation (4 of 12 cells) in experiments with TTX did not differ significantly (Fisher's exact probability test) from the control experiments performed either at room temperature (2 of 7 cells; p = 0.33) or at 32°C (6 of 13 cells; p = 0.26), indicating that in all cases, the same mechanism (TTX-independent GABA release) is recruited by TRH.
Figure 7.

Unimodal presynaptic modulation of spontaneous IPSCs by TRH is seen either in the presence of TTX or the antagonist at the κ-opioid receptor, nor-binaltorphimine (1 μm each). A, Neuron with an increased miniature IPSC frequency in response to TRH and summary of frequency changes in all cells recorded in the presence of TTX (at right). Kolmogorov–Smirnov Z (K–S Z) and p values are given on corresponding cumulative fraction histograms, illustrating no change in amplitude but increase of frequency (decrease in interevent intervals) in the presence of TRH. B, Neuron with a decreased sIPSC frequency in response to TRH in the presence of nor-binaltorphimine and summary of frequency changes in all cells belonging to the same group (at right). co, Control; wa, washout.
Can the reduction of sIPSC frequency under TRH be attributed to the action-potential-mediated release of endogenous dynorphin, which suppresses GABA release in TMN at the presynaptic site (Eriksson et al., 2004)? We addressed this question in experiments where TRH was applied in the presence of the kappa (κ)-opioid receptor antagonist nor-binaltorphimine: in two of six investigated neurons, TRH decreased the sIPSC frequency from 2.1 ± 0.76 Hz to 1.2 ± 0.25 Hz; in four cells, the frequency of sIPSCs was not affected by TRH. Facilitation of sIPSC frequency by TRH was not seen in these experiments (Fig. 7B).
Montirelin (CG3703) induces waking in mice
During the lights on period, montirelin (TRH analog) markedly enhanced wakefulness (Fig. 8) at the expense of slow-wave sleep and paradoxical sleep in wild-type mice (HDC+/+) in a dose-dependent manner. Our in vitro experiments (local application) showed no difference between responses of TMN neurons to 2.5 and 10 μm of montirelin; these concentrations correspond to ∼1 mg/kg and 3 mg/kg doses administered in vivo. The larger behavioral response to 3 mg/kg dose of montirelin can be explained by the higher occupancy of brain TRH Rs at this concentration, which was shown for the rat after intravenous injections of different montirelin concentrations by Urayama et al. (2001). After montirelin injection in our study, HDC+/+ mice demonstrated a suppression of cortical slow activity (δ + θ ranges) and spindles (8–14 Hz), which resulted in a state of total cortical activation, i.e., low-voltage electrical activity with dominant waves in the β and γ bands (20–60 Hz). These effects on the cortical EEG were manifested on polygraphic scoring as an almost total waking state, characterized by significantly delayed sleep latency (Fig. 8) during more than 2 h with 3 mg/kg. In HDC−/− mice, the same injections of montirelin had no significant effect on the wake duration compared with saline injection of the same animals.
Figure 8.

The TRH analog montirelin lacks waking effect in histamine deficient mice (HDC−/−). A, Waking effect of montirelin (1 mg/kg, i.p.) in wild-type mice (HDC+/+) and its absence in HDC−/− mice (EEG, EMG, hypnogram). B, Hourly cumulative values of waking during 1 h before and 4 h after the injection of vehicle (NaCl; no symbols), montirelin at 1 mg/kg (open symbols), or 3 mg/kg (filled symbols) in HDC+/+ and HDC−/− mice. C, Latencies to SWS and PS after compound injection (n = 8 and 6 in 4 and 3 animals for HDC+/+ and HDC−/− mice, respectively). Note the significant effect induced by montirelin in HDC+/+ mice and its absence in HDC−/− mice [a, b: montirelin vs NaCl in the same group, p < 0.01, 0.05, Dunnett's t test; c, d: p < 0.05 HDC+/+ vs HDC−/−, montirelin 1 mg/kg plus α-FMH ((S)-α-fluoromethylhistidine) 60 mg/kg vs montirelin 1 mg/kg, two-tailed t test after ANOVA for repeated measures].
In HDC−/− mice, 3 mg/kg montirelin increased sleep latency compared with the saline control; however, this increase represented only 23% of that seen in WT (HDC+/+) (Fig. 8). As HDC−/− mice have perturbed wakefulness and a straightforward conclusion about histamine's role in TRH-mediated arousal may suffer from compensatory changes in other aminergic systems of the brain or other transmitters in TMN neurons in these mice, we performed experiments with the acute depletion of TMN neurons from histamine by the intraperitoneal injection of (S)-α-fluoromethylhistidine (Parmentier et al., 2002) 3 h before the montirelin injection in wild-type mice. This depletion blocked the montirelin-mediated increase in waking (Fig. 8).
TRH receptor-expression in HDC knock-out mice
We investigated the relative abundance of mRNAs encoding for the TRH receptors with the help of semiquantitative real-time RT-PCR. All data points in each amplification were normalized on the probe WT#1 (showing lowest receptor expression). No significant difference between HDC KO and WT mice was obtained: the levels of mRNA represented, for TRH R1, 2.8 ± 0.44 versus 2.39 ± 0.5 (p = 0.47) and for TRH R2, 1.38 ± 0.4 versus 0.53 ± 0.26 (p = 0.11), respectively.
Discussion
The present study elucidates mechanisms of TRH receptor-mediated responses and expression of TRHRs in individual posterior hypothalamic histaminergic TMN neurons. We show a direct depolarization of most histaminergic cells (ca 70%) by maximal concentrations of TRH through the activation of electrogenic Na+/Ca2+ exchange. This depolarization was not affected by cesium or barium, indicating that block of a K+ conductance is not involved in the TRH-mediated excitation of TMN neurons. This effect reversed at the potential typical for nonselective cation channels. The depolarization was not accompanied by an obvious change in membrane resistance. One possible explanation is the origin of the response on remote dendrites, whereas the membrane resistance is mainly measured from the soma.
In several previous studies on mechanisms of the TRH action in the nervous system, inhibition of a resting K+ conductance was suggested as a major mechanism (Bayliss et al., 1992; Deng et al., 2006). However, immature motoneurons displayed activation of a nonselective cationic conductance by TRH and lack of interaction with potassium channels (Bayliss et al., 1994); moreover, their depolarization by TRH was not accompanied by a change in input resistance. This “immature” type of the response, partially explored by Bayliss et al. (1992, 1994), bears similarity with the TRH responses in TMN neurons, which were, however, recorded in young adult rodents.
Single-cell RT-PCR revealed expression of one or both TRH receptors in most TMN neurons, whereas a quarter of them lacked detectable amount of encoding for the TRH receptors mRNAs. The expression of these receptors corresponded to the ratio (70–75%) of TMN neurons responding to TRH in electrophysiological recordings. However, as we did not perform single-cell RT-PCR from the same neurons from which we recorded, we cannot say for sure whether the lacking (or low) receptor expression in some cells or receptor internalization limits neuronal excitability by TRH. Previous in situ hybridization studies demonstrated that TRH R1 is predominantly distributed in hypothalamic areas (Heuer et al., 2000), whereas TRHR2 seems to be widely distributed throughout the brain (Heuer et al., 2000; O'Dowd et al., 2000). An immunohistochemical analysis by Gotoh et al. (2007) suggested that TRHR2, and to a lower extent TRHR1, are expressed in the histaminergic TMN neurons of rats. These data, however, should be considered with care, as the polyclonal antibodies (obtained from Santa Cruz) were not tested for specificity in TRHR KO mice. What is the functional meaning of the TMN neuron heterogeneity with respect to the expression of TRH receptors? TRHR1- and TRHR2-expressing cells may belong to different functional systems (feeding/neuroendocrine vs arousal and cognition), as these receptors are expressed in distinct pathways (Heuer et al., 2000). However, because of the lack of specific pharmacological tools targeting one or the other receptor, the functional meaning of the heterogeneous expression of TRHRs in TMN neurons remains, at present, unclear.
The benzodiazepine CDZ is a competitive TRHR antagonist, which dose dependently attenuated and blocked the TRH effect in TMN neurons. Interestingly, higher doses of CDZ produce a decrease of TMN neuron's spontaneous firing. This effect can occur because of a decrease of spontaneous TRH tone on histaminergic cells or attributable to lowering constitutive activity of TRHRs (Heinflink et al., 1995; Jinsi-Parimoo and Gershengorn, 1997). High rates of turnover of TRHRs have been described in various cell types (Ashworth et al., 1995; Drmota et al., 1998; O'Dowd et al., 2000). Therefore, rapid agonist-induced internalization of TRHRs may be responsible for the fast desensitization kinetics observed in the presence of TRH in our experiments and our failure to get a second response to TRH in the same slice. At maximal concentrations (2–10 μm in bath perfusion or 1 μm in local application experiments), TRH caused depolarization or enhanced firing rate in ∼70% of all recorded TMN neurons. Responses to 2 and 10 μm of TRH (bath perfusion) were statistically indistinguishable from each other and from 1 μm concentration used for the local application; therefore, we assume that the real acting concentration was ∼1 μm in all our experiments.
Experiments with two NCX inhibitors, benzamil and KB-R7943, indicated that TMN neurons are excited via NCX activation that causes depolarization. The presence of a residual depolarization after NCX blockade in some sharp electrode experiments, and only weak block of TRH-induced increase of firing in a small fraction of extracellularly recorded neurons, may be explained by NCX antagonists not reaching their maximal effective concentrations at the depth of recording site in some slices. This is in keeping with the fact that TRH-evoked inward currents could not be observed at room temperature. The majority of studies using in vitro slice recordings are done, like the present one, at 32°C, many at room temperature. Considering that the physiological temperature in rodents may vary between 35.9 and 39°C (Harkness and Wagner, 1989; Kiyatkin and Mitchum, 2003) the in vivo NCX activation and TRH effects are likely even larger than in the in vitro situation. Although benzamil blocks not only NCX but also the Na+/H+ exchanger, KB-R7943 was long considered a highly specific antagonist of NCX (Iwamoto et al., 1996). Recent data revealing its inhibitory action toward nonselective cation channels (TRPC family) challenged this opinion. Rosker et al. (2004) explain this inhibitory action through the direct interaction between the cytosolic C terminus of the heterologously expressed TRPC3 channel and the endogenous NCX1, indicating that intact Na+/Ca 2+ exchange must be essential for the proper TRPC channel function.
The NCX is electrogenic, with an exchange ratio of three Na+ in for every Ca2+ that is pushed out and is expressed throughout the brain (Quednau et al., 1997). We have shown that serotonin and orexin peptides induce depolarization of TMN neurons by activation of the NCX (Eriksson et al., 2001a,b). In the same way, H1-receptor-mediated depolarization of rat vasopressin neurons in the supraoptic nucleus occurs through the activation of NCX (Smith and Armstrong, 1996). These receptors are all coupled to phospholipase C, and this is also true for TRHRs (Gershengorn and Osman, 1996). The activation of NCX is likely secondary to a surge in the intracellular Ca2+ concentration, released from the intracellular stores; as during activation of the receptors coupled to inositol 1,4,5-trisphosphate production, no obvious Ca2+-channel component was seen after NCX activation in previous studies (Smith and Armstrong, 1996; Eriksson et al., 2001a,b).
In contrast to the hippocampus, where TRH enhances the frequency of sIPSCs in CA1 pyramidal neurons (Atzori and Nistri, 1996; Deng et al., 2006), TRH bidirectionally modulates the frequency of sIPSCs in TMN neurons. This bidirectional nature of TRH modulation is preserved under room temperature and AMPA/NMDA receptor antagonists. Interestingly, TTX-insensitive GABA release is facilitated by TRH, whereas TTX-sensitive (action potential-dependent release) is inhibited. One of the possible explanations for the TRH-mediated suppression of GABA release would be depolarization block of firing (inactivation of Nav channels) because of the strong and long-lasting neuronal depolarization of GABAergic neurons or their axons. Depolarization block of firing was seen in some of our sharp electrode recordings from TMN neurons (10 μm of TRH); we assume it can also occur in the GABAergic neurons projecting to the TMN. Another possibility would be the action-potential-dependent release of some inhibitory neurotransmitter (modulator) acting presynaptically. One possible candidate for such an action is dynorphin. VLPO (ventrolateral preoptic) neurons (the major GABAergic input to the TMN) express both μ- and κ-opioid receptors (Mitchell et al., 1997), and agonists of both receptors can suppress sIPSCs in TMN neurons (Eriksson et al., 2004). Interestingly, dynorphin (κ-opioid receptor agonist)-positive fibers are found at high density in the TMN region (Lantos et al., 1995), and dynorphin expression is restricted to the orexin neurons in the lateral hypothalamic area (Chou et al., 2001). Orexinergic neurons are excited by TRH (Hara et al., 2007) and may corelease dynorphin and orexin. Whereas orexin enhances the frequency of sIPSCs recorded from TMN neurons, dynorphin inhibits it; when both peptides are coapplied, the effect of dynorphin dominates (Eriksson et al., 2001a). Presynaptic κ-opioid receptors inhibit transmitter release from a variety of neurons activating voltage-dependent K+ channels (Schlicker and Kathmann, 2008). Our experiments with the κ-opioid receptor antagonist nor-binaltorphimine indicated that endogenous dynorphin is unlikely responsible for the TRH-mediated inhibition of presynaptic GABA release, as such inhibition was observed in two of six neurons exposed to nor-binaltorphimine. Interestingly, facilitation of GABA release by TRH was not observed in these experiments, indicating that facilitation by TRH may demand preconditioning by dynorphin (hyperpolarization of presynaptic terminals).
Does TRH receptor deficiency affect wakefulness, controlled by histamine? TRHR1 knock-out mice display a central hypothyroidism (plasma T4 and T3 levels are reduced by a factor of three) (Rabeler et al., 2004) with growth retardation, similar to the clinical manifestation of central hypothyroidism seen in a patient with an inactivating mutation in TRH R1 gene (Collu et al., 1997). As hypothyroidism in general is accompanied by fatigue and daytime sleepiness, the few studies on central hypothyroidism did not detect cognitive deficits (Collu et al., 1997) or changes in sleep–wake cycle (Zeng et al., 2007) during TRH R1 deletion, indicating that the arousal effects of TRH may be compensated by other transmitter systems, for example, by histamine (present study) or orexin (Hara et al., 2007; Gonzalez et al., 2009). Conditional cell-type-specific TRH R KO mice will be necessary to determine the role of TRH in the regulation of waking and attention.
The TRH analog montirelin induces robust waking in wild-type mice but not in mice lacking histamine synthesis or under acute disruption of histamine synthesis. TRH receptor expression in the posterior hypothalamus does not differ significantly between WT and KO mice. Therefore, the difference in the behavioral response can be attributed to the lack of histamine. In conclusion, TRH can control arousal through excitation of TMN neurons ascribing an important role in this action to the brain histaminergic system.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Grants SE 1767, SFB 575/3, and SFB 575/8, and an Inserm grant to J.-S.L. and R.P. We thank Dr. Hiroshi Ohtsu for the generous donation of HDC knock-out mice. We are grateful to Annette Scherer and Claudia Wittrock for excellent technical assistance.
References
- Airaksinen MS, Alanen S, Szabat E, Visser TJ, Panula P. Multiple neurotransmitters in the tuberomammillary nucleus: comparison of rat, mouse, and guinea pig. J Comp Neurol. 1992;323:103–116. doi: 10.1002/cne.903230109. [DOI] [PubMed] [Google Scholar]
- Ashworth R, Yu R, Nelson EJ, Dermer S, Gershengorn MC, Hinkle PM. Visualization of the thyrotropin-releasing hormone receptor and its ligand during endocytosis and recycling. Proc Natl Acad Sci U S A. 1995;92:512–516. doi: 10.1073/pnas.92.2.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atzori M, Nistri A. Effects of thyrotropin-releasing hormone on GABAergic synaptic transmission of the rat hippocampus. Eur J Neurosci. 1996;8:1299–1305. doi: 10.1111/j.1460-9568.1996.tb01298.x. [DOI] [PubMed] [Google Scholar]
- Bayliss DA, Viana F, Berger AJ. Mechanisms underlying excitatory effects of thyrotropin-releasing hormone on rat hypoglossal motoneurons in vitro. J Neurophysiol. 1992;68:1733–1745. doi: 10.1152/jn.1992.68.5.1733. [DOI] [PubMed] [Google Scholar]
- Bayliss DA, Viana F, Kanter RK, Szymeczek-Seay CL, Berger AJ, Millhorn DE. Early postnatal development of thyrotropin-releasing hormone (TRH) expression, TRH receptor binding, and TRH responses in neurons of rat brainstem. J Neurosci. 1994;14:821–833. doi: 10.1523/JNEUROSCI.14-02-00821.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broberger C, McCormick DA. Excitatory effects of thyrotropin-releasing hormone in the thalamus. J Neurosci. 2005;25:1664–1673. doi: 10.1523/JNEUROSCI.3198-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charli JL, Joseph-Bravo P, Palacios JM, Kordon C. Histamine-induced release of thyrotropin releasing hormone from hypothalamic slices. Eur J Pharmacol. 1978;52:401–403. doi: 10.1016/0014-2999(78)90298-4. [DOI] [PubMed] [Google Scholar]
- Chou TC, Lee CE, Lu J, Elmquist JK, Hara J, Willie JT, Beuckmann CT, Chemelli RM, Sakurai T, Yanagisawa M, Saper CB, Scammell TE. Orexin (hypocretin) neurons contain dynorphin. J Neurosci. 2001;21:RC168. doi: 10.1523/JNEUROSCI.21-19-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collu R, Tang J, Castagné J, Lagacé G, Masson N, Huot C, Deal C, Delvin E, Faccenda E, Eidne KA, Van Vliet G. A novel mechanism for isolated central hypothyroidism: inactivating mutations in the thyrotropin-releasing hormone receptor gene. J Clin Endocrinol Metab. 1997;82:1561–1565. doi: 10.1210/jcem.82.5.3918. [DOI] [PubMed] [Google Scholar]
- Deng PY, Porter JE, Shin HS, Lei S. Thyrotropin-releasing hormone increases GABA release in rat hippocampus. J Physiol. 2006;577:497–511. doi: 10.1113/jphysiol.2006.118141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drmota T, Gould GW, Milligan G. Real time visualization of agonist-mediated redistribution and internalization of a green fluorescent protein-tagged form of the thyrotropin-releasing hormone receptor. J Biol Chem. 1998;273:24000–24008. doi: 10.1074/jbc.273.37.24000. [DOI] [PubMed] [Google Scholar]
- Ericson H, Watanabe T, Köhler C. Morphological analysis of the tuberomammillary nucleus in the rat brain: delineation of subgroups with antibody against L-histidine decarboxylase as a marker. J Comp Neurol. 1987;263:1–24. doi: 10.1002/cne.902630102. [DOI] [PubMed] [Google Scholar]
- Eriksson KS, Sergeeva O, Brown RE, Haas HL. Orexin/hypocretin excites the histaminergic neurons of the tuberomammillary nucleus. J Neurosci. 2001a;21:9273–9279. doi: 10.1523/JNEUROSCI.21-23-09273.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson KS, Stevens DR, Haas HL. Serotonin excites tuberomammillary neurons by activation of Na(+)/Ca(2+)-exchange. Neuropharmacology. 2001b;40:345–351. doi: 10.1016/s0028-3908(00)00175-1. [DOI] [PubMed] [Google Scholar]
- Eriksson KS, Sergeeva OA, Selbach O, Haas HL. Orexin (hypocretin)/dynorphin neurons control GABAergic inputs to tuberomammillary neurons. Eur J Neurosci. 2004;19:1278–1284. doi: 10.1111/j.1460-9568.2004.03243.x. [DOI] [PubMed] [Google Scholar]
- Faucard R, Armand V, Héron A, Cochois V, Schwartz JC, Arrang JM. N-methyl-D-aspartate receptor antagonists enhance histamine neuron activity in rodent brain. J Neurochem. 2006;98:1487–1496. doi: 10.1111/j.1471-4159.2006.04002.x. [DOI] [PubMed] [Google Scholar]
- Gershengorn MC, Osman R. Molecular and cellular biology of thyrotropin-releasing hormone receptors. Physiol Rev. 1996;76:175–191. doi: 10.1152/physrev.1996.76.1.175. [DOI] [PubMed] [Google Scholar]
- Gonzalez A, Horjales-Araujo E, Fugger L, Broberger C, Burdakov D. Stimulation of orexin/hypocretin neurones by thyrotropin-releasing hormone. J Physiol. 2009;587:1179–1186. doi: 10.1113/jphysiol.2008.167940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh K, Fukagawa K, Fukagawa T, Noguchi H, Kakuma T, Sakata T, Yoshimatsu H. Hypothalamic neuronal histamine mediates the thyrotropin-releasing hormone-induced suppression of food intake. J Neurochem. 2007;103:1102–1110. doi: 10.1111/j.1471-4159.2007.04802.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas H, Panula P. The role of histamine and the tuberomamillary nucleus in the nervous system. Nat Rev Neurosci. 2003;4:121–130. doi: 10.1038/nrn1034. [DOI] [PubMed] [Google Scholar]
- Haas HL, Reiner PB. Membrane properties of histaminergic tuberomammillary neurones of the rat hypothalamus in vitro. J Physiol. 1988;399:633–646. doi: 10.1113/jphysiol.1988.sp017100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas HL, Sergeeva OA, Selbach O. Histamine in the nervous system. Physiol Rev. 2008;88:1183–1241. doi: 10.1152/physrev.00043.2007. [DOI] [PubMed] [Google Scholar]
- Hara J, Xie XM, Sakurai T, Kilduff TS. Thyrotropin releasing hormone excites hypocretin/orexin neurons via pre and postsynaptic mechanisms. Soc Neurosci Abstr. 2007;33:471–16. [Google Scholar]
- Harkness JE, Wagner JE. Ed 3. Philadelphia: Lea and Febiger; 1989. The biology and medicine of rabbits and rodents. [Google Scholar]
- Heinflink M, Nussenzveig DR, Grimberg H, Lupu-Meiri M, Oron Y, Gershengorn MC. A constitutively active mutant thyrotropin-releasing hormone receptor is chronically down-regulated in pituitary cells: evidence using chlordiazepoxide as a negative antagonist. Mol Endocrinol. 1995;9:1455–1460. doi: 10.1210/mend.9.11.8584022. [DOI] [PubMed] [Google Scholar]
- Heuer H, Schäfer MK, O'Donnell D, Walker P, Bauer K. Expression of thyrotropin-releasing hormone receptor 2 (TRH-R2) in the central nervous system of rats. J Comp Neurol. 2000;428:319–336. [PubMed] [Google Scholar]
- Iwamoto T, Watano T, Shigekawa M. A novel isothiourea derivative selectively inhibits the reverse mode of Na+/Ca2+ exchange in cells expressing NCX1. J Biol Chem. 1996;271:22391–22397. doi: 10.1074/jbc.271.37.22391. [DOI] [PubMed] [Google Scholar]
- Jinsi-Parimoo A, Gershengorn MC. Constitutive activity of native thyrotropin-releasing hormone receptors revealed using a protein kinase C-responsive reporter gene. Endocrinology. 1997;138:1471–1475. doi: 10.1210/endo.138.4.5059. [DOI] [PubMed] [Google Scholar]
- Kiyatkin EA, Mitchum RD., Jr Fluctuations in brain temperature during sexual interaction in male rats: an approach for evaluating neural activity underlying motivated behavior. Neuroscience. 2003;119:1169–1183. doi: 10.1016/s0306-4522(03)00222-7. [DOI] [PubMed] [Google Scholar]
- Kubek MJ, Garg BP. Thyrotropin-releasing hormone in the treatment of intractable epilepsy. Pediatr Neurol. 2002;26:9–17. doi: 10.1016/s0887-8994(01)00321-6. [DOI] [PubMed] [Google Scholar]
- Lantos TA, Görcs TJ, Palkovits M. Immunohistochemical mapping of neuropeptides in the premamillary region of the hypothalamus in rats. Brain Res Brain Res Rev. 1995;20:209–249. doi: 10.1016/0165-0173(94)00013-f. [DOI] [PubMed] [Google Scholar]
- Lin JS, Sakai K, Jouvet M. Evidence for histaminergic arousal mechanisms in the hypothalamus of cat. Neuropharmacology. 1988;27:111–122. doi: 10.1016/0028-3908(88)90159-1. [DOI] [PubMed] [Google Scholar]
- Mitchell V, Prevot V, Jennes L, Aubert JP, Croix D, Beauvillain JC. Presence of mu and kappa opioid receptor mRNAs in galanin but not in GnRH neurons in the female rat. Neuroreport. 1997;8:3167–3172. doi: 10.1097/00001756-199709290-00032. [DOI] [PubMed] [Google Scholar]
- Monti JM, Jantos H, Boussard M, Altier H, Orellana C, Olivera S. Effects of selective activation or blockade of the histamine H3 receptor on sleep and wakefulness. Eur J Pharmacol. 1991;205:283–287. doi: 10.1016/0014-2999(91)90911-9. [DOI] [PubMed] [Google Scholar]
- Nillni EA, Sevarino KA. The biology of pro-thyrotropin-releasing hormone-derived peptides. Endocr Rev. 1999;20:599–648. doi: 10.1210/edrv.20.5.0379. [DOI] [PubMed] [Google Scholar]
- Nishino S, Arrigoni J, Shelton J, Kanbayashi T, Dement WC, Mignot E. Effects of thyrotropin-releasing hormone and its analogs on daytime sleepiness and cataplexy in canine narcolepsy. J Neurosci. 1997;17:6401–6408. doi: 10.1523/JNEUROSCI.17-16-06401.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Dowd BF, Lee DK, Huang W, Nguyen T, Cheng R, Liu Y, Wang B, Gershengorn MC, George SR. TRH-R2 exhibits similar binding and acute signaling but distinct regulation and anatomic distribution compared with TRH-R1. Mol Endocrinol. 2000;14:183–193. doi: 10.1210/mend.14.1.0407. [DOI] [PubMed] [Google Scholar]
- Ohtsu H, Tanaka S, Terui T, Hori Y, Makabe-Kobayashi Y, Pejler G, Tchougounova E, Hellman L, Gertsenstein M, Hirasawa N, Sakurai E, Buzás E, Kovács P, Csaba G, Kittel A, Okada M, Hara M, Mar L, Numayama-Tsuruta K, Ishigaki-Suzuki S, et al. Mice lacking histidine decarboxylase exhibit abnormal mast cells. FEBS Lett. 2001;502:53–56. doi: 10.1016/s0014-5793(01)02663-1. [DOI] [PubMed] [Google Scholar]
- Parmentier R, Ohtsu H, Djebbara-Hannas Z, Valatx JL, Watanabe T, Lin JS. Anatomical, physiological, and pharmacological characteristics of histidine decarboxylase knock-out mice: evidence for the role of brain histamine in behavioral and sleep–wake control. J Neurosci. 2002;22:7695–7711. doi: 10.1523/JNEUROSCI.22-17-07695.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quednau BD, Nicoll DA, Philipson KD. Tissue specificity and alternative splicing of the Na+/Ca2+ exchanger isoforms NCX1, NCX2, and NCX3 in rat. Am J Physiol. 1997;272:C1250–C1261. doi: 10.1152/ajpcell.1997.272.4.C1250. [DOI] [PubMed] [Google Scholar]
- Rabeler R, Mittag J, Geffers L, Rüther U, Leitges M, Parlow AF, Visser TJ, Bauer K. Generation of thyrotropin-releasing hormone receptor 1-deficient mice as an animal model of central hypothyroidism. Mol Endocrinol. 2004;18:1450–1460. doi: 10.1210/me.2004-0017. [DOI] [PubMed] [Google Scholar]
- Riehl J, Honda K, Kwan M, Hong J, Mignot E, Nishino S. Chronic oral administration of CG-3703, a thyrotropin releasing hormone analog, increases wake and decreases cataplexy in canine narcolepsy. Neuropsychopharmacology. 2000;23:34–45. doi: 10.1016/S0893-133X(99)00159-1. [DOI] [PubMed] [Google Scholar]
- Rosker C, Graziani A, Lukas M, Eder P, Zhu MX, Romanin C, Groschner K. Ca(2+) signaling by TRPC3 involves Na(+) entry and local coupling to the Na(+)/Ca(2+) exchanger. J Biol Chem. 2004;279:13696–13704. doi: 10.1074/jbc.M308108200. [DOI] [PubMed] [Google Scholar]
- Schlicker E, Kathmann M. Presynaptic neuropeptide receptors. Handb Exp Pharmacol. 2008;184:409–434. doi: 10.1007/978-3-540-74805-2_13. [DOI] [PubMed] [Google Scholar]
- Sergeeva OA, Eriksson KS, Sharonova IN, Vorobjev VS, Haas HL. GABA(A) receptor heterogeneity in histaminergic neurons. Eur J Neurosci. 2002;16:1472–1482. doi: 10.1046/j.1460-9568.2002.02221.x. [DOI] [PubMed] [Google Scholar]
- Sergeeva OA, Chepkova AN, Doreulee N, Eriksson KS, Poelchen W, Mönnighoff I, Heller-Stilb B, Warskulat U, Häussinger D, Haas HL. Taurine-induced long-lasting enhancement of synaptic transmission in mice: role of transporters. J Physiol. 2003a;550:911–919. doi: 10.1113/jphysiol.2003.045864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeeva OA, Amberger BT, Eriksson KS, Scherer A, Haas HL. Co-ordinated expression of 5-HT2C receptors with the NCX1 Na+/Ca2+ exchanger in histaminergic neurones. J Neurochem. 2003b;87:657–664. doi: 10.1046/j.1471-4159.2003.02036.x. [DOI] [PubMed] [Google Scholar]
- Sergeeva OA, Klyuch BP, Fleischer W, Eriksson KS, Korotkova TM, Siebler M, Haas HL. P2Y receptor-mediated excitation in the posterior hypothalamus. Eur J Neurosci. 2006;24:1413–1426. doi: 10.1111/j.1460-9568.2006.05027.x. [DOI] [PubMed] [Google Scholar]
- Sergeeva OA, Parmentier R, Vandael D, Klyuch BP, Haas HL. Excitation of histaminergic neurons by thyrotropin-releasing hormone. Soc Neurosci Abstr. 2007;33:199–2. doi: 10.1523/JNEUROSCI.2976-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BN, Armstrong WE. The ionic dependence of the histamine-induced depolarization of vasopressin neurones in the rat supraoptic nucleus. J Physiol. 1996;495:465–478. doi: 10.1113/jphysiol.1996.sp021607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steininger TL, Alam MN, Gong H, Szymusiak R, McGinty D. Sleep-waking discharge of neurons in the posterior lateral hypothalamus of the albino rat. Brain Res. 1999;840:138–147. doi: 10.1016/s0006-8993(99)01648-0. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Lin JS, Sakai K. Neuronal activity of histaminergic tuberomammillary neurons during wake–sleep states in the mouse. J Neurosci. 2006;26:10292–10298. doi: 10.1523/JNEUROSCI.2341-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urayama A, Yamada S, Hirano K, Deguchi Y, Kimura R. Brain receptor binding characteristics and pharmacokinetic-pharmacodynamic analysis of thyrotropin-releasing hormone analogues. Life Sci. 2001;70:647–657. doi: 10.1016/s0024-3205(01)01445-x. [DOI] [PubMed] [Google Scholar]
- Valatx JL, Bugat R. Genetic factors as determinants of the waking-sleep cycle in the mouse. Brain Res. 1974;69:315–330. doi: 10.1016/0006-8993(74)90009-2. [DOI] [PubMed] [Google Scholar]
- Vanni-Mercier G, Gigout S, Debilly G, Lin JS. Waking selective neurons in the posterior hypothalamus and their response to histamine H3-receptor ligands: an electrophysiological study in freely moving cats. Behav Brain Res. 2003;144:227–241. doi: 10.1016/s0166-4328(03)00091-3. [DOI] [PubMed] [Google Scholar]
- Yarbrough GG. On the neuropharmacology of thyrotropin releasing hormone (TRH) Prog Neurobiol. 1979;12:291–312. doi: 10.1016/0301-0082(79)90012-1. [DOI] [PubMed] [Google Scholar]
- Zeng H, Schimpf BA, Rohde AD, Pavlova MN, Gragerov A, Bergmann JE. Thyrotropin-releasing hormone receptor 1-deficient mice display increased depression and anxiety-like behavior. Mol Endocrinol. 2007;21:2795–2804. doi: 10.1210/me.2007-0048. [DOI] [PubMed] [Google Scholar]