

Abstract
A collection of new reversible glycosidase inhibitors of the iminoalditol type featuring N-substituents containing perfluorinated regions has been prepared for evaluation of physicochemical, biochemical and diagnostic properties. The vast variety of feasible oligofluoro moieties allows for modular approaches to customised structures according to the intended applications, which are influenced by the fluorine content as well as the distance of the fluorous moiety from the ring nitrogen. The first examples, in particular in the D-galacto series, exhibited excellent inhibitory activities. A preliminary screen with two human cell lines showed that, at subinhibitory concentrations, they are powerful pharmacological chaperones enhancing the activities of the catalytically handicapped lysosomal D-galactosidase mutants associated with GM1 gangliosidosis and Morquio B disease.
Keywords: chaperones, enzymes, fluorine, glycosidases, inhibitors
Introduction
Organic products containing more than one fluorine atom, in particular oligofluoroalkyl compounds with three or (preferably) more fluorine atoms have recently attracted pronounced interest as so-called “fluorous” substances. Their unique properties can be exploited by applying “fluorous technologies”. Perfluorocarbons such as perfluorinated butyltetrahydrofuran were initially developed as respiratory gas carriers and blood substitutes; this was most impressively exemplified by the “submerged mouse” breathing a perfluoroalkyl solution of oxygen.[1] The term “fluorous chemistry”, that is, the chemistry of such oligofluoro compounds, was introduced in a seminal paper by Horvath and Rabai[2] in 1994 in context with the bi-phasic separation techniques employing a third phase, the “fluorous phase”, in addition to the common aqueous and organic media usually utilised. Pioneered by groups such as Curran’s,[3] fluorous chemistry has developed prolifically, and oligofluoro substituents in reagents or substrates and starting materials have been exploited for the easy and convenient separation of intermediates or the removal of excess reagents and undesired side-products from complex reaction mixtures in a vast range of fluorous-compound-based isolation and purification techniques.[4] Noteworthy, the noncovalent attachment of small molecules to surfaces by fluorous interactions to form microarrays for the discovery of histone deacetylase inhibitors was recently reported by Schreiber and his group.[5] Such fluorous microarrays for function-selective protein “fishing” have since attracted considerable interest. For example, Spring and coworkers attached fluorous-tagged biotin to a fluorous-surface-based microarray and could demonstrate the biotin–avidine interaction with Cy3- and Cy5-labelled avidine.[6] By the same token, Pohl and her group have shown the interactions of sugars that had been attached to a fluorous surface with concanavalin A (conA) exploiting fluorescein-tagged conA[7] and have nicely extended the technology since then.[8] Trifluoromethionine was recently employed to produce a highly active fluorous DNA polymerase as a 19F NMR spectroscopic probe.[9] Likewise, engineering protein properties with other fluorous amino acids provided more stable analogues due to potent packing effects.[10]
One of the truly fascinating fields of glycochemistry and biochemistry are the iminosugar-based natural and synthetic glycosidase inhibitors[11] resulting from the formal replacement of the ring oxygen by a basic nitrogen atom, which is able to form powerful ionic bonds with carboxyl groups in the enzymes’ active sites. Due to their notable biological activities and pharmacological properties of selected derivatives, iminoalditols such as compounds 1–3 have enjoyed great interest over the past three decades. Surprisingly, in the light of the above, fluorous iminoalditols featuring alkyl groups containing more than three fluorine atoms have not been investigated systematically as biological probes for glycosidases to date.
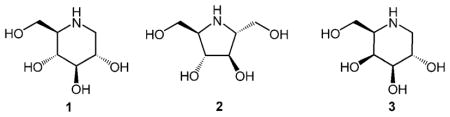
We are interested in investigating the opportunities coming with this new class of “fluorous iminoalditols” as glycosidase probes. Not only could the “fluorous properties” as mentioned in the introduction, be interesting for analytical and likely also diagnostic purposes, but the oligofluoro moieties would also provide extreme hydrophobicity and possibly lead to improved activities or selectivities as compared to the already quite potent “simple” lipophilic N-alkyl derivatives internationally investigated and exploited thus far.[12] Indeed, lipophilic glycosidase inhibitors have recently been used as pharmacological chaperones to enhance lysosomal enzyme activities that are deficient in one of the more than 40 different lysosomal storage disorders.[13] In many of these diseases, mutant lysosomal enzymes that cannot obtain and/or retain their functional conformation are recognised as misfolded by the quality control machinery in the endoplasmatic reticulum (ER) and are eventually targeted for degradation; this results in decreased intracellular activity of the enzyme. Some inhibitors can act as pharmacological chaperones in cells and bind to and stabilise the functional conformation of the mutant enzymes, enabling their exit from the ER (their site of synthesis, folding and assembly) and subsequent transport to the lysosome.[14] Although successful, only a limited number of β-galactosidase inhibitors have been shown to enhance the activity of mutant lysosomal β-galactosidase in fibroblast cell lines from patients with the lysosomal storage disorders GM1 gangliosidosis and Morquio B.[15] Here we show that some of the “fluorous iminoalditols” have the potential to act as efficient pharmacological chaperones.
Results and Discussion
Syntheses and structural variability
To acquire an initial picture on the viability of the new class of fluorous iminoalditols, a set of quite different structures (5–8, 10, 12, 15) was selected to cover a range of configurations, polarities, steric demands, as well as linker moieties such as ethers, amides and carbamates.
For the exploratory studies described, due to our current main interest in galactosidases, emphasis was laid on the D-galacto series represented by compounds 5–8 and 12. As an internal standard for probing the selectivity of these galactosidase inhibitors, N-acetylhexosaminidase inhibitor 10 was prepared. Glucosidase inhibitor 15 was chosen for comparison with previously prepared unsubstituted or nonfluorous analogues.
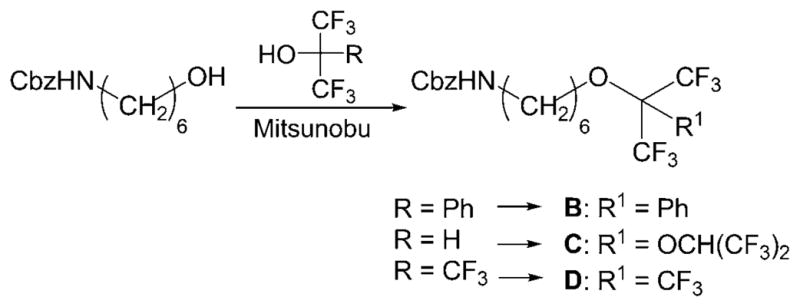
For easy access to the desired structures, a set of fluorous reagents A–D was prepared by exploiting the unique properties of oligofluoro compounds,[16] in particular, the strong acidity (1,1,1,3,3,3-hexafluoropropan-2-ol, pKa = 9.3) of fluorous alcohols.[17] For the synthesis of compound 5, fluorous hexyl ethers 6 and 8, as well as acetal 7, reductive amination of easily available, partially protected L-lyxo-hexos-5-ulose 4 was the key step. Reaction of compound 4 with the amine generated from fluorous azide A (available by reaction of the commercial iodo compound with sodium azide under phase-transfer conditions[16]) under hydrogenation conditions gave desired iminogalactitol derivative 5 (Scheme 1).
Scheme 1.

Synthesis of iminogalactitol derivative 5. a) H2, Pd/C, MeOH; b) H3O +
Fluorous amines B–D were made available by Mitsunobu reaction of the commercial N-benzyloxycarbonyl-6-aminohexanol with the respective oligofluoro alcohols providing the corresponding ethers in good yields (Scheme 2). Interestingly, in the case of 1,1,1,3,3,3-hexafluoropropan-2-ol, under the reaction conditions employed, the initially formed corresponding ether reacted with another molecule of the fluorous alcohol to give stable acetal C, which bears four trifluoromethyl moieties.
Scheme 2.
Synthesis of fluorous amines B–D.
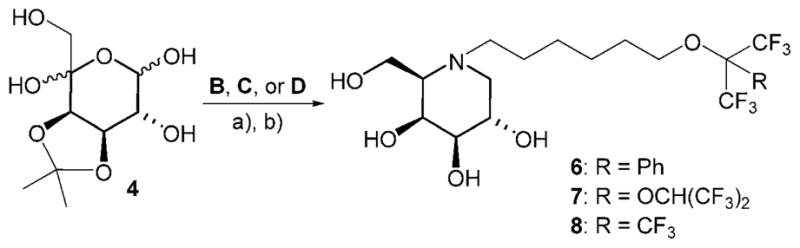
Reaction of ulosose 4 with B, C and D, respectively, provided galactosidase inhibitors 6–8 in fair yields (Scheme 3).
Scheme 3.
Synthesis of galactosidase inhibitors 6–8. a) H2, Pd/C, MeOH; b) H3O +
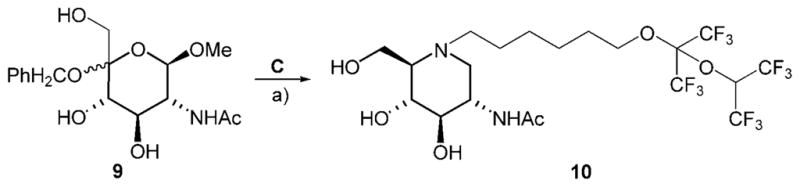
Analogously, treatment of GlcNAc-derived ulososide 9[18] with fluorous amine C gave N-acetylhexosaminidase inhibitor 10 (Scheme 4).
Scheme 4.
Synthesis of N-acetylhexosaminidase inhibitor 10. a) H2, Pd/C, MeOH
The reaction of N-(6-Amino)hexyl-1-deoxy-D -galactonojirimycin[19] (11) with commercial reagent E yielded the corresponding fluorous urethane 12 (Scheme 5).
Scheme 5.

Synthesis of fluorous urethane 12. c) NEt3, DMF.
In the L-ido series, compound 13[20] with methyl 6-oxocaproate gave intermediate 14 under standard hydrogenation conditions. Saponification of the ester 14 followed by reaction of the free carboxylate 14 a with freshly prepared nonafluorohexyl amine A furnished fluorous amide 15 (Scheme 6).
Scheme 6.

Synthesis of fluorous amide 15. a) H2, Pd/C, MeOH; d) NaOH; e) HBTU, NEt3, DMF.
Final products 5–8, 10, 12 and 15 were screened by employing two standard D-galactosidases (β-galactosidase from E. coli and α-galactosidase from green coffee beans) as well as the β-glucosidase/β-galactosidase from Agrobacterium sp. and compared with the structural parent compounds 3 (for 5–12) and 13 (for compound 15). All compounds were nicely soluble under the respective conditions employed and did not show significant differences to related inhibitors devoid of the fluorous substructures under consideration. N-Acetyl-β-D-hexosaminidase inhibitor 10 was screened with human lysosomal hexosaminidase A and exhibited the same IC50 of 6 μM as the N-unsubstituted parent compound, 2-acetamino-1,2,5-trideoxy-1,5-imino-D-glucitol.[21] Glucosidase inhibitor 15 turned out slightly better (by a factor of two) than its parent 13 (Table 1).
Table 1.
Ki values [μM] of compounds.
| Compounds | Enzymes[a] | ||||||
|---|---|---|---|---|---|---|---|
| ABG | E. c. | GCB | HLβG[b] | HLβG[c] | HLαG[c] | HexA | |
| 3 | 100 | 13 | 0.013 | 0.6 | 5.1 | 0.02 | n.i. |
| 5 | 450 | 3.5 | 3.2 | 15 | 0.36 | 16 | n.i. |
| 6 | 0.7 | 0.37 | 0.36 | 0.8 | 0.10 | 28 | n.i. |
| 7 | 12 | 0.39 | 2.9 | 3.5 | 0.39 | 15 | n.i. |
| 8 | 2.1 | 1.1 | 1.4 | 0.8 | 0.12 | 33 | n.i. |
| 10 | n.d. | n.d. | n.d. | n.i. | n.i. | n.d. | 6 |
| 12 | 5 | 0.60 | 11 | n.i. | n.i. | n.d. | n.i. |
| 13 | 26 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d |
| 15 | 11 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
ABG =β-glucosidase/β-galactosidase Agrobacterium sp. (pH 7); E.c. =β-galactosidase E. coli (pH 7); GCB =α-galactosidase green coffee beans (pH 6.5); HLβG =human lysosomal β-galactosidase (pH 4.5); HLαG=human lysosomal α-galactosidase (pH 4.5). For N-acetylhexosaminidase A (HexA), IC50 is given.
From skin fibroblasts;
From placenta (Km = 0.2 mM).[28] n.d. =not determined, n.i. =no inhibition.
Gratifyingly, in the D-galacto series all new compounds except 5 exhibited pronouncedly improved activities with the β-galactosidases probed and exhibited Ki values of one up to two orders of magnitude smaller than parent compound 3. Inhibitors 5–8 showed good solubility and suitable activities for the intended purpose and were screened with human lysosomal α-and β-galactosidases from placenta as well as human lysosomal β-galactosidase from skin fibroblasts. Inhibitory activities with β-galactosidase were better than or ranging around the value of parent compound 3 with very good selectivities when compared to the values with the corresponding α-specific enzyme (Table 1). Compounds 6–8 were thus selected for preliminary pharmacological chaperoning experiments.
Previously, Tominaga and co-workers[15a] showed that compound 3 acted as a pharmacological chaperone in GM1 gangliosidosis patient fibroblasts, increasing the enzyme activity of mutant β-galactosidase bearing mutations I51T, R201H or R457Q, by 2.2, 2.6 and 6.1-fold, respectively. It is important to bear in mind that a maximal response was achieved only after using 500 μM of compound 3. Inhibitors 6 and 8 were also evaluated as potential pharmacological chaperones in GM1 patient fibroblasts heterozygous for the mutation R148S/D332N in β-galactosidase. Although both compounds exhibited similar strength of binding to human β-galactosidase, only compound 8 significantly increased enzyme activity (>1.6-fold) relative to DMSO-treated cells. The differences in enzyme enhancement efficacy might be due to differences in intracellular bioavailability and/or metabolism, as has been postulated for some β-glucocerebrosidase pharmacological chaperones.[14c] Whereas 3 gave maximal enzyme enhancement response in patient cells at a concentration of 500 μM, compound 8 gave a maximal response at a more than tenfold-lower concentration of 30 μM (Figure 1).
Figure 1.
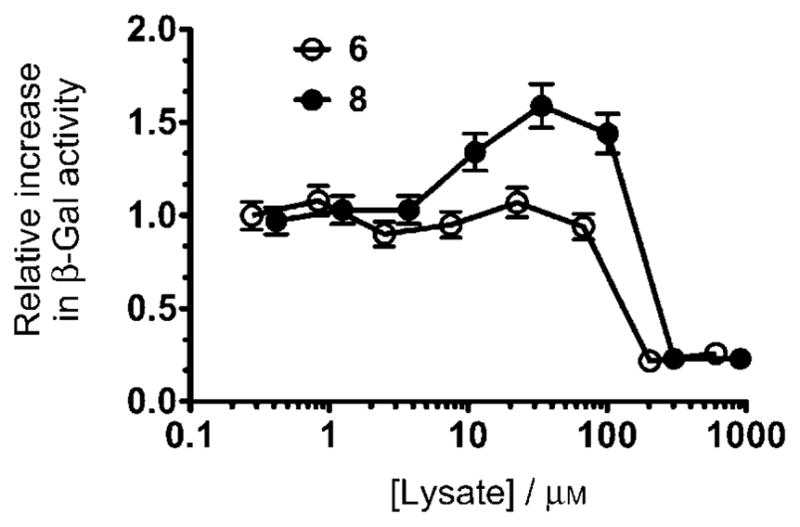
Compound 6 increases β-Gal activity in a GM1 Gangliosidosis patient fibroblast. β-Gal activity (hydrolysis of 4-methylumbelliferyl β-D-galacto-pyranoside fluorogenic substrate) in lysates from 6 or 8; treated cells were expressed relative to DMSO-treated cells. Cells were grown for five days in the presence of increasing concentration of 6 or 8. Error bars correspond to an average reading error of 7.4 %.
Furthermore, compounds 6–8 were screened in a juvenile GM1 gangliosidosis mutation cell line (p.R201C), which is known to be chaperone-sensitive.[15a,22] For this particular cell line, compounds 6–8 exhibited very similar efficacy between 5 and 50 μM. A fivefold increase of residual β-galactosidase activity representing 35 % of normal cells was obtained at 5 and 20 μM with compound 6. Compound 7 led to a fourfold increase (ca. 40 % of normal activity) at 20 and 50 μM. With inhibitor 8, the observed increase at 20 μM was also fourfold (37 % of normal cells). Compound 3 did not exhibit any appreciable chaperone activity across the entire concentration range screened but acted as an inhibitor.
Conclusions
From this first set of structural variations and configurations it may be expected that fluorous iminoalditol derivatives will be powerful glycosidase inhibitors with a variety of options as provided by the nature and the lengths of the spacer arms and by the number and distribution of fluorine atoms in the fluorous substituents, both of which are expected to alter and possibly improve the interactions with lipophilic pockets adjacent to the enzymes’ active sites.[12c,d,f] Interesting selectivities such as observed in the D-galacto series (HLβG/HLαG) might also be found with other configurations. Thus, some of these derivatives might offer worthwhile chemical and biological properties for further exploitation.
Experimental Section
General methods
Optical rotations were measured on a Perkin–Elmer 341 polarimeter at the wavelength of 589 nm and a path length of 10 cm at 20 °C. NMR spectra were recorded on a Varian INOVA 500 operating at 599.82 MHz (1 H), and at 125.894 MHz (13C) or on a Bruker Ultrashield spectrometer at 300.36 and 75.53 MHz, respectively. CDCl3 was employed for protected compounds and [D4]MeOH or D2O for unprotected inhibitors. Chemical shifts are listed in ppm by employing residual, nondeuterated solvent as the internal standard. The signals of the protecting groups were found in the expected regions and are not listed explicitly. Mass spectra were recorded on an Agilent Systems Quadrupole LC–MS in the positive mode. Electron impact (EI, 70 eV) HRMS spectra were recorded on Waters GCT Premier equipped with direct insertion (DI). MALDI-TOF mass spectrometry was performed on a Micromass Tof-Spec 2E time-of-flight mass spectrometer. Analytical TLC was performed on precoated aluminium plates silica gel 60 F254 (E. Merck 5554), detected with UV light (254 nm), 10 % vanillin/sulfuric acid as well as ceric ammonium molybdate (100 g ammonium molybdate/8 g ceric sulfate in 1 L 10 % H2SO4) and heated on a hotplate. Preparative TLC was performed on precoated glass plates silica gel 60 F254, 0.5 mm (E. Merck 5744). For column chromatography silica gel 60 (230–400 mesh, E. Merck 9385) was used.
Kinetic studies
Agrobacterium sp. β-galactosidase/-glucosidase: was purified and assayed as described.[23] Kinetic studies were performed at 37 °C in pH 7.0 sodium phosphate buffer (50 mM) containing 0.1% bovine serum albumin by using 7.20×10−5 mg mL−1 enzyme. Approximate values of Ki were determined by using a fixed concentration of substrate, 4-nitrophenyl β-D-glucopyranoside (0.11 mM=1.5 Km) and inhibitor concentrations ranging from 0.2 to 5-times the Ki value that was ultimately determined. A horizontal line drawn through 1/Vmax in a Dixon plot of this data (1/V vs [I]) intersects the experimental line at an inhibitor concentration equal to −Ki. Full Ki determinations when required, were performed by using the same range of inhibitor concentrations while also varying substrate (4-nitrophenyl glucoside) concentrations from approximately 0.015 mM to 0.6 mM. Data were analysed by direct fit to the Michaelis–Menten equation describing reaction in the presence of inhibitors by using the program GraFit.
E. coli β-galactosidase
(Sigma) kinetic studies were performed at 37 °C in pH 7.0 sodium phosphate buffer (50 mM ), using 1 nM enzyme. Approximate values of Ki were determined by using a fixed concentration of substrate, 2-nitrophenyl β-D-galactopyranoside (0.15 mM=1.5Km) and inhibitor concentrations ranging from 0.2 times to five times the Ki value ultimately determined. A horizontal line drawn through 1/Vmax in a Dixon plot of this data (1/V vs [I]) intersects the experimental line at an inhibitor concentration equal to −Ki.
Green coffee bean α-galactosidase
(Sigma) kinetic studies were performed at 37 °C in pH 6.5 sodium phosphate buffer (50 mM) by using 2 nM enzyme. Approximate values of Ki were determined by using a fixed concentration of substrate, 4-nitrophenyl β-D-galactopyranoside (0.7 mM=1.5Km) and inhibitor concentrations ranging from 0.2 to 5-times the Ki value that was ultimately determined. A horizontal line drawn through 1/Vmax in a Dixon plot of this data (1/V vs [I]) intersects the experimental line at an inhibitor concentration equal to −Ki.
Human β-galactosidase
Human skin fibroblasts were grown in minimal essential medium (MEM) with Earle’s Salts (PAA, Pasching, Austria) containing 10 % foetal bovine serum, 400 μM L-glutamine and 50 μg mL−1 gentamicin at 37 °C and 5 % CO2. All cells used in this study were between the third and nineteenth passages. Cells were harvested by trypsinisation or scraping as described earlier and cell homogenates were prepared in Eppendorf tubes in a 0.9 % NaCl solution containing 0.01 % Triton. Modified β-gal assays were used to estimate the IC50 values of putative β-gal inhibitors. For triplicate assays, confluent fibroblast cells from healthy patients (3×0 75 mL flasks) were harvested by trypsinisation, resuspended in 0.9 % NaCl (1.5 mL) containing 0.01 % Triton, sonicated and centrifuged. For inhibition assays, cell homogenate (20 μL; 40 μL total protein) was mixed with prewarmed (90 μL; 37 °C) β-gal substrate solution and inhibitor solutions (10 μL) in final concentrations ranging from 0–100 μM. Substrate blanks contained 0.9 % NaCl (20 μL) instead of cell homogenate, whereas enzyme blanks were made with substrate buffer without β-gal substrate. The samples were incubated for 30 min at 37 °C in a water bath, and the reaction was stopped by addition of stop buffer (2.5 mL). The amount of hydrolysed 4-methylumbelliferone was determined with a Luminescence Spectrometer (LS50B, Perkin–Elmer) with an excitation wavelength of 360 nm and emission at 450 nm. Data analysis was performed with Microcal Origin v6.0 by using the IC50 module based on sigmoid curve fitting. For enzyme kinetic experiments cells from healthy controls were prepared as described for IC50 determination. To determine Ki values, fixed concentrations of each inhibitor were used while varying the β-gal substrate concentration from 100–450 μM. Data analysis was performed with Microcal Origin v6.0 by using a nonlinear curve-fitting module based on the Michaelis–Menten equation for competitive inhibitors.
The inhibitory activity of compounds 3 and 5–8 against human lysosomal α- as well as β-galactosidase was also evaluated by using a lysosomal-enzyme-enriched conA fraction from human placenta as the enzyme source.[24] For the β-galactosidase, 4-nitrophenyl β-D-galactopyranoside (2.5 mM) as the substrate (Km = 0.2 mM). Reactions were performed at 37 °C in pH 4.5 citrate phosphate buffer (100 mM) as described previously.[25] IC50 values (μM) were extracted from the enzyme activity curves in presence of increasing concentrations of the inhibitor using nonlinear regression as implemented within Graphpad Prism 5.
IC50 of hexosaminidase inhibitor 10 was determined with purified human placental N-acetyl-β-D -hexosaminidase A (hexA; final 0.3 mg mL−1) and methylumbelliferyl N-acetyl-β-D-glucosaminide (final 1.6 mM) in citrate phosphate buffer (100 mM) at pH 4.5 in the presence of 0.025 % human serum albumin at 37°C for 30 min.
Intracellular β-galactosidase activity
Infantile GM1 gangliosidosis (R148S/D332N) fibroblasts[26] were treated with escalating doses of compounds 6 and 8 (dissolved in DMSO) for 5 d. Intracellular β-galactosidase activity was measured by using the fluorogenic substrate 4-methylumbelliferyl β-D-galactopyranoside as previously described.[25]
Juvenile GM1 gangliosidosis (p.R201C) human skin fibroblasts were grown until semiconfluency in 6-well plates. Compounds were added at concentrations from 0–500 μM and incubated for four more days at 37 °C. Intracellular β-galactosidase activity was determined as described.[22]
General procedure for intramolecular reductive amination
One equivalent of azide A or the respective N-benzyloxycarbonyl component B, C or D was added to a 0.03 M solution of 4 (or 9) in MeOH/H2O (15:1, v/v), Pd(OH)2/C (20 %, 0.1 equiv) and the heterogeneous reaction mixture was stirred under an atmosphere of H2 at ambient pressure and RT until TLC indicated completed conversion of the starting material. After filtration and removal of the solvent under reduced pressure, the residue was dissolved in MeOH/H2O 1:1 (v/v) and the pH value was adjusted to pH 1 with aq conc HCl to remove the isopropylidene group. The solution was concentrated under reduced pressure and the remaining residue was subjected to fluorous-phase extraction and subsequently purified by column chromatography on silica gel (CHCl3/MeOH/concd NH4OH, 500:100:6; v/v/v), yielding the target compound as a colourless syrup. The formation of L-altro-configured products from D-galactose derived ulosose 4 and L-ido configuration from ulososide 9, respectively, was not observed.
General procedure for Mitsunobu reactions on N-benzyloxycarbonyl-6-aminohexanol; Reagents B, C, and D
Triphenylphosphane (3.15 g, 12 mmol, 3 equiv) was added to a 2% solution of commercial N-benzyloxycarbonyl-6-aminohexanol (1.0 g, 4.0 mmol) in dry THF. The mixture was cooled to 0°C and diethylazodicarboxylate (DEAD, 1.84 mL, 12 mmol, 3 equiv) was added dropwise. The reaction mixture was brought to RT, and the respective fluorous alcohol (3 equiv) was added. When TLC indicated completed conversion of the starting material, the solvent was removed under reduced pressure and the crude residue was partly purified by fluorous solid-phase extraction by employing MeOH/H2O for loading and washing followed by THF as the eluent. Subsequent purification on silica gel provided compounds B, C, and D in yields of 91 %, 82 %, and 80 %, respectively. B: 1H NMR (CDCl3): δ = 5.19 (d, J =11.2 Hz, 2H), 4.74 (br s; NH), 3.98 (t, J =5.9 Hz, 2 H), 3.18 (dd, J = 13.2 Hz, J =6.9 Hz, 2H), 1.67 (m, 2 H), 1.51 (m, 2 H), 1.45–1.30 ppm (m, 4H); 13C NMR (CDCl3): δ = 156.6, 136.9, 130.3, 128.8 (2 C), 128.6, 128.2, 122.7 (q, JC,F = 291 Hz), 82.8 (hep, JC,F = 28.1 Hz), 66.7, 66.4, 41.1, 30.0, 29.8, 26.6, 25.5 ppm; MS: m/z calcd for C23H25NO3F6Na: 500.1636 [M+Na]+, found: 500.1612.
C
1H NMR (CDCl3): δ = 5.12 (s, 2 H), 4.92 (q, JH,CF3 5.2 Hz, 1 H), 4.47 (br s, 1H; NH), 4.02 (t, J =6.2 Hz, 2 H), 3.19 (q, J =6.7 Hz, 2 H), 1.68 (m, 2 H), 1.52 (m, 2 H), 1.44–1.30 ppm (m, 4H); 13C NMR (CDCl3): δ = 124.2–116.6 (m, JC,F = 293 Hz, 4C; CF3), 95.2 (p, JC,F = 32.8 Hz), 69.7, 69.4 (m, JC,F = 34.6 Hz; CHCF3), 66.7, 40.9, 29.9, 29.4, 26.3, 25.1 ppm; MS: m/z calcd for C20H21NO4F12Na: 590.1177 [M+Na] +, found: 590.1153.
D
1H NMR (CDCl3): δ = 5.19 (d, J = 11.2 Hz, 2H), 4.76 (brs, 1H; NH), 3.55 (t, J =6.8 Hz, 2 H), 3.18 (m, 2 H), 1.70 (m, 2H), 1.53 (m, 2 H), 1.43 (m, 2H), 1.35 (m, 2H); 13C NMR (CDCl3): δ=120.7 (q, JC,F = 293 Hz, 3 C), 80.0 (m, JC,F = 29.6 Hz), 69.9, 66.9, 41.2, 30.1, 29.8, 26.5, 25.3; MS: m/z calcd for C18H20NO3F9Na: 492.1197 [M+Na] +, found: 492.1172.
3,4-O-Isopropylidene-L arabino-hexos-5-ulose (4)
A 20 % solution of 1,2:3,4-di-O-isopropylidene-5-deoxy-hex-5-eno-L-arabino-pyranose[27] (10.0 g, 41.3 mmol) in CH2Cl2 was added to a 10 % solution of 3-chloroperbenzoic acid (13.8 g, 61.6 mmol, 77 %) in CH2Cl2, and the reaction was stirred at RT for 4 h. After completed conversion of the starting material, the mixture was cooled to −18 °C, and a white precipitate formed, which was removed by filtration. The filtrate was washed consecutively with sat. aq NaHCO3 and H2O until the pH value of the aqueous layer was neutral. The organic phase was separated, dried (Na2SO4) filtered and concentrated under reduced pressure. The crude residue was taken up in dry MeOH and the solution was cooled to −30 °C. NaOMe (1 M in MeOH) was added carefully to adjust the pH value to 10. After completed conversion of the intermediate, the reaction mixture was neutralised with acidic ion exchange resin Amberlite IR 120 [H+]. Following removal of the resin by filtration, the solution was concentrated under reduced pressure. Chromatography of the residue provided the title compound (mixture of several pyranoid and furanoid tautomers, 6.54 g, 67 %) as off-white foam, which was immediately used in the next step.
N-(3,3,4,4,5,5,6,6,6-Nonafluoro)hexyl-1,5-dideoxy-1,5-imino-D-galactitol (5)
3,3,4,4,5,5,6,6,6-Nonafluorohexyl azide A (290 mg, 1.00 mmol) and Pd/C (10 %, 50 mg) were added a to a 1 % methanolic solution of 3,4-O-isopropylidene-L-arabino-hexos-5-ulose (4, 406 mg, 1.72 mmol), and the mixture was stirred under an atmosphere of H2 at ambient pressure for 36 h. The catalyst was removed by filtration, and the filtrate was concentrated under reduced pressure. The crude residue was taken up in a mixture of 1:1 MeOH/H2O (15 mL), and CHCl3 (6 mL) was added to provide a clear solution. The pH value was adjusted to 1 by addition of conc. HCl. After completed removal of the isopropylidene group as indicated by TLC, the reaction mixture was concentrated under reduced pressure. Chromatography (CHCl3/MeOH/NH4OH 600:100:7 v/v/v) gave free inhibitor 5 (163 mg, 23 % from 4) as a white powder. (c = 1.9 in MeOH); 1H NMR ([D4]MeOH): δ= 3.93 (m, 1H; H4), 3.81 (m, 3 H; H2, H6a, H6b), 3.24 (dd, J2,3 = 8.8 Hz, J3,4 = 2.5 Hz, 1H; H3), 3.10 (m, 1 H; H1′a), 3.03 (m, 1 H; H1′b), 2.92 (dd, J1ax,1eq = 11.3 Hz, J1eq,2 = 2.7 Hz, 1 H; H1eq), 2.60–2.28 (m, JC,F 17.3 Hz, 2 H; H2′), 2.18 ppm (dd, J1ax,2 = 10.1 Hz, 1H; H1ax); 13C NMR ([D4]MeOH): δ = 122.5–107.4 (m, 4 C, C3′, C4′, C5′, C6′), 77.0 (C3), 72.6 (C4), 68.9 (C2), 64.8 (C5), 63.2 (C6), 57.8 (C1), 45.2 (t, JC,F = 4 Hz, C1′), 26.9 ppm (t, JC,F = 21.4 Hz, C2′); MS m/z calcd for C12H16N2O4F9H: 410.1014 [M+H] +, found: 410.1026, 432.0850 [M+Na]+.
N-(α,α-Di-trifluoromethyl)benzyloxyhexyl-1,5-dideoxy-1,5-imino-D-galactitol (6)
By following the general procedure for the Mitsunobu reactions, the reaction of ulosose 4 (100 mg, 0.42 mmol) and reagent B gave fluorous inhibitor 6 (147 mg, 71 %) as a white foam. (c = 0.5 in MeOH); 1H NMR ([D4]MeOH): δ = 3.98 (m, 1 H; H4), 3.84–2.76 (m, 3H; H2, H6a, H6b), 3.57 (m, 2H; H6′a, H6′b), 3.21 (dd, J2,3 = 9.7 Hz, J3,4 = 3.3 Hz, 1 H; H3), 2.98 (dd, J1eq,2 = 4.7 Hz, J1eq,1ax = 11.2 Hz, 1 H; H1eq), 2.68 (m, 1 H; H1′ax), 2.51 (m, 1 H; H1′b), 2.37 (m, 1H; H5), 2.12 (dd, J1ax,2 = 10.8 Hz, 1H; H1eq), 1.73 (m, 2 H; H5′), 1.58–1.49 (m, 2 H; H-2′), 1.49–1.42 (m, 2H), 1.36–1.27 ppm (m, 2 H); 13C NMR ([D4]MeOH): δ=131.6, 129.9, 129.3, 129.2, 124.0 (q, 2 C, JC,F = 290 Hz, C2″), 84.1 (m, JC,F = 29 Hz, C1″), 77.3 (C3), 72.3 (C4), 69.0 (C2), 67.6, 65.2, 62.4 (C5, C6, C6′), 58.1, 54.0 (C1, C1′), 30.7, 28.3, 26.8, 24.9 ppm (C2′, C3′, C4′, C5′); MS: m/z calcd for C21H29NO5F6H: 490.4674 [M+H] +, found: 490.50.
N-[(1,1,1,3,3,3-Hexafluoropropyl-2-oxy)-1,1,1,3,3,3-hexafluoropropyl-2-oxy]hexyl-1,5-dideoxy-1,5-imino-D-galactitol (7)
By following the general procedure for Mitsunobu reactions, the reaction of ulosose 4 (415 mg, 1.76 mmol) and reagent C gave fluorous inhibitor 7 (306 mg, 30 %) as a white foam. (c = 0.8 in MeOH); 1H NMR ([D4]MeOH): δ=5.76 (m, J1″F = 5.3 Hz, 1 H; H1″), 4.09 (m, 2H; H6′), 3.98 (m, 1 H; H4), 3.79 (m, 3 H; H2, H6a, H6b), 3.20 (m, J2,3 = 9.3 Hz, 1 H; H3), 2.98 (dd, J1eq,2 = 2.8 Hz, J1ax,1eq = 10.3 Hz, 1 H; H1eq), 2.72 (m, 1H; H1′a), 2.37 (m, 1H; H1′b), 2.11 (dd, J1ax,2 = 10.9 Hz, 1 H; H1ax), 1.80–1.69 (m, 2 H), 1.60–1.48 (m, 2 H), 1.48–1.40 (m, 2 H), 1.37–1.25 ppm (m, 2 H); 13C NMR ([D4]MeOH): δ=125.7–117.9 (m, JC,F = 286 Hz, 2 C), 120.2 (m, JC,F = 293 Hz, 2C), 96.5 (m, JC,F = 32.7 Hz), 77.3 (C3), 72.2 (C4), 71.2 (C2), 70.2 (m, JC,F = 34.1 Hz), 69.0 (C5), 65.2 (C6′), 62.4 (C6), 58.0 (C1′), 53.9 (C1), 30.3, 28.1, 26.3, 25.0 ppm (C2′, C3′, C4′, C5′); MS: m/z calcd for C18H25NO6F12H: 580.3918 [M+H] +, found: 580.3956.
N-(Nonafluoro-tert-butyloxy)hexyl-1,5-dideoxy-1,5-imino-D-galactitol (8)
By following the general procedure for Mitsunobu reactions, the reaction of ulosose 4 (100 mg, 0.42 mmol) with reagent D gave fluorous inhibitor 8 (158 mg, 78 %) as a white foam. (c =1.8 in MeOH); 1H NMR ([D4]MeOH): δ=4.08 (dd, J = 5.3 Hz, J =5.9 Hz, 2H; H6′), 3.98 (m, 1 H; H4), 3.80 (m, 3 H; H2, H6a, H6b), 3.21 (dd, J2,3 = 9.3 Hz, J3,4 = 2.8 Hz, 1 H; H3), 2.98 (dd, J1ax,1eq = 11.2 Hz, J1eq,2 4.4 Hz, 1H; H1eq), 2.72 (m, 1H; H1′a), 2.51 (m, 1H; H1′b), 2.40 (m, 1H; H5), 2.11 (dd, J1ax,2 10.1 Hz, 1H; H1ax), 1.70 (m, 2 H; H2′), 1.51 (m, 2 H; H5′), 1.43 (m, 2H), 1.32 ppm (m, 2H); 13C NMR ([D4]MeOH): δ = 121.9 (m, 3 C, JC,F = 299 Hz, 3 C2″), 81.1 (m, JC,F 29.5 Hz, C1″), 77.2 (C3), 72.2 (C4), 71.5 (C2), 69.0 (C6′), 65.2 (C5), 62.4 (C6), 58.0 (C1), 53.9 (C1′), 30.8, 28.1, 26.4, 24.9 ppm (C2′, C3′, C4′, C5′); MS: m/z calcd for C16H24NO5F9H: 482.1589 [M+H]+, found: 482.1638.
N-[(1,1,1,3,3,3-Hexafluoropropyl-2-oxy)-1,1,1,3,3,3-hexafluoropropyl-2-oxy]hexyl-2-acetamino-1,2,5-trideoxy-1,5-imino-D-glucitol (10)
H2O (1 mL), Pd/C (10 %, 50 mg) and compound C (0.750 mg, 7.87 mmol) were added to a 2 % MeOH solution of ulososide 9[18] (530 mg, 1.55 mmol), and the mixture was stirred under an atmosphere of H2 at ambient pressure for 24 h, when TLC indicated completed conversion of the starting material 9. The catalyst was removed by filtration, and the filtrate was concentrated under reduced pressure. Chromatography (CHCl3/MeOH/NH4OH 80:10:1, v/v/v) furnished 10 (376 mg, 39 %) as a colourless syrup. (c =1.4 in H2O); 1H NMR ([D4]MeOH): δ = 5.81 (hept, J1″,F = 5.3 Hz, H1″), 4.09 (t, J5′,6′ = 5.9 Hz, 2 H; H6′), 3.88 (dd, J5,6a = 1.5 Hz, J6a,6b = 11.5 Hz, 1H; H6a), 3.84–3.77 (m, 2 H; H2, H6b), 3.38 (dd, J3,4 = 9.2 Hz, 1 H; H4), 3.20 (dd, J2,3 = 10.0 Hz, 1 H; H3), 3.00 (dd, J1ax,1eq = 11.3 Hz, J1eq,2 = 4.2 Hz, 1 H; H1eq), 2.80 (m, 1 H; H1′a), 2.55 (m, 1H; H1′b), 2.13 (m, 1H; H5), 2.08 (dd, J1ax,2 = 11.0 Hz, 1H; H1ax), 1.95 (s, 3 H; NAc), 1.78–1.70 (m, 2 H), 1.53–1.39 (m, 4H), 1.36–1.26 ppm (m, 2 H); 13C NMR ([D4]MeOH): δ = 173.6 (NHAc), 125.8–117.9 (m, JC,F = 293 Hz, 4CF3), 96.5 (m, JC,F = 32.9 Hz), 77.7 (C3), 72.8 (C4), 71.2 (C6′), 70.2 (m, JC,F = 34.1 Hz), 67.5 (C5), 59.8 (C6), 55.6, 53.4, 51.9 (C1, C1′, C2), 30.3 (C5′), 28.1, 26.3, 25.5 (C2′, C3′, C4′), 22.7 ppm (NHAc); MS: m/z calcd for C20H28N2O6F12Na: 643.1653 [M+Na]+, found: 643.1652.
N-[4-(3,3,4,4,5,5,6,6,6-Nonafluorohexyl)benzyloxycarbonyl-amino]hexyl-1,5-dideoxy-1,5-imino-D-galactitol (12)
Commercially available 2,5-dioxopyrrolidin-1-yl 4-(3,3,4,4,5,5,6,6,6-nonafluorohexyl)benzyl carbonate E (204 mg, 0.42 mmol) was added to a solution of N-(6-amino)hexyl-1-deoxy-D-galactonojirimycin 11[19] (108 mg, 0.41 mmol) and Et3N (86 mg, 0.82 mmol) in dry DMF (15 mL), and the reaction mixture was stirred at RT for 90 min. After completed conversion, MeOH (5 mL) was added, and the solvents were removed under reduced pressure. Chromatography of the residue provided 12 (128 mg, 49 %). (c = 0.7 in MeOH); 1H NMR ([D4]MeOH): δ = 7.40–7.26 (m, 4H), 5.08 (s, 2 H; CH2Ph), 4.03 (dd, J3,4 = 3.0 Hz, J4,5 = 1.9 Hz, 1H; H4), 3.86 (ddd, J1ax,2 = 10.5 Hz, J1eq,2 = 4.7 Hz, J2,3 = 9.2 Hz, 1 H; H2), 3.86–3.81 (m, 2 H; H6a, H6b), 3.27 (dd, 1 H; H3), 3.14 (t, J =6.9 Hz, 2 H; H6′), 3.05 (dd, J1ax,1eq = 11.2 Hz, 1H; H1eq), 3.00–2.92 (m, 2H), 2.78 (m, 1H; H1′a), 2.59 (m, 1 H; H1′b), 2.54–2.38 (m, 3 H; H5, 2 H2″), 2.21 (dd, 1H; H1ax), 1.65–1.47 (m, 4 H), 1.46–1.26 ppm (m, 4H); 13C NMR ([D4]MeOH): δ = 158.9, 140.3, 136.9, 129.5, 129.3, 122.1–108.0 (m, C4F9), 77.1 (C3), 72.1 (C4), 68.8 (C2), 67.0 (CH2Ph), 65.4 (C5), 62.2 (C6), 57.9 (C1), 54.0 (C1′), 41.7 (C6′), 33.6 (t, JC,F = 22 Hz, C2″), 30.9, 28.2, 27.7, 27.0 ppm (t, JC,F = 5 Hz, C1″), 25.0; MS: m/z calcd for C26H35N2O6F9H: 643.5722 [M+H]+, found: 643.5703.
N-Methoxycarbonylpentyl-1,5-dideoxy-1,5-imino-L-iditol (14)
Adipic acid hemialdehyde methylester (1.1 mL, 7.9 mmol) was added to a 10 % methanolic solution of 1,5-dideoxy-1,5-imino-L-iditol[20] 13 (1.26 g, 7.22 mmol), and the mixture was stirred with Pd/C (10%, 260 mg) under an atmosphere of H2 at ambient pressure until TLC indicated completed conversion of the starting iminosugar. The catalyst was removed by filtration, the filtrate was concentrated under reduced pressure, and the remaining residue was chromatographed to give 14 as a colourless syrup (1.10 g, 52 %). (c = 6.5 in MeOH); 1H NMR ([D4]MeOH): δ = 3.86–3.79 (m, 2 H; H6a, H6b), 3.71–3.69 (m, 1 H; H4), 3.65 (s, 3H; OCH3), 3.58–3.50 (m, 1H; H2), 3.39 (dd, J2,3 = J3,4 = 8.3 Hz, 1H; H3), 3.05 (m, 1 H; H5), 2.82–2.74 (m, 2H; H1eq, H1′a), 2.68–2.63 (m, 1 H; H1′b), 2.59 (dd, J1ax,1eq = 11.2 Hz, J1ax,2 = 10.8 Hz, 1H; H1ax), 2.33 (t, 2H; H5′), 1.63, 1.53, 1.33 ppm (3 m, 2 H each, H2′, H3′, H4′); 13C NMR ([D4]MeOH): δ = 174.8 (C6′), 74.6 (C3), 71.5, 70.0 (C2, C4), 63.2 (C5), 56.5 (C6), 54.2 (C1), 51.7 (C1′), 51.0 (OMe), 35.6 (C5′), 27.0, 26.6, 24.7 ppm (C2′, C3′, C4′); MS: m/z calcd for C13H25NO6H: 292.3553 [M+H]+, found: 292.3600 [M+H]+.
N-Carboxypentyl-1,5-dideoxy-1,5-imino-L-iditol (14 a) and N-[(3,3,4,4,5,5,6,6,6-Nonafluoro)hexylaminocarbonyl]pentyl-1,5-di-deoxy-1,5-imino-L-iditol (15)
NaOH (0.5 M, 2.0 mL) was added to an ice-cold 1% solution of 14 (237 mg, 0.81 mmol) in dioxane/H2O (1:1, v/v), and the mixture was stirred until TLC (CHCl3/MeOH/NH4OH, 500:100:6) indicated completed saponification. The mixture was brought to pH 6 by addition of ion-exchange resin IR 120 [H+]. After removal of the resin by filtration, the filtrate was concentrated under reduced pressure to give carboxylic acid 14 a, which was immediately used in the next step. Et3N (120 μL, 0.9 mmol), and HBTU (320 mg, 0.9 mmol) were added to a 1 % DMF solution of crude 14a (230 mg, 0.83 mmol), compound A (3 mmol, 3.3 equiv), and the reaction mixture was stirred at RT until TLC indicated quantitative consumption of the iminoalditol. The solvent was removed under reduced pressure and the remaining residue was chromatographed (CHCl3/MeOH/NH4OH 700:100:8) to provide 15 (200 mg, 47 %) as a colourless syrup. (c = 1.1 in MeOH); 1H NMR ([D4]MeOH): δ = 4.01–3.92 (m, 3H; H3, H6a, H6b), 3.91–3.84 (m, 1H; H4), 3.78 (dd, J1ax,2 = 4.5 Hz, 1H; H2), 3.53 (t, 2H; H1″), 3.46 (m, 1 H; H5), 3.38 (m, 1H; H1ax), 3.28–3.18 (m, 3 H; H1eq, H1′), 2.54–2.34 (m, 2 H; H2″), 2.26 (t, 2H; H5′), 1.88–1.63 (m, 4H; H2′, H4′), 1.49–1.36 (m, 2 H; H3′); 13C NMR ([D4]MeOH): δ = 176.1 (CONH), 122.5–107.6 (m, 4 C, C4F9), 72.2 (C3), 70.7 (C2), 69.6 (C4), 63.9 (C5), 59.6 (C6), 54.9 (C1), 53.7 (C1′), 36.6 (C5′), 32.6 (t, JC,F = 4.9 Hz, C1″), 31.2 (t, JC,F = 21.1 Hz, C2″), 27.3, 26.3 (3 C, C2′, C3′ C4′); MS: m/z calcd for C18H27N2O5F9H: 523.1854 [M+H]+, found: 523.1802.
Acknowledgments
Financial support by the Austrian Ministry of Science and Research (Project GZ 200.156/2-VI/1a/2006), the Austrian Research Society for Mucopolysaccharidoses and Related Diseases as well as by the Canadian Institutes of Health Research (CIHR Team Grant CTP-82944) is gratefully acknowledged.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cbic.201000192.
References
- 1.Clark LC, Gollan F. Science. 1966;152:1755–1756. doi: 10.1126/science.152.3730.1755. [DOI] [PubMed] [Google Scholar]
- 2.Horvath IT, Rabai J. Science. 1994;266:72–75. doi: 10.1126/science.266.5182.72. [DOI] [PubMed] [Google Scholar]
- 3.Curran DP. J Fluor Chem. 2008;129:898–902. doi: 10.1016/j.jfluchem.2008.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Zhang W, Cai C. Chem Commun. 2008:5686–5694. doi: 10.1039/b812433g. [DOI] [PubMed] [Google Scholar]; b) Dandapani S. QSAR Comb Sci. 2006;25:681–688. [Google Scholar]; c) Yoder NC, Y4ksel D, Dafik L, Kumar K. Curr Opin Chem Biol Curr Op Chem Biol. 2006;10:576–583. doi: 10.1016/j.cbpa.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 5.Vargas AJ, Bradner JE, Tang W, McPherson OM, Greenberg EF, Koehler AN, Schreiber SL. Angew Chem. 2007;119:8106–8110. doi: 10.1002/anie.200703198. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2007;46:7960–7964. doi: 10.1002/anie.200703198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nicholson RL, Ladlow ML, Spring DR. Chem Commun. 2007:3906–3908. doi: 10.1039/b712906h. [DOI] [PubMed] [Google Scholar]
- 7.Pohl NL. Angew Chem. 2008;120:3930–3932. [Google Scholar]; Angew Chem Int Ed. 2008;47:3868–3870. doi: 10.1002/anie.200704801. [DOI] [PubMed] [Google Scholar]
- 8.a) Chen GS, Pohl NL. Org Lett. 2008;10:785–788. doi: 10.1021/ol702915e. [DOI] [PubMed] [Google Scholar]; b) Song EH, Pohl NL. Fut Med Chem. 2009;1:889–896. doi: 10.4155/fmc.09.76. [DOI] [PubMed] [Google Scholar]; c) Collet BYM, Nagashima T, Yu MS, Pohl NLB. J Fluor Chem. 2009;130:1042–1048. doi: 10.1016/j.jfluchem.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holzberger B, Rubini M, Mçller HM, Marx A. Angew Chem Int Ed. 2010;49:1324–1327. doi: 10.1002/anie.200905978. [DOI] [PubMed] [Google Scholar]
- 10.Buer BC, de La Salud-Bea R, Al Hashimi HM, Marsh ENG. Biochemistry. 2009;48:10810–10817. doi: 10.1021/bi901481k. [DOI] [PubMed] [Google Scholar]
- 11.a) Compain P, Martin OR, editors. Iminosugars: From Synthesis to Therapeutic Applications. Wiley; New York: 2007. [Google Scholar]; b) St4tz AE, editor. Iminosugars as Glycosidase Inhibitors. Wiley-VCH; Weinheim: 1999. [Google Scholar]
- 12.For example: Overkleeft HS, Renkema GH, Neele J, Vianello P, Hung IO, Strijland A, van der Burg AM, Koomen GJ, Pandit UK, Aerts JMFG. J Biol Chem. 1998;273:26522–26527. doi: 10.1074/jbc.273.41.26522.Sawkar AR, Cheng WC, Beutler E, Wong CH, Balch WE, Kelly JW. Proc Natl Acad Sci USA. 2002;99:15428–15433. doi: 10.1073/pnas.192582899.Wennekes T, van den Berg RJBHN, Donker W, van der Marel GA, Strijland A, Aerts JMFG, Overkleeft HS. J Org Chem. 2007;72:1088–1097. doi: 10.1021/jo061280p.Yu Z, Sawkar AR, Whalen LJ, Wong CH, Kelly JW. J Med Chem. 2007;50:94–100. doi: 10.1021/jm060677i.Brumshtein B, Aguilar-Moncayo M, Garcia-Moreno MI, Ortiz Mellet C, Garcia Fernandez JM, Silman I, Shaaltiel Y, Aviezer D, Sussman JL, Futerman AH. ChemBioChem. 2009;10:1480–1485. doi: 10.1002/cbic.200900142.Suzuki Y, Ogawa S, Sakakibara Y. Perspect Med Chem. 2009;3:7–19. doi: 10.4137/pmc.s2332.
- 13.a) Fan JQ. Trends Pharmacol Sci. 2003;24:355–360. doi: 10.1016/S0165-6147(03)00158-5. [DOI] [PubMed] [Google Scholar]; b) Tropak MB, Mahuran DJ. FEBS J. 2007;274:4951–4961. doi: 10.1111/j.1742-4658.2007.06040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Steet RA, Chung S, Wustman B, Powe A, Do H, Kornfeld SA. Proc Natl Acad Sci USA. 2006;103:13813–13818. doi: 10.1073/pnas.0605928103. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hamanaka R, Shinohara T, Yano S, Nakamura M, Yasuda A, Yokoyama S, Fan JQ, Kawasaki K, Watanabe M, Ishii S. Biochim Biophys Acta, Mol Basis Dis. 2008;1782:408–413. doi: 10.1016/j.bbadis.2008.03.001. [DOI] [PubMed] [Google Scholar]; c) Maegawa GHB, Tropak MB, Buttner JD, Rigat BA, Fuller M, Pandit D, Tang L, Kornhaber GJ, Hamuro Y, Clarke JTR, Mahuran DJ. J Biol Chem. 2009;284:23502–23516. doi: 10.1074/jbc.M109.012393. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Khanna R, Benjamin ER, Pellegrino L, Schilling A, Rigat BA, Soska R, Nafar H, Ranes BE, Feng J, Lun Y, Powe AC, Palling DJ, Wustman BA, Schiffmann R, Mahuran DJ, Lockhart DJ, Valenzano KJ. FEBS J. 2010;277:1618–1638. doi: 10.1111/j.1742-4658.2010.07588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Tominaga L, Ogawa Y, Taniguchi M, Ohno K, Matsuda J, Oshima A, Suzuki Y, Nanba E. Brain Dev. 2001;23:284–287. doi: 10.1016/s0387-7604(01)00216-9. [DOI] [PubMed] [Google Scholar]; b) Matsuda J, Suzuki O, Oshima A, Yamamoto Y, Noguchi A, Takimoto K, Itoh M, Matsuzaki Y, Yasuda Y, Ogawa S, Sakata Y, Nanba E, Higaki K, Ogawa Y, Tominaga L, Ohno K, Iwasaki H, Watanabe H, Brady RO, Suzuki Y. Proc Natl Acad Sci USA. 2003;100:15912–15917. doi: 10.1073/pnas.2536657100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szonyi F, Cambon A. J Fluor Chem. 1989;42:59–68. [Google Scholar]
- 17.a) Dyatkin BL, Mochalina EP. Tetrahedron. 1965;21:2991–2995. [Google Scholar]; b) Falck JR, Yu J, Cho HS. Tetrahedron Lett. 1994;35:5997–6000. [Google Scholar]; c) Sebesta DP, O’Rourke SS, Pieken WA. J Org Chem. 1996;61:361–362. [Google Scholar]
- 18.Steiner AJ, Schitter G, St4tz AE, Wrodnigg TM, Tarling CA, Withers SG, Mahuran DJ, Tropak MB. Tetrahedron: Asymmetry. 2009;20:832–835. doi: 10.1016/j.tetasy.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greimel P, H3usler H, Lundt I, Rupitz K, St4tz AE, Tarling CA, Withers SG, Wrodnigg TM. Bioorg Med Chem Lett. 2006;16:2067–2070. doi: 10.1016/j.bmcl.2006.01.095. [DOI] [PubMed] [Google Scholar]
- 20.Fowler PA, Haines AH, Taylor RJK, Chrystal EJT, Gravestock MB. Carbohydr Res. 1993;246:377–381. [Google Scholar]
- 21.Fleet GWJ, Smith PW, Nash RJ, Fellows LE, Parekh RB, Rademacher TW. Chem Lett. 1986:1051–1054. [Google Scholar]
- 22.a) Hofer D, Paul K, Fantur K, Beck M, B4rger F, Caillaud C, Fumic K, Ledvinova J, Lugowska A, Michelakakis H, Radeva B, Ramaswami U, Plecko B, Paschke E. Hum Mutat. 2009;30:1214–1221. doi: 10.1002/humu.21031. [DOI] [PubMed] [Google Scholar]; b) Iwasaki H, Watanabe H, Iida M, Ogawa S, Tabe M, Higaki K, Nanba E, Suzuki Y. Brain Dev. 2006;28:482–486. doi: 10.1016/j.braindev.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 23.a) Prade H, Mackenzie LF, Withers SG. Carbohydr Res. 1997;305:371–381. [Google Scholar]; b) Kempton JB, Withers SG. Biochemistry. 1992;31:9961–9969. doi: 10.1021/bi00156a015. [DOI] [PubMed] [Google Scholar]
- 24.Mahuran DJ, Lowden JA. Can J Biochem. 1980;58:287–294. doi: 10.1139/o80-038. [DOI] [PubMed] [Google Scholar]
- 25.Tropak MB, Reid SP, Guiral M, Withers SG, Mahuran DJ. J Biol Chem. 2004;279:13478–13487. doi: 10.1074/jbc.M308523200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang S, Bagshaw R, Hilson W, Oho Y, Hinek A, Clarke JT, Callahan J. Biochem J. 2000;348:621–632. [PMC free article] [PubMed] [Google Scholar]
- 27.Helferich B, Himmen E. Chem Ber. 1929;62:2136–2141. [Google Scholar]
- 28.Meisler M. Methods Enzymol. 1972;28:820–824. [Google Scholar]