

Abstract
Inference of biological networks from high-throughput data is a central problem in bioinformatics. Particularly powerful for network reconstruction is data collected by recent studies that contain both genetic variation information and gene expression profiles from genetically distinct strains of an organism. Various statistical approaches have been applied to these data to tease out the underlying biological networks that govern how individual genetic variation mediates gene expression and how genes regulate and interact with each other. Extracting meaningful causal relationships from these networks remains a challenging but important problem. In this article, we use causal inference techniques to infer the presence or absence of causal relationships between yeast gene expressions in the framework of graphical causal models. We evaluate our method using a well studied dataset consisting of both genetic variations and gene expressions collected over randomly segregated yeast strains. Our predictions of causal regulators, genes that control the expression of a large number of target genes, are consistent with previously known experimental evidence. In addition, our method can detect the absence of causal relationships and can distinguish between direct and indirect effects of variation on a gene expression level.
Key words: algorithms, gene networks, machine learning, regulatory regions
1. Introduction
INFERENCE OF BIOLOGICAL NETWORKS from high-throughput genomic data is a central problem in bioinformatics where many different types of methods have been proposed and applied to a wide diversity of datasets (Markowetz and Spang, 2007). Several recent studies have collected data in model organisms such as yeast and mouse which contain both genetic variations as well as gene expressions from a set of genetically distinct group of individuals. Originally, these “genetical genomics” datasets were used to identify genetic variations located at specific genomic locations that affect expression levels in the form of linkages or associations (Brem et al., 2002; Brem and Kruglyak, 2005). These studies treated expression levels as quantitative traits and each associated genomic location is called an expression quantitative trait locus (eQTL). More recently, various statistical approaches have been applied to these datasets demonstrating them as being particularly powerful for teasing out the underlying biological networks that govern how genetic variations mediate differential gene expression and how genes regulate and interact with each other (Lee et al., 2006; Robinson et al., 2002; Ghazalpour et al., 2006; Subramanian et al., 2005). Some of these methods build on pioneering work in using graphical models to model gene regulatory networks (Friedman et al., 2000; Pe'er et al., 2001; Segal et al., 2003; Friedman, 2004; Hartemink et al., 2001).
Extracting meaningful causal relationships from these networks has been a challenging but important area of genetical genomics. What differs genetical genomics studies from traditional microarray analysis and what makes causal inference possible is the idea to model genetic variations as random perturbations to the underlying regulatory network. A principled way of representing the causal relationships in a biologicla network is using graphical causal models (Pearl, 1988, 2000). Such models represent causal relationships between random variables by means of a directed acyclic graph called a causal graph, where a directed edge between two variables represents direct causal influence. The data-generating process represented by a causal graph imposes a variety of constraints, such as conditional independence constraints, on the observed data. A rich theory of causal inference has been developed (Pearl, 2000; Spirtes et al., 2000) which attempts to reconstruct aspects of the graph from the pattern of constraints in the observations. Causal relationships can then be read off directly from the reconstructed graph.
The advantage of the causal inference paradigm is that predictions made are in fact causal, and so can be directly verified with knockout, siRNA or allele swap experiments. Compared to other methods such as co-expression networks which aim to capture the global structure in the regulatory network, causal inference methods attempt to identify the actual biological mechanisms regulating gene expression. Furthermore, for many applications where the final goal is to perturb the biological system in some way, causal networks are advantageous because they naturally predict the effect of possible interventions. The resulting models can be perturbed in silico to help guide which experimental perturbations to apply.
The disadvantage of these methods is that existing causal inference theory is a large sample theory, and is only guaranteed to work asymptotically. Unfortunately, in the case of inferring biological networks from gene expression data, there are far fewer samples than genes, which means practical applications must be a successful synthesis of ideas from both causal inference and small sample statistics.
There are two main approaches to learning causal graphs in biological networks. Score-based methods assign scores to models which both produce high likelihood of the observed data, and have limited complexity, and search for the highest scoring model (Suzuki, 1993; Lam and Bacchus, 1994). These methods have been used in identifying causal regulators in yeast (Friedman et al., 2000; Pe'er et al., 2001; Segal et al., 2003; Zhu et al., 2008; Bing and Hoeschele, 2005; Kulp and Jagalur, 2006) and causal mediators of disease in mice (Schadt et al., 2005). Constraint-based methods rule out those causal graphs inconsistent with patterns of conditional independence constraints in the observations. These methods have been applied to discovering causal relationship between pairs of genes (Chen et al., 2007).
In this article, we discover the presence and absence of causal relationships between genes in yeast by examining their expression levels over a set of individuals with random genetic variations. Causal discovery is challenging in our case because there are several thousand genes, while the number of samples is very limited. In particular, most conventional conditional independence tests or model selection algorithms are not reliable in the small sample case, since conditioning severely reduces the number of samples available, and as a result we cannot infer independence with high confidence, limiting our ability to induce features of the causal graph.
Our approach is to rely on basic properties of graphical models to infer or exclude edge directionality based on either simple unconditional independence tests which are possible to perform even in the small sample case, or on results of simple model selection amongst small causal sub-graphs of the overall causal model which have particularly strong signals. Our philosophy is that due to the small number of samples, it is impossible to accurately recover the complete causal graph. We opt to predict only the subset of the network where our predictions are likely to be correct.
We take advantage of prior biological knowledge that genetic variations affect gene expressions, but not vice versa. This knowledge can be expressed graphically as forbidding directed paths from gene expressions to genetic variations. While in general it is not possible to recover most causal structures based on unconditional independence tests, the availability of prior knowledge allows us to “bootstrap” certain edge orientations, which in turn allows us to orient more paths as causal using basic properties of d-separation (described below). Moreover, we can also rule out certain edge orientations using the same principals, thus identifying the absence of certain causal relationships.
Our method is inherently conservative, only predicting the existence and orientation of edges in the causal graph if there is strong support from the sample data. As expected, our approach predicts only a small fraction of the complete causal regulatory network of yeast. However, the actual predictions made by the method are surprisingly consistent with previous experimentally validated knowledge of yeast gene regulation.
We demonstrate the utility of our method by analyzing the Brem et al. (2002) yeast strains. The 112 yeast strains in this dataset was created by crossing a laboratory strain with a wild strain of S. cerevisiae. Both genetic variations and gene expressions from each offspring have been collected. We focus our analysis on an interesting feature of this dataset known as “regulatory hotspots” or regions in the genome in which a genetic variation is correlated with the expression levels of many genes. Compared to traditional eQTL mapping techniques that first identified these “regulatory hotspots,” our method provides much richer causal information that simple correlation can not capture. First, our method allows us to infer causal relationships between pairs of genes to identify global regulators that control the gene expression of target genes correlated with a “regulatory hotspot”. Second, our method can exclude causal relationships between genes. Third, even when considering only variation-expression pairs, our method can distinguish whether a variation has a direct or an indirect effect on expression. While several other methods attempt to infer causal relationships between genes (Zhu et al., 2008; Chen et al., 2007), our method is the first to be able to exclude causal relationships and distinguish between direct and indirect effects of a variation.
We evaluate our method's ability to infer regulatory relationships by comparing it directly to two other competing methods as well as verifying our results with previous experimental validations. Using our method, of the 12 genes for which there is some experimental evidence that they behave as master regulators (Yvert et al., 2003; Zhu et al., 2008), we recover 9 of them. Furthermore, for one of our predictions, ILV 6, a competing method by Zhu et al., 2008 (2008) was not able to identify the gene as a causal regulator based on expression data alone. The gene was only identified when additional transcription factor binding data was incorporated. Combined with our method's ability to exclude specific causal relationships, we used gene set enrichment analysis to find that gene targets not causally affected by a regulator to be enriched for different pathways and biological processes than gene targets affected by the same regulator.
To evaluate our ability to distinguish between direct and indirect effects of a genetic variation on gene expression, we take advantage of the fact that most expression transcripts are affected directly by a few variations close to the gene through a mechanism called cis-regulation. Since our method does not rely on information about the relative positions of a genetic variation and its effected gene, an enrichment of cis-effects in our predictions for direct causal effects validates our method.
A shorter version of this article has previously been published as part of a conference proceeding (Kang et al., 2009). In this article, we provide more details on our causal inference procedure by providing the exact likelihoods for each gene in a triplet. We also updated our results by systematically identifying “regulatory hotspots” using a previously published method based on dividing the genome into discrete sections and approximating the appearance of a linkage as a Poisson process (Brem et al., 2002). Using this method, we identified 9 “regulatory hotspots” and 38 regulator genes which mediate the genetic variations. Finally, we provide two additional visualizations for the causal relationships we discover. We use a spring embedded algorithm to construct the yeast causal network and show that the “regulatory hotspots” overlap well with the inherent hub structures. We also use a representation grouped by the “regulatory hotspots” to show that there is significant cross talk between hotspots.
2. Methodology
2.1. Causal graphs for genetical genomics
We first introduce the machinery of causal inference needed to formalize our approach to inferring causal relationships between a genetic variation (a SNP) and the expression of a pair of genes. Our primary object of study is the probabilistic causal model (Pearl, 2000).
Definition 1
A probabilistic causal model (PCM) is a tuple M = 〈U, V, F, P(u)〉, where
U is a set of background or exogenous variables, which cannot be observed or experimented on, but which can influence the rest of the model.
V is a set
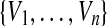 of observable or endogenous variables. These variables are considered to be functionally dependent on some subset of U ∪ V.
of observable or endogenous variables. These variables are considered to be functionally dependent on some subset of U ∪ V.F is a set of functions
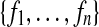 such that each fi is a mapping from a subset of U ∪ V \{Vi} to Vi, and such that ∪ F is a function from U to V.
such that each fi is a mapping from a subset of U ∪ V \{Vi} to Vi, and such that ∪ F is a function from U to V.P(u) is a joint probability distribution over the variables in U.
PCMs represent causal relationships between observable variables in V by means of the functions in F: a given variable Vi is causally determined by fi using the values of the variables in the domain of fi. Causal relationships entailed by a given PCM have an intuitive visual representation using a graph called a causal diagram. In this graph, each node is represented by a vertex, and a directed edge is drawn from a variable X to a variable Vi if X appears in the domain of fi. A graph obtained in this way from a model is said to be induced by said model.
A node Y is an ancestor of node Z in a causal diagram G if there is a directed path from Y to Z. Causal diagrams are generally assumed to be acyclic. While we expect the full yeast regulatory network to have causal cycles (they serve as common regulatory mechanisms), in this paper we concentrate our efforts on the fragments of the overall network where acyclicity holds.
One advantage of causal graphs, and graphical models in general (Pearl, 1988; Jordan and Weiss, 2002) is their ability to represent conditional independence relations between variables in a qualitative and intuitive way using the notion of path blocking known as d-separation (Pearl, 1988). Two variables X, Y are d-separated if all causal and confounding paths from X to Y contain at least one variable whose value is known, and the value of no common effect of both X and Y is known. Every d-separation statement involving two nodes (or sets of nodes) in the graph corresponds to a conditional independence among the corresponding sets of variables. That is, if every path from X to Y is blocked or d-separated by Z in a causal diagram G, then X and Y are conditionally independent given Z in every probability distribution compatible with G (Pearl, 1988). Furthermore in stable (Pearl and Verma, 1991) or faithful (Spirtes et al., 1993) models the converse is also true: conditional independencies in the observations imply the corresponding d-separation statement holds in the underlying causal graph. The faithfulness assumption thus allows us to infer aspects of the generating causal graph from conditional independence constraints apparent in the data, and is crucial for inductive causal inference. Faithfulness holds in “most” causal models, and can thus be justified on Occam's Razor grounds (Pearl, 2000).
Constraint-based inference of correct edge orientations in a causal diagram has two fundamental limits in practical applications. The first is that it can be difficult to collect sufficient samples to perform reliable conditional independence tests, and the second is that some causal diagrams may disagree on orientations of particular edges while entailing the same set of conditional independence constraints (such causal diagrams are called Markov-equivalent [Verma and Pearl, 1990]).
In this article, we will use causal graphs to represent causal interactions between genetic variations and gene expression levels in yeast. In our case, the genetic variations is the set of single nucleotide polymorphisms (SNPs). In this article, we limit our focus to inferring the presence or absence of a causal relationship between gene expression levels based on independence tests and model selection we can actually perform. We will be relying on the following three (elementary) theorems in graphical models.
Theorem 1
Let G be a causal graph where X is d-connected to Y via a path ending in an arrow pointing to Y, X is d-connected to Z, and X and Z are d-separated by Y. Then Y is an ancestor of Z.
If we assume faithfulness, this theorem implies we can infer causal directionality based on the result of two unconditional independence tests, and one conditional independence test. In our case, X is a SNP, Y is the expression level of a gene and Z is the expression level of a second gene. We are using our prior knowledge that expression levels do not affect SNP values to satisfy one of the preconditions of the theorem, namely that the d-connected path must end in an arrow pointing to Y. In particular, if Y is a gene expression value, and X is a SNP value correlated with on Y, then Y cannot cause X. Using this theorem, we are able to infer a causal relationship between the expression levels of genes Y and Z.
Unfortunately, testing whether X is conditionally independent of Z given Y in the small sample case is not feasible. An alternative approach which is more appropriate in our case is to use a model selection method, that is rather than performing the independence test, find the causal model over the local variables of interest, and read off causal directionality from its graph. In general, if we restrict ourselves to a small part of a large causal model which contains three variables X, Y, Z, the causal diagram which captures conditional independencies in the corresponding marginal distribution, that is P(x, y, z), will be a mixed graph containing both directed and bidirected arcs, called a latent projection (Verma and Pearl, 1990). In latent projections, a directed arc from X to Y corresponds to a d-connected path which starts with an arrow pointing away from X and ends with an arrow pointing towards Y in the original, larger graph such that every node on the path other than X and Y is marginalized out or latent. Similarly, a bidirected arc from X to Y corresponds to a d-connected path in the larger graph which starts with an arrow pointing to X, ends with an arrow pointing to Y, and every node on the path other than X and Y is marginalized out or latent.
If we restrict ourselves to local models of three variable marginal distributions, where certain causal relationships are excluded due to prior knowledge (e.g., genes cannot cause SNPs), the complete set of causal hypotheses is captured by a small set of latent projections.
Theorem 2
Let G be a causal graph where X is an ancestor of Y and Z. Then the latent projection which represents conditional independencies of P(x, y, z) is one of the graphs in Figure 3a.
FIG. 3.

Possible causal graphs relating a triplet considering a SNP S with the level of gene expression for genes Ei and Ej. Bidirected edges denote hidden common causes. (a) Nine possible causal models consistent with S being a causal ancestor of Ei and Ej (models H4 through H9 are indistinguishable from observations of the triplet). (b) Four possible causal models consistent with S being a causal ancestor of Ei while being uncorrelated with Ej.
Theorem 2 allows us to select the graph in Figure 3a which best fits the available data (we use a version of the likelihood ratio test), and use this graph to conclude causal directionality. The next theorem allows us to conclude the opposite, that a variable cannot be a causal ancestor of another.
Theorem 3
Let G be a causal graph where X is d-connected to Y, and X and Z are d-separated. Then Y cannot be an ancestor of Z.
As before, faithfulness allows us to apply this theorem to conclude the absence of causal directionality based on the results of two unconditional independence tests. In our case SNP X is associated with the expression level of gene Y, and SNP X is either independent of the expression level of gene Z or conditionally independent given some other gene. In this case we can rule out a direct causal relation between expression levels of genes Y and Z. In our case, the possible models are shown in Figure 3b. In the small sample case, we again use a maximum likelihood method to perform such tests.
In the next section, we describe our statistical methodology in more detail.
2.2. Inference algorithm overview
Our algorithm for inferring the presence or absence of causal relationships of gene expression proceeds in four steps. First, we find for every gene expression, the set of potential causal SNPs using the standard F-test. Second, we infer the presence of causal relationships between pairs of genes correlated with the same SNP by comparing the likelihoods of possible models. Third, we distinguish between direct and indirect effects of genetic variation on gene expression. Fourth, we infer the absence of causal relationships based on the results of step one and Theorem 3.
2.3. Finding potential causal SNPs
In the first step, we attempt to find, for every gene expression level, the set of potential causal SNPs, in other words the set of SNPs which are either causal or which are confounded with causal SNPs.
To examine the (potential) causal relationship between SNP Si and expression level Ej in our small sample case, we assume the following linear relationship between the two: Ej = αSi +ɛ. We use an arrow notation to signify potential causality (→) and the negation ↛ as no potential causality. Under the null hypothesis of no potential causal relationship between the SNP and expression levels (Si ↛ Ej), we expect α = 0 (H0). Under the alternate hypothesis of a potential causal relationship (Si → Ej), we expect α = 0 (H1). To decide between these hypotheses, one could calculate the likelihood ratio statistic 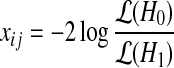 or use the standard F-test which is related to the likelihood ratio statistic
or use the standard F-test which is related to the likelihood ratio statistic 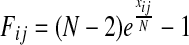 and follows asymptotically the F distribution with 1, k − 2 degrees of freedom where k is the number of samples. We calculate the F statistic Fij for every SNP/expression pair (Si, Ej). To assign significance, we shuffle the labels of the individuals B times to obtain the null statistics
and follows asymptotically the F distribution with 1, k − 2 degrees of freedom where k is the number of samples. We calculate the F statistic Fij for every SNP/expression pair (Si, Ej). To assign significance, we shuffle the labels of the individuals B times to obtain the null statistics  . Then the p-value of each SNP and expression pair can be calculated by looking at the ranking of the statistic of the pair in the permuted null statistic distribution.
. Then the p-value of each SNP and expression pair can be calculated by looking at the ranking of the statistic of the pair in the permuted null statistic distribution.
We can easily estimate the false discovery rate (FDR) for our statistic using previous approaches (Storey and Tibshirani, 2003). To limit the number of potential causal networks to evaluate in subsequent steps, we filter the SNP/expression pairs for those with a FDR of q < 0.01.
Due to linkage disequilibrium or local correlation of variation, the SNPs which are correlated with expressions are not likely to be actually causal, but instead correlated with causal SNPs in the same genomic region. Since all of the SNPs are correlated in a region, this does not affect our ability to make inferences about the causal regulatory network, but we must keep in mind that the SNPs which we predict to have direct effects are likely proxies for the true causal variants.
2.4. Finding causal relationships between genes
The next stage of our algorithm consists of inferring causal directionality between gene expressions by using Theorems 1 and 2. Since the two unconditional independence tests have already been performed in the first step, all that remains is to test conditional independence. Unfortunately, conditional tests present a problem in the small sample case since conditioning further limits the number of samples we have to test. An alternative approach is to consider multiple models consistent with the results of the unconditional independence tests where in some models the conditional independence holds, and in others it does not. If a model where the conditional independence test holds is the best fit for the data, and moreover accounts for more of the fit compared to a “default” model making no conditional independence assumptions, then we assume the conditional independence is likely true.
In our case, we are considering fragments of the causal graph consisting of a single SNP S and two expression levels Ei, Ej dependent on S (due to step 1). Figure 3a shows the nine possible causal models in the case that all of the elements are pairwise correlated. In H1 the SNP affects both expression levels independently. In H2 and H3, there is a direct causal relationship between the two expression levels. The “default” models H4 through H9 impose no constraints on the data and are indistinguishable based on conditional independence tests. Since they are all equivalent, for simplicity, we only consider H4 below.
We obtain information about the network whenever we predict a triplet to have a model H1, H2, or H3. To distinguish between the three hypothesis H1, H2 and H3, we compute likelihood ratio statistics for each hypothesis against the alternative H4, and conclude that a hypothesis is likely true if the corresponding ratio exceeds the other ratio (e.g., fits better than the other simple hypothesis) and is close to unity (e.g., a simpler hypothesis accounts for the observations). The fact that the likelihood ratio is close to unity means that the missing edge in the triplet does not hurt the likelihood of the model compared to “default” model (H4). This is equivalent to the standard approach of performing a likelihood ratio test for model selection taking into account a complexity penalty. In this case, the complexity penalty would be applied to H4 since the model has an additional degree of freedom. We also pairwise compare the likelihoods between H1, H2 and H3 against each other and only consider triplets where the most likely hypothesis is more likely than the others using a threshold.
We compute the likelihood for each model by computing the likelihoods at each target node. Since we are interested only in the causal effects on individual genes, we can represent the causal effects on an individual gene using a linear model assuming Gaussian noise. For every triplet, we can write the following linear model for genes g1 and g2 and the common associated SNP s.
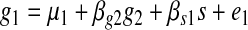 |
(1) |
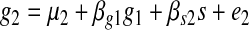 |
(2) |
where μ1 and μ2 are the means for g1 and g2 repectively, and βg2, βs1, βg1, and βs2 are causal coefficients for g2, s, g1, and s to their causal target nodes respectively, and e1 and e2 represent noise terms which follow Gaussian distribution. In this model, all coefficients are estimated by maximum likelihood estimation. The regression coefficients are interpreted as the Wright's rule (Wright, 1921) sum of path products of coefficients in the underlying (and unknown) true causal graph.
Since we assume that each gene expression is independently sampled from a underlying generative model, computing the likelihood of the model given data is done by multiplying all the gaussian density of errors calculated by least square method. We can represent this mathematically as follows:
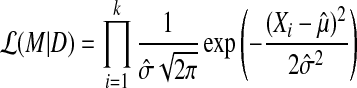 |
(3) |
where, Xi is the data sample, and k is the number of samples, and  and
and 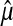 is computed by maximum likelihood estimation from given data.
is computed by maximum likelihood estimation from given data.
2.5. Distinguishing between direct and indirect effects of variation
If a SNP S is associated with two genes Ei and Ej, the nine possible models are H1, H2, H3 and the “default” models H4 through H9. The models H2 and H3 explain the associations as a direct effect of the SNP on one gene and an indirect effect on the other. Model H1 suggests that the SNP directly affects the expression levels of both genes. Since our statistical methodology uses H4 as the default model, we are unable to distinguish between direct and indirect effects if we can not classify a triplet as one of either H1, H2, or H3.
Establishing direct and indirect effects in causal analysis is always done with respect to particular model granularity. This is because it is generally always possible to observe intermediate variables between any direct cause and its effect—finer granularity removes directness of causation. In our case, when distinguishing direct versus indirect effects, the specific triplet that we are observing determines whether or not an effect is direct or indirect. Consider the following motivating example of a SNP S and three genes with the underlying network S → E1 → E2 → E3. If we consider the triplet (S, E2, E3), the correct structure of the subgraph is H2 and S will have a direct effect on E2 and an indirect effect on E3. Now if we consider the triplet (S, E1, E2), the correct structure is again H2 and S will have a direct effect on E1 and an indirect effect on E2. Intuitively, this is because when we consider the triplet (S, E2, E3), E1 is unobserved. Thus each prediction of a triplet as H2 or H3 induces a partial order on the causal relationships between gene pairs. After examining all pairs, we return the minimum set of causal relationships which are consistent with all of the triplet predictions.
More complicated networks introduce ambiguity into our ability to distinguish between direct and indirect effects. For example, if we add the edge S → E3 in our example, we can still identify E1 → E2 as a direct effect from the triplet (S, E1, E2), but are unable to identify E2 → E3 as a direct effect. This is because we will predict the structure of each triplet containing E3 and either E1 or E2 as H4 where the effects are ambiguous. However, if there is an additional edge in the graph E3 → E4, the triplet (S, E3, E4) would identify E3 → E4 as a direct effect.
2.6. Excluding causal relationships between genes
The ability to exclude certain causal relationships between genes, an inherent advantage of causal analysis, is important to obtaining a more complete understanding of genetic regulation. For example, a gene might be causal to a number of genes enriched for a biological process but not causal to a number genes enriched for a different biological process even though it is correlated with both sets of genes. We attempt to determine the absence of causal relationships by looking at a SNP and a pair of genes where the SNP is the potential cause of one gene, but not the other. In this case, basic properties of d-separation (Theorem 3) guarantee that there are only four possibilities H10 through H13 (Fig. 3b).
In H10, the SNP affects gene expression Ei, but gene expression Ej is completely independent from both the SNP and gene expression Ei. In H11, both the SNP and gene expression Ej affect gene expression Ei simultaneously. In H12, the SNP affects gene expression Ei, and gene expression Ei and gene expression Ej has a hidden common causal parent. In H13, both the SNP and gene expression Ej affect gene expression Ei and at the same time, gene expression Ei and gene expression Ej has a hidden common causal parent. In none of these models, gene expression Ei affects gene expression Ej.
We model the association between a SNP and a gene expression using a linear gaussian model as in Section 3.2. We correct multiple hypothesis testing problem by computing the false discovery rate (FDR) (Storey and Tibshirani, 2003). We identify pairs of genes Ei and Ej where we can exclude causal relationships using the following criterion: a SNP is significantly associated with Ei (FDR of q < 0.01) and not associated with Ej (FDR of q > 0.9).
3. Results
We applied our method to an expression dataset of 5534 genes and a genotyping dataset of 2956 SNPs collected over 112 genetic segregants of yeast. After step 1, we found 42331 (SNP, expression) pairs where the SNP is causal to the expression at a FDR of q < 0.01. We constructed triplets from these causal pairs to significantly reduce the number of possible causal models to evaluate for causal relationships between the genes in step 2. For each triplet, we considered the four possible models H1, H2, H3 and H4 and identified the most likely as described above. We find the most likely of H1, H2 and H3 and required that the log likelihood difference of the best model be within 2 of H4. This is equivalent to penalizing the likelihood of H4 and applying using the likelihood ratio for model selection. Inferring causal relationships with few samples can result in directional and causal conflicts. A directional conflict occurs when the direction of causation predicted between two genes is inconsistent using different SNPs. A causal conflict occurs when the presence and absence of a causal relationship predicted between two genes is inconsistent using different SNPs. We examined the robustness of our method by quantifying the number of directional and causal conflicts. Directional conflicts result when triples containing the same pair of genes and different SNPs predict different directional causal relations between the genes. Causal conflicts result when different triples both predict and exclude the same causal edge. As Table 1 shows, consistent across complexity penalties, fewer than 3% of predicted causal relationships are in conflict. These prediction conflicts are due to the limited number of samples available. We exclude all conflict predictions from our final result.
Table 1.
Summary Statistics for Different Likelihood Thresholds
| Complexity penalty | No. causal regulators | No. affected genes | No. causal relationships | No. direction conflicts | No. causal conflicts |
|---|---|---|---|---|---|
| 1 | 146 | 1106 | 2135 | 34 (1.6%) | 7 (.3%) |
| 1.5 | 183 | 1272 | 2794 | 30 (1.1%) | 11 (.4%) |
| 2 | 212 | 1396 | 3370 | 44 (1.3%) | 13 (.4%) |
| 2.5 | 240 | 1524 | 3983 | 59 (1.5%) | 18 (.5%) |
| 3 | 266 | 1615 | 4571 | 81 (1.8%) | 20 (.4%) |
The genetic variations inherent in the individuals we study can be seeing as naturally occurring random perturbations to the underlying regulatory networks that ultimately give rise to subtle differences in gene expression. We present our results in the context of these regulatory networks by identifying genes that are directly effected by the SNPs, regulators and those genes that are controlled by the regulators, targets. Formally, we call a gene a regulator if there exists a directed edge from a cis SNP to the gene and a gene a target if there exists a directed edge from a regulator to the gene. Intuitively, the requirement for a causal cis SNP ensures a high probability that the SNP directly perturbs the gene expression of the regulator. In our data, we found 3370 causal relationships consisting of 212 causal regulator genes and 1396 affected target genes. Table 1 shows the number of causal relationships, causal regulators and affected target genes discovered using various model complexity penalties for H4.
One way to make sense of the large number of causal relationships detected is to look for causal regulators that affect a number of genes or “causal hubs.” Of particular interest is identifying causal regulators that are associated with “regulatory hotspots,” defined as regions of the yeast genome linked to the expression of a large number of genes. Presumably, these “causal hubs” are important regulatory elements that lead to subtle changes in expression of genes belonging to a number of different biological processes and functions. Previous analyses have identified several “regulatory hotspots” in the yeast genome but very little is known about the corresponding “causal hubs” because of the limited resolution of genotyping studies. In a few isolated cases, several groups have performed experimental knock out studies to confirm the existence of causal regulators and allele swap studies to further show that these regulators are perturbed by the corresponding “regulatory hotspot” (Yvert et al., 2003; Zhu et al., 2008).
We first identified 9 “regulatory hotspots” similar to previous methods (Brem et al., 2002) by dividing the genome into 611 bins and approximating the number of linkages expected in each bin as a Poisson process. Figures 1 and 2 show the complete causal network inferred by our method with regulators and targets colored by the “regulatory hotspots” they belong to. Gray nodes indicate that a gene does not belong to any identified “regulatory hotspot.” Figure 1 shows the spring embedded network where the position of the nodes are determined so that the Euclidean distance is approximately proportional to the geodesic distance between two nodes (Kamada and Kawai, 1989). Several regulatory hotspots overlap remarkably well with the inherent hub structures that are present in this representation including hotspot 2 (bright red), hotspot 3 (bright green), and hotspot 9 (light blue). Figure 2 shows the same causal network but with the nodes grouped in a circle by the “regulatory hotspot” they belong to. This representation shows that there is significant cross talk between the regulatory hotspots and there is a significant number of genes, indicated by the gray nodes, that are not part of any regulatory hotspot in our causal network.
FIG. 1.

Complete causal network in yeast with the nine regulatory hotspots colored. Circles designate regulators, squares designate targets and diamonds designate genes that are both regulators and targets. The spring-embedded view of the causal network shows that some hotspots, hotspots 2 (red), 3 (bright green), and 9 (light blue), overlap well with the hub like structures of the network where regulators are positioned in the middle and targets surround the causal hub.
FIG. 2.

Complete causal network in yeast with the nine regulatory hotspots colored. Circles designate regulators, squares designate targets and diamonds designate genes that are both regulators and targets. Causal network grouped by hotspot shows that some regulators and targets (indicated by gray) are not part of known regulatory hotspots.
We further summarize our results by examining each regulatory hotspot in detail. The 38 genes which are involved in the nine regulatory hotspots among the top 45 genes which have the largest number of targets are summarized in Table 2. The seven genes which don't belong to nine hotspots include SDS24(60), LYS2(17), URA3*(17), GAS2(19), NMA111(20), NAM9+(14), and YML133C(30). Both Chen et al. (2007) and Zhu et al. (2008) applied causal inference methods to the same data allowing us to perform a direct comparison of the results. Among the genes suspected to be global regulators in the hotspots, there are a total of 12 causal regulators with some experimental evidence. Nine were proposed by the original group that collected the data: AMN1, MAK5, LEU2, MATALPHA1, URA3, GPA1, HAP1, SIR3, and CAT5 (Yvert et al., 2003). Three additional were validated in Zhu et al. (2008): ILV6, SAL1, and PHM7. Our method discovers all but 3 of these (MAK5, SIR3, and CAT5). We note that SIR3 and CAT5 have much weaker experimental evidence than the others and none of the comparison methods—neither Chen et al. (2007) nor Zhu et al. (2008)—were able to find these three. The best validation of our method is that we were able to find ILV6 which was experimentally validated in Zhu et al. (2008). However, Zhu et al. (2008) used additional types of data (incorporating TFBS data from ChIP-chip experiments, phylogenetic conservation, and protein protein interaction data (PPI)) in order to discover ILV6 and they claim that they would not have been able to discover ILV6 if they used only the data that we used. We note that ILV6 was also suggested as a regulator for this hotspot by Kulp et al. (2006). We recover the highlighted genes from Chen et al. (2007) including NAM9 which was not found by Zhu et al. (2008) and is supported by “bioinformatics type evidence” (GO analysis, etc). A direct comparison to Chen et al. (2007) is difficult because their results are organized in a different way, yet our results are consistent with Chen et al. (2007) in that they highlight their discovery of 6 of the experimentally validated regulators which we also discover.
Table 2.
Regulatory Hotspots and Corresponding Regulators
| Hot spot | SNP Chr | SNP Loc | Regulators | No. targets |
|---|---|---|---|---|
| 1 | 2 | 370000 | TAT1(49) | 49 |
| 2 | 2 | 530000 | AMN1*+(226),YSW1+ (190), TBS1*(177), CNS1*+(166), ARA1*(162), SUP45*(31), AGP2(17), TOS1*(16), YBR137W(16) | 303 |
| 3 | 3 | 90000 | NFS1 *(106), CIT2*(100), LEU2*+(77), HIS4(66), ILV6*+(29) | 169 |
| 4 | 3 | 200000 | MATALPHA1*(40), MATALPHA2 (24) | 41 |
| 5 | 8 | 110000 | GPA1*+(15) | 15 |
| 6 | 12 | 640000 | HAP1*(22), MAP1*(22) | 40 |
| 7 | 12 | 1060000 | YLR464W*(32), YRF1-4* (30), YRF1-5*(22) | 33 |
| 8 | 14 | 503000 | SAL1*+(138), LAT1(77), COG6(69), TOP2*(62), MSK1(38), YNL035C(38), SWS2(17) | 320 |
| 9 | 15 | 150000 | PHM7*+(227), RFC4(96), NDJ1*(69), HAL9*(66), ZEO1(55), WRS1(38), SKM1(28), YOL092W(18) | 263 |
Table 2 summarizes our results. Experimentally validated predictions are shown in bold. Regulators with an asterisk (*) were found by Zhu et al. (2008). Regulators marked with a plus (+) were found in Chen et al. (2007) study and unlabeled regulators are novel predictions. In parentheses after the name of the regulator is the number of targets that we found. We note that in most cases the experimentally validated regulator is at the top of the list. We also observed that with various model complexity cut off, the ranking of predicted genes is maintained, if the model complexity cut off is less than a certain threshold. Of particular interest are a group of regulators linked to chromosome 14 which is enriched for mitochondrial genes. Previous published studies in yeast did not identify any putative regulators in this region (Yvert et al., 2003). We found a number of genes in this region (including three previously identified genes, SAL1, NAM9 and TOP2) and several proteins of unknown function (including NMA111 and YNL035C).
We validate our ability to distinguish between direct and indirect effects of variation by considering the genomic positions of SNPs and the locations of genes that they are associated with. Variation that affects expression can be classified into two broad categories: cis-regulation which is an effect of a variation near a gene that affects expression of the gene and trans-regulation which is an effect of variation located in one region of the genome affecting expression of genes in other regions. It is suspected that most cis-regulation is direct while trans-regulation may be either direct or indirect. Of the 42, 331 SNP gene pairs where the SNP is associated with the expression of the gene, 11, 328 are predicted as cis-regulated gene while 31, 003 are trans-regulated gene. Using our approach, out of the 11, 328 cis-regulated genes, we predict 9, 385 of them to have a direct effect on expression. Out of 31,003 the trans-regulated genes, 20,509 of the SNP gene pairs have indirect effects. Thus cis-regulated genes are enriched in our predicted set of directly affected genes, while trans-regulated genes are enriched in indirectly affected genes.
We speculate that the identified causal regulators are likely to either directly control or perturb biological processes. However, step 3 of our analysis also identifies a collection of genes that are causally irrelevant to other genes. Combining results from these two steps can help us identify specific biological processes that are either regulated or not regulated by these causal regulators. We examined those eight significant causal regulators from our results with previous experimental validation. For each regulator, we construct two sets of genes, those that are causal targets and those that are causally irrelevent. We then use the hypergeometric distribution to assess the statistical significance of overlap of each gene set to known gene sets. Table 3 shows the different GO pathways that are enriched when we performed this analysis. The eight regulators appear to be involved in very different biological processes. For example, AMN1 is a causal regulator for ribosome biogenesis and assembly while four other regulators LEU2, MATALPHA1, URA3, and ILV6 are causally irrelevant for the process. Similarly, SAL1 is a causal regulator for the process of translation while HAP1 and PHM7 are causally irrelevant for the process. We notice that all significant processes are crucial for cell growth and survival but are controlled by different global regulators. The causal analysis shows that most of these global regulators participate specifically in certain biological processes. The one example of multiple regulators from the same regulatory hotspot includes LEU2 and ILV6. In this case, these two regulators participate in similar biological processes of organic acid metabolic process and amine biosynthetic process respectively. We further confirmed the specificity of these global regulators by enrichment analysis for localization of the causal and causally irrelevant targets. For example, SALl's causal targets are enriched for localization to the ribosome while HAPl's targets are enriched for localization to the mitochondrial membrane. Furthermore, both PHM7 and HAP1's causally irrelevant targets localized to cytosolic region of the cell where translation takes place. Similarly, although LEU2 and ILV6's causal targets are not enriched for a specific cellular compartment, their causally irrelevant targets are both enriched for the nucleolus where ribosome biogenesis and assembly takes place.
Table 3.
Significantly Enriched Processes for Causal and Not Causal Genes
|
Gene |
Regulated targets |
Unregulated targets |
||
|---|---|---|---|---|
| GO pathway | p value | GO Pathway | p value | |
| AMN1 | Ribosome biogenesis and assembly | 1.7×10−34 | Establishment of localization | 2.3×10−7 |
| LEU2 | Organic acid metabolic process | 3.0×10−7 | Ribosome biogenesis and assembly | 3.5×10−10 |
| MATα1 | Biological regulation | 5.9×10−6 | Ribosome biogenesis and assembly | 3.6×10−11 |
| URA3 | De novo pyrimidine base biosynthetic process | 4.6×10−6 | Ribosome biogenesis and assembly | 5.0×10−6 |
| HAP1 | Mitochondrial electron transport chain | 2.4×10−10 | Translation | 3.6×10−13 |
| ILV6 | Amine biosynthetic process | 2.4×10−17 | Ribosome biogenesis and assembly | 2.9×10−16 |
| SAL1 | Translation | 7.9×10−30 | Chromosome organization and biogenesis | 1.3×10−6 |
| PHM7 | Carbohydrate metabolic process | 3.7×10−9 | Translation | 2.0×10−8 |
4. Discussion
In this article, we combined a principled representation of causality using graphical causal models with small sample statistical methods to infer the presence and absence of causal relationships between yeast genes. Working with a dataset of genetically identical yeast strains allowed us to make strong causal assumptions about edge directionality in the underlying causal model. These assumptions, in turn, allowed us to take maximum advantage of the limited samples we had available by employing either unconditional independence tests, or simple model selection to discover or exclude causal directionality between gene expressions. This work motivates theoretical questions about the limits of causal inference based on either restricting or eliminating conditional independence tests, and relying strictly on unconditional tests. In addition, our method does not explicitly account for hidden confounding effects and could potentially make erroneous predictions. Detecting causal relationships with latent variables is a challenging and active area of both theoretical and applied research. Promising new techniques have been suggested and can potentially be incorporated into our method.
We demonstrated the usefulness of our method by examining yeast expressions collected over a segregated population derived from two parental strains to identifying many experimentally validated causal regulators. In addition, our approach is able to distinguish between direct and indirect variations and exclude causal relationships between genes. These results provide a rich description of the yeast gene regulation network beyond any previous results from mapping studies, coexpression analysis and competing causal methods.
Several interesting extensions can be applied to our method. One can either empirically or theoretically characterize the strength of effects recoverable by our method to hypothesize about the strength of regulation between genes. Many biological networks are in fact cyclical in nature and the assumption of certain type of noise structures has been shown to be useful in identifying cycles in causal graphs. Finally, incorporating additional phenotype information can potentially help us understand the genetic basis of complex phenotypes.
5. Acknowledgments
E.Y.K., C.Y., I.S., and E.E. are supported by the National Science Foundation (grants 0513612, 0731455, and 0729049) and the National Institutes of Health (grant 1K25HL080079). This research was supported in part by the UCLA subcontract of contract N01-ES-45530 from the National Toxicology Program/National Institute of Environmental Health Sciences to Perlegen Sciences.
Disclosure Statement
No competing financial interests exist.
References
- Bing N. Hoeschele I. Genetical genomics analysis of a yeast segregant population for transcription network inference. www.genetics.org/cgi/content/abstract/170/2/533. Genetics. 2005;170:533–542. doi: 10.1534/genetics.105.041103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem R. Kruglyak L. The landscape of genetic complexity across 5,700 gene expression traits in yeast. www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve\&db=pubmed\&dopt=Abstract\&list\_uids=15659551\&query\_hl=17\&itool=pubmed\_docsum. Proc. Natl. Acad. Sci. USA. 2005;102:1572–1577. doi: 10.1073/pnas.0408709102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem R.B. Yvert G. Clinton R., et al. Genetic dissection of transcriptional regulation in budding yeast. www.sciencemag.org/cgi/content/abstract/296/5568/752. Science. 2002;296:752–755. doi: 10.1126/science.1069516. [DOI] [PubMed] [Google Scholar]
- Chen L. Emmert-Streib F. Storey J. Harnessing naturally randomized transcription to infer regulatory relationships among genes. http://genomebiology.com/2007/8/10/R219. Genome Biol. 2007;8:R219. doi: 10.1186/gb-2007-8-10-r219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman N. Inferring cellular networks using probabilistic graphical models. Science's STKE. 2004;303:799. doi: 10.1126/science.1094068. [DOI] [PubMed] [Google Scholar]
- Friedman N. Linial M. Nachman I., et al. Using Bayesian networks to analyze expression data. J. Comput. Biol. 2000;7:601–620. doi: 10.1089/106652700750050961. [DOI] [PubMed] [Google Scholar]
- Ghazalpour A. Doss S. Zhang B., et al. Integrating genetic and network analysis to characterize genes related to mouse weight. http://genetics.plosjournals.org/perlserv/?request=get-document\&doi=10.1371/journal.pgen.0020130. PLoS Genet. 2006;2 doi: 10.1371/journal.pgen.0020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartemink A. Gifford D. Jaakkola T., et al. Using graphical models and genomic expression data to statistically validate models of genetic regulatory networks. Pac. Symp. Biocomput. 2001;6:422–433. doi: 10.1142/9789814447362_0042. [DOI] [PubMed] [Google Scholar]
- Jordan M.I. Weiss Y. Graphical models: probabilistic inference. In: Arbib M., editor. The Handbook of Brain Theory and Neural Networks. 2nd. MIT Press; Cambridge, MA: 2002. [Google Scholar]
- Kamada T. Kawai S. An algorithm for drawing general undirected graphs. Inf. Process. Lett. 1989;31:7–15. [Google Scholar]
- Kang E.Y. Shpitser I. Ye C., et al. Detecting the presence and absence of causal relationships between expression of yeast genes with very few samples. Proc. 13th Annu. Conf. Res. Comput. Mol. Biol. 2009:466–481. doi: 10.1089/cmb.2009.0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulp D. Jagalur M. Causal inference of regulator-target pairs by gene mapping of expression phenotypes. www.biomedcentral.com/1471-2164/7/125. BMC Genomics. 2006;7:125. doi: 10.1186/1471-2164-7-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam W. Bacchus F. Learning bayesian belief networks: an approach based on the MDL principle. Comput. Intell. 1994;10 [Google Scholar]
- Lee S.-I. Pe'er D. Dudley A.M., et al. Identifying regulatory mechanisms using individual variation reveals key role for chromatin modification. www.pnas.org/cgi/content/abstract/103/38/14062. Proc. Natl. Acad. Sci. USA. 2006;103:14062–14067. doi: 10.1073/pnas.0601852103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowetz F. Spang R. Inferring cellular networks-a review. BMC Bioinform. 2007;8:S5. doi: 10.1186/1471-2105-8-S6-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearl J. Probabilistic Reasoning in Intelligent Systems. Morgan and Kaufmann; San Mateo: 1988. [Google Scholar]
- Pearl J. Causality: Models, Reasoning, and Inference. Cambridge University Press; New York: 2000. [Google Scholar]
- Pearl J. Verma T.S. A theory of inferred causation. Principles Knowl. Represent. Reason. Proc. 2nd Int. Conf. 1991:441–452. [Google Scholar]
- Pe'er D. Regev A. Elidan G., et al. Inferring subnetworks from perturbed expression profiles. Bioinformatics. 2001;17(S1):S215–S225. doi: 10.1093/bioinformatics/17.suppl_1.s215. [DOI] [PubMed] [Google Scholar]
- Robinson M. Grigull J. Mohammad N., et al. Funspec: a web-based cluster interpreter for yeast. www.biomedcentral.com/1471-2105/3/35. BMC Bioinform. 2002;3:35. doi: 10.1186/1471-2105-3-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt E.E. Lamb J. Yang X., et al. An integrative genomics approach to infer causal associations between gene expression and disease. http://dx.doi.org/10.1038/ng1589. Nat. Genet. 2005;37:710–717. doi: 10.1038/ng1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E. Shapira M. Regev A., et al. Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat. Genet. 2003;34:166–176. doi: 10.1038/ng1165. [DOI] [PubMed] [Google Scholar]
- Spirtes P. Glymour C. Scheines R. Causation, Prediction, and Search. Springer Verlag; New York: 1993. [Google Scholar]
- Spirtes P. Glymour C. Scheines R. Causation, Prediction, and Search. MIT Press; Cambridge, MA: 2000. [Google Scholar]
- Storey J. Tibshirani R. Statistical significance for genomewide studies. www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve\&db=pubmed\&dopt=Abstract\&list\_uids=12883005. Proc. Natl. Acad. Sci. USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A. Tamayo P. Mootha V.K., et al. From the cover: gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. www.pnas.org/cgi/content/abstract/102/43/15545. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki J. A construction of bayesian networks from databases based on an MDL scheme. Proc. UAI1993. 1993:266–273. [Google Scholar]
- Verma T.S. Pearl J. Equivalence and synthesis of causal models [Technical report R-150] Department of Computer Science, University of California; Los Angeles: 1990. [Google Scholar]
- Wright S. Correlation and causation. J. Agric. Res. 1921;20:557–585. [Google Scholar]
- Yvert G. Brem R. Whittle J., et al. Trans-acting regulatory variation in Saccharomyces cerevisiae and the role of transcription factors. Nat. Genet. 2003;35:57–64. doi: 10.1038/ng1222. [DOI] [PubMed] [Google Scholar]
- Zhu J. Zhang B. Smith E., et al. Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat. Genet. 2008;40:854. doi: 10.1038/ng.167. [DOI] [PMC free article] [PubMed] [Google Scholar]