

Abstract
The innate immune response is the first line of host defense against infections. This system employs a number of different types of cells, which in turn activate different sets of genes. Microarray studies of human and mouse cells infected with various pathogens identified hundreds of differentially expressed genes. However, combining these datasets to identify common and unique response patterns remained a challenge. We developed methods based on probabilistic graphical models to combine expression experiments across species, cells, and pathogens. Our method analyzes homologous genes in different species concurrently overcoming problems related to noise and orthology assignments. Using our method, we identified both core immune response genes and genes that are activated in macrophages in both human and mouse but not in dendritic cells, and vice versa. Our results shed light on immune response mechanisms and on the differences between various types of cells that are used to fight infecting bacteria. For supporting website, see www.cs.cmu.edu/∼lyongu/pub/immune/.
Key words: computational molecular biology, expression analysis, Gaussian random field, gene expression, gene networks, immune response, machine learning, sequence analysis
1. Introduction
Innate immunity is the first line of antimicrobial host defense in most multi-cellular organisms and is instructive to adaptive immunity in higher organisms (Fearon et al., 1996). There are multiple types of immune cells, including macrophages and dendritic cells. Depending on their role, each type of cell may respond by activating a different set of genes, even to the same bacteria (Chaussabel et al., 2003). In addition to the cell type, innate immune response differs based on the specific pathogen in question (Nau et al., 2002). To date, gene expression profiling has been used to investigate transcriptional changes in human and mouse macrophages and dendritic cells during infection with several different pathogens (Chaussabel et al., 2003; Detweiler et al. 2001; Draper et al., 2006; Hoffmann et al., 2004; Huang et al., 2001; Lang et al., 2002; McCaffrey et al., 2004; van Erp et al., 2006). In each of these studies, a list of genes involved in the response is determined by first ranking the genes based on their expression changes and then selecting the top-ranked genes based on a score or p-value cutoff. While some articles have analyzed data from multiple cell types or multiple pathogens, a large-scale comparison of these datasets across cells, pathogens, and different species has not yet been performed.
Microarray expression experiments that study immune response to bacteria infection can be divided along several lines. Here we focus on three such divisions: cell types, bacteria types, and host species.
Innate immunity is the result of the collective responses of different immune cells, which are differentiated from multipotential hematopoietic stem cells (Keller and Snodgrass, 1990). To understand the roles of and possible interplays between different types of immune cells, it is important to identify both the common responses of different immune cells, as well as responses unique to a certain cell type. Identification of genes differentially expressed in macrophages but not in dendritic cells, and vice versa, may highlight their specific functions and help us understand mechanisms leading to their different immune response roles. In addition to the different cells, specific bacteria types are known to trigger very different innate immune responses (Nau et al., 2002). Specifically, response to Gram-positive and Gram-negative bacteria is activated by different membrane receptors that recognize molecules associated with these bacteria. Finally, many of the key components in the innate immune system are highly conserved (Hoffmann et al., 1999). For example, the structure of Toll-like receptors (TLRs), a class of membrane receptors that recognizes molecules associated with bacteria, is highly conserved from Drosophila to mammals. It is less known though to what extent the immune response program is conserved and what other genes play a role in this conserved response.
While each of these subsets of experiments (e.g., macrophages vs. dendritic, human vs. mouse) can be analyzed separately using ranking methods and then compared, due to noise in gene expression data methods that rely on a score cutoff become much less reliable for genes closer to the threshold (Lu et al., 2007). Thus, analyzing responses to different pathogens and then examining the overlap between the lists derived for each experiment may not identify a comprehensive list of immune response genes. Similarly, while comparing the expression changes triggered by similar bacteria in human and mouse may lead to the identification of conserved immune response patterns, direct comparison of these profiles across experiments is sensitive to noise and orthology assignments, leading to unreliable results and underestimation of conservation (Liu et al., 2007).
In previous work (Lu et al., 2006, 2007), we combined expression datasets from several species to identify conserved cell cycle genes. The underlying idea is that pairs of orthologous genes are more likely than random pairs to be involved in the same cellular system. Thus, if one of the genes in the pair has a high microarray expression score while the other has a medium score, we can use the high scoring gene to elevate our belief in its ortholog, and vice versa. We used discrete Markov random fields (MRFs) to construct a homology graph between genes in different species. We developed a belief propagation algorithm to propagate information across species allowing orthologous genes to be analyzed concurrently.
Here we extend this method in several ways so that it can be applied to analyzing immune response data. Unlike the cell cycle, which we assumed worked in a similar way in all cell types of a specific species, here we are interested in both common responses and distinguishing responses for each dividing factor. This requires a different analysis of the posterior values assigned to nodes in the graph. In addition, for the immune response analysis, genes are represented multiple times in the graph (once for each cell and bacteria type) leading to a new graph topology. We are also interested in multiple labels for immune response (up, down, not changing) compared to the binary labels we used for cell cycle analysis. Finally, in this article, we use a Gaussian random field instead of a discrete Markov random field leading to faster updates and improved analysis. Instead of simply connecting genes with high protein sequence similarity, the edges in the graph are determined in a novel way that enables us to utilize the information contained in sequence homology in a global manner, leading to improved prediction performance.
We have used our method to combine data from expression experiments across all three dividing factors. Our method identified a core set of genes containing many of the known immune response genes and a number of new predictions. In addition, our method successfully highlighted differences between conserved responses in macrophages and dendritic cells, shedding new light on the functions of these types of cells.
A number of articles have used Markov random field models to integrate biological data sources. These include work on protein function prediction (Deng et al., 2004; Letovsky and Kasif, 2003) and functional orthology prediction (Bandyopadhyay et al., 2006). Our method has different goals and uses different data sources. In addition, our work differs from these previous articles in several important aspects. Our method propagates information from different cell types and species to improve gene function prediction, while previous work either did not use cross-species information, or only used it to align networks from different species. We do so by defining our model on a network that explicitly represents genes from different species and cell types. In contrast, previous work either focused on a single species (Deng et al., 2004; Letovsky and Kasif, 2003), or on an aligned network where each node represents an orthologous group (Bandyopadhyay et al., 2006). Finally, our model is defined on continuous random variables instead of discrete variables, which enables us to predict three-class labels (up/unchanged/down), while most previous models only handle two-class labels.
2. Computational Model: Gaussian Random Field
We formulate the problem of identifying immune response genes using a probabilistic graphical model. In a probabilistic graphical model, random variables are represented by nodes in a graph, and conditional dependency relations are represented by edges. Probabilistic graphical models can be based on directed graphs or on undirected graphs. The model we use here is based on undirected graphs, where special functions (termed “potential functions”) are defined on nodes and edges of the graph, and the joint probability distribution is represented by the product of these potential functions. The form of the potential functions encodes our prior knowledge as well as modeling preferences.
We use Gaussian random fields (GRFs) to model the assignment of gene labels. Gaussian random fields are a special type of Markov random fields. In a GRF, every node follows a normal distribution, and all nodes jointly follow a multivariate normal distribution. There are two types of nodes in our graphical model (Fig. 1). The first type is a gene node; it represents the status of a gene in a certain cell type, from a certain host species, in response to a certain type of pathogen. Here we consider two cell types (macrophages and dendritic cells), two host species (humans and mice), and two pathogen types (Gram-negative and Gram-positive bacteria), although the model is general and can accommodate other types as well. The set of possible labels for each gene can be either two (involved in immune response or not), or three (suppressed, induced, or unchanged during immune response). For simplicity, we will describe our model using binary labels, but will present the results based on both sets of possible labels.
FIG. 1.

Diagram of the Gaussian random field (GRF) model. (a) A subgraph in the GRF containing homologous human and mouse genes. The white node hm+ represents the (latent) label of the human gene h in macrophages under infection of Gram-positive bacteria. hm− represents the gene's label in macrophages under infection of Gram-negative bacteria. hd+ and hd− represent the labels of the same genes in dendritic cells under the infection of Gram-positive or Gram-negative bacteria. mm+, mm−, md+, and md− are similarly defined for the homologous mouse gene m. Two white nodes are connected by an edge if they represent the same gene in two experiments, either on the same cell type or under the infection of the same type of bacteria. We also connect two white nodes if they represent homologous genes in the same cell type and under the infection of the same type of bacteria. The black nodes represent the observation from the expression data in a certain cell type and under the infection of the appropriate bacteria. They are connected with the white nodes representing the corresponding genes under the same condition. (b) A high level diagram of the GRF model. Each dotted box represents a subgraph of four nodes related to the same gene as those shown in (a), and each “edge” represents four edges connecting the nodes of homologous genes in the two dotted boxes, in the same way as shown in (a).
Corresponding to each gene node is a score node, representing the observed expression profile of the corresponding gene. Together, the GRF jointly models the labels of all genes in all cell types, all species, and under both types of infection conditions. The edges in the GRF represent the conditional dependencies between gene labels. We put an edge between two gene nodes when they are a priori more likely to have the same label. Specifically, there are two cases where we add an edge. In the first case, for each gene node in the graph, we connect it with another gene node if the protein sequence similarity between these two genes is high and the experiments related to both nodes are in the same cell and bacteria types. The assumption is that genes with similar sequence are more likely to have similar function in the same type of cell and for the same bacteria. The edge potential function defined on these edges introduces a penalty when two genes with high sequence similarity are assigned different labels. In the second case, we connect a gene node with another gene node if the two nodes represent the same gene in the same type of cell or bacteria. Here we assume the genes are likely to function similarly in the same type of cell, or under the same type of infection. Again, the potential function penalizes the case where a gene is assigned a different label under different conditions for the same cell. The size of the penalty depends on the strength or weight attached to the edge. Different edges may have different weights. The joint probability is defined as the product of the node potential functions and edge potential functions, divided by a normalization function. We can infer the label of individual genes by estimating the joint maximum a posteriori (MAP) assignment of all nodes.
2.1. Computing the weight matrix
An important issue in random field models is the assignment of edge weights. Employing a similar approach but in a simpler setting, Lu et al. (2006) use a Markov random field to jointly model gene statuses in multiple species, where edges in the graph are weighted by BLASTP (Altschul et al., 1990) scores between pairs of genes. Given two genes connected in the graph, the edge weight (BLASTP bit score) represents the sequence similarity between the two genes, which in turn captures the a priori dependency between their labels. While this is a useful strategy, in a Markov random field model, edges represent the dependency between the two nodes conditioned on the labels of all other nodes (Bishop, 2006). In contrast, sequence similarity is computed for a pair of genes regardless of other genes. In other words, what a BLASTP score captures is the marginal dependency between the two genes' labels rather than the conditional dependency.
To address this issue we compute new edge weights using the BLASTP score matrix, which captures the marginal covariance of the Gaussian random field. It has been shown that for GRFs the appropriate weight matrix is equal to the inverse of the marginal covariance matrix (Zhu, 2005).
Using this observation, we can build a similarity matrix based on BLASTP scores, and use its inverse as the weight matrix for the GRF. Each row (and each column) in the similarity matrix corresponds to a gene. If the BLASTP bit score between two genes is above a cutoff, we set the corresponding elements in the similarity matrix to that score. Otherwise, it's set to zero. We use a stringent cutoff so that we are fairly confident of the functional conservation when we add a non-zero element. Because the similarity matrix contains scores for all genes in two species, the computational cost to invert it is very high. We thus compute an approximate inverse. We first convert the matrix into a diagonal block matrix by Markov clustering algorithm (Enright et al., 2002), then compute the approximate inverse by inverting each block independently. The matrix inversion is done using the Sparse Approximate Inverse Preconditioner (Grote and Huckle, 1997).
Finally, we assign edge weights based on this inverse matrix. Note that each gene is represented by four nodes in the graph, because it is present in different experiments on two cell types and two types of pathogens. For edges connecting gene nodes in different species, we set the weight according to the inverse similarity matrix. For edges connecting the same gene in different types of cells and bacteria, we use a single hyperparameter as their edge weight for cell and bacteria relationships.
2.2. Expression score distribution
The gene expression score is a numeric summary computed from the gene's microarray time series, which we will define in Results. We assume that the scores of genes with the same label follow a Gaussian distribution with an experiment specific mean and variance. Due to noise in microarray experiments, these distributions are highly overlapping, making it hard to separate labels by expression score alone.
2.3. Node potential function
The node potential functions capture information from gene expression data. For each gene i, let Ci denote its (hidden) label, Si denote its expression score, yi denote the random variable in the GRF associated with this gene. As mentioned above Ci can be a binary variable or a ternary variable if we consider three gene labels. Si and yi are both real variables. Because each yi follows a normal distribution, we need to have a way to link a gene's probability of belonging to each class with the corresponding normal distribution. This is achieved by the probit link function. In the binary labels case, let pi be the probability of gene i being an immune response gene conditioned on its expression score Si,
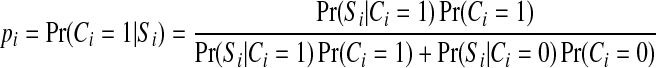 |
For the GRF the node potential function is defined as
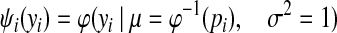 |
1 |
where ψ(yi | μ, σ2) is the probability density function for the normal distribution with mean μ and variance σ2, and φ−1(x) is the probit function, i.e., the inverse cumulative distribution function for the standard normal distribution. In other words, the information from a gene's expression score is encoded by a normal distribution of yi such that pi = Pr(yi > 0).
In the case of three labels for genes (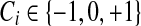 ), we can use the following formulas to link the probabilities of Ci and yi:
), we can use the following formulas to link the probabilities of Ci and yi:
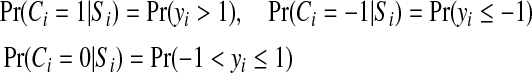 |
(2) |
It can be proven that given any (non-zero) probability mass function on Ci, we can find a normal distribution N(μ, σ2) such that these formulas are satisfied when yt ∼ N(μ, σ2). In fact, if we denote α = Pr (Ci = −1 |Si), b = Pr (Ci = 0|Si), and c = Pr (Ci = 1 |Si), it can be verified that σ = 2 / (Φ−1(a + b) − Φ−1(a)) and μ = 1 + σ · Φ−1(a) gives the distribution that satisfies Eq (2).
2.4. Edge potential function
The edge potential functions capture the conditional dependencies between pairs of gene nodes. The assumptions here are that (1) genes with higher sequence similarity are more likely than otherwise to have the same or similar functions; and (2) a given gene is likely to have the same function across cell types and across pathogens.
First we will define the edge potential functions for edges connecting homologous genes in the same cell type and under infection of the same type of bacteria. In this case, the edge potential function depends on the weight matrix we introduced above. Note that although all elements in the BLAST score matrix are non-negative (sequence similarities are non-negative), its inverse matrix may have negative elements. As a consequence, edge weights can be either positive or negative. A positive edge weight indicates that the labels of the two gene are positively correlated, conditioned on the labels of all other gene nodes. A negative edge weight means that they are negatively correlated, conditioned on the other gene nodes.
The following edge potential function captures this dependency (=0 is a positive hyperparameter):
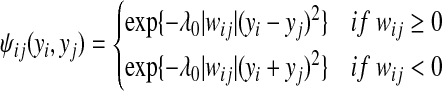 |
When the edge weight wij is positive, the edge potential function places a penalty if yi and yj are different. The larger the difference, the higher the penalty. Likewise, when wij is negative, the edge potential function introduces a penalty based on how close yi and yj are to each other. The penalty becomes higher when we become more confident in yi and yj and the two are close.
For edges connecting the same gene in the same cell type but under infection of different types of bacteria, the edge potential function is defined as
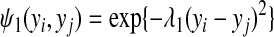 |
where λ1 is a positive hyperparameter. Similarly for edges connecting the same gene under the infection of the same type of bacteria but in different cell types, the edge potential is defined as
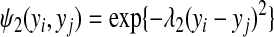 |
where λ2 is a positive hyperparameter. Together, the joint likelihood function is defined as
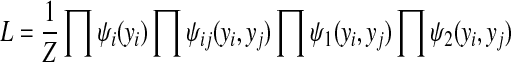 |
3. Learning The Model Parameters
In this section, we will present our algorithm based on two gene classes. The algorithm can be extended to three gene classes by using different node potential functions (see Section 2.3). For our model, we need to learn the hyperparameters λ. We also need to learn the parameters of the expression score distributions for each combination of cell types, host species, and pathogen types. In each case, there are four parameters 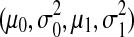 , i.e., the means and variances of the two different Gaussian distributions, one corresponding to the scores of immune response genes, the other corresponding to the scores of the remaining genes.
, i.e., the means and variances of the two different Gaussian distributions, one corresponding to the scores of immune response genes, the other corresponding to the scores of the remaining genes.
We learn these parameters in an iterative manner, by an EM-style algorithm. We start from an initial guess of the parameters. Based on these parameters, we infer “soft” posterior assignments of labels to the genes using a version of the belief propagation algorithm on the GRF. The posterior assignments are in turn used to update the score distribution parameters. We repeat the belief propagation algorithm based on the new parameters to infer updated assignments of labels. This procedure goes on iteratively until the parameters and the assignments do not change anymore. We discuss these steps in detail below.
3.1. Iterative step 1: inference by belief propagation
Given the model parameters, we want to compute the posterior marginal distribution for each latent variable yi, from which we can derive for each gene node the posterior probability of being involved in immune response. It is hard to compute the posteriors directly because the computational complexity of the normalization function in the joint likelihood function scales exponentially. However, due to the dependency structure in the GRF, we can adapt the standard Belief Propagation algorithm (Yedidia et al., 2003) for GRF, and use it to compute all the posteriors efficiently.
Unlike MRFs defined on discrete variables, variables in GRFs are continuous and follow normal distributions. The current estimation of the marginal posterior (“belief”) of every latent variable yi in the GRF is a normal distribution. Similarly, the “messages” passed between nodes are also normal distributions.
The Belief Propagation algorithm consists of the following two steps: “message passing,” where every node in the GRF passes its current belief to all its neighbors, and “belief update,” where every node updates its belief based on all incoming messages. The algorithm starts from a random guess of the beliefs and messages, and then repeats these two steps until the beliefs converge.
(1) Message passing. In this step, every node yi computes a message for each of its neighbors yj, sending yi's belief of yj's distribution. The message is based on the potential functions, which represent local information (node potential) and pairwise constraints (edge potential), as well as incoming messages from all yi's neighbors except yj.
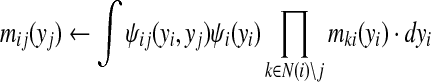 |
(3) |
(2) Belief update. Once node yi has received messages from all its neighbors, it updates the current belief incorporating all these messages and the local information from the node potential. The update rule is as follows
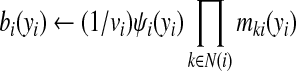 |
(4) |
where vi is a normalization constant to make bi(yi) a proper distribution.
Because all the messages and beliefs come from a normal distribution, they can be represented by the corresponding means and variances. Thus, in this case the message update rule and belief update rule above can be formulated into rules updating the means and variances directly, completely avoiding these computationally expensive integration operations.
3.1.1. Derivation of the update rules
We now derive the update rules used in the belief propagation algorithm. Note that the operations carried out in Eqs (3) and (4) are multiplication of univariate Gaussian distributions and marginalization of bivariate Gaussian distributions. For multiplication of univariate Gaussian distributions with mean μi and variance 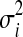 , we have
, we have
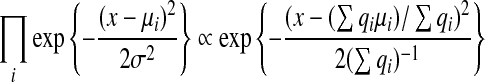 |
where 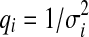 . The resulting product is a Gaussian distribution with the following mean and variance
. The resulting product is a Gaussian distribution with the following mean and variance
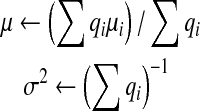 |
We can get belief update rules for Eq (4) by substituting μi and 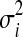 with the mean and variance of mki(yi) and Ψi(yi) where k belongs to the set of the neighbors of i excluding j.
with the mean and variance of mki(yi) and Ψi(yi) where k belongs to the set of the neighbors of i excluding j.
Next we derive the rules for marginalization of bivariate Gaussian distributions in Eq (3). Let
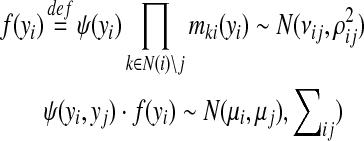 |
(5) |
and
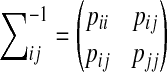 |
We can compute the mean and variance of message mij(yj), which is the result of marginalization of the bivariate Gaussian distribution in Eq (3), by matching the left-hand side (LHS) and right-hand side (RHS) of Eq (5). By expanding the exponent of the RHS of Eq (5), we get
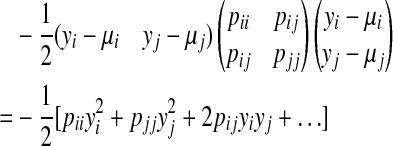 |
(6) |
Substituting and expanding the exponent of the LHS of Eq (5), we get
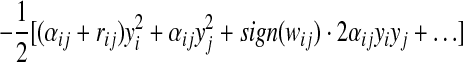 |
(7) |
where
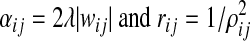 |
Equating (6) and (7), we can get the following update rules for computing the mean and variance of message mij(yj)
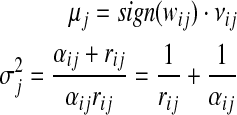 |
3.2. Iterative step 2: updating the score distribution
The posterior computed in step 1 is based on the current (the g'th iteration) estimation of parameters, collectively denoted by θ(g). The goal now is to determine the parameters that maximize the expected log-likelihood of the complete data over the observed expression scores given the parameters θ(g) = (μ0(g), σ0(g), μ1(g), σ1(g)).
To update the parameters of the score distributions, we first compute the posterior probability of a gene being involved in immune response, based on the posterior of yi. This is the same as applying the reverse probit function:
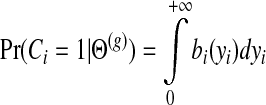 |
For simplicity, we use the following notations
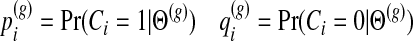 |
The updated distribution parameters for a Gaussian mixture are computed by standard rules
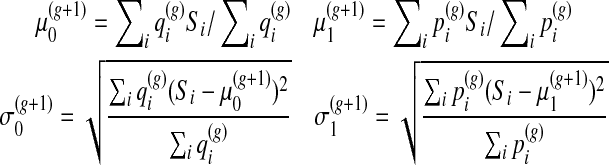 |
Our algorithm is summarized in Table 1.
Table 1.
Algorithm for Combining Immune Response Gene Expression Data
| Input |
|---|
| 1. Expression score Si for each gene in each cell type, host species, and pathogen type |
| 2. Graph structure (edge weights) |
| Output |
| For each gene node, its posterior probability of belonging to each class |
| Initialization |
| For each combination of host species, cell type, and pathogen type, compute estimates μ0, σ0, μ1, and σ1 using permutation analysis |
| Iterate until convergence |
| 1. Use Belief Propagation to infer a posterior for each gene node |
| 2. Use the estimated posterior to re-estimate the Gaussian expression score distributions |
3.3. Learning the hyperparameters
To learn the hyperparameters λ0, λ1, and λ2, we use a list of known immune genes. These serve as training data for our algorithm. Following convergence of the belief propagation algorithm, we optimize the prediction accuracy using the Nelder-Mead algorithm (Nelder and Mead, 1965). Note that this list is not used for the Results below. We divided our list of known immune genes and only used a third to learn the parameters. The other two thirds were used for the comparisons discussed below.
4. Results
4.1. Immune response data
Immune response microarray experiments were retrieved from supporting websites (Chaussabel et al., 2003; Detweiler et al., 2001; Draper et al., 2006; Granucci et al., 2001; Hoffmann et al., 2004; Huang et al., 2001; Lang et al., 2002; McCaffrey et al., 2004; van Erp et al., 2006), totaling 39 data sets. The data sets include experiments on macrophages and dendritic cells in humans and mice. For each cell type, we have included experiments using Gram-positive and Gram-negative bacteria, except for mouse dendritic cells, for which we only found Gram-negative bacteria datasets. Human and mouse orthologs were downloaded from Mouse Genome Database (Eppig et al., 2005). Table 2 summarizes the datasets used in this article.
Table 2.
Summary of Datasets Used
| Host/cell type | Gram-negative datasets | Gram-positive datasets |
|---|---|---|
| Human macrophages | 5 | 2 |
| Human dendritic cells | 9 | 2 |
| Mouse macrophages | 7 | 7 |
| Mouse dendritic cells | 7 | 0 |
4.2. Computing expression scores and edge weights
For each gene in each experiment, an expression score is computed from the gene expression time series data. The score is based on the slope of the time series to capture both the change in expression levels and the time between infection and response. Specifically, we first compare the absolute values of the highest and the lowest expression levels. The score is positive if the former is higher, or negative if the latter is higher. Denote the time point that corresponds to the highest absolute value of the expression level as ti. The score is computed as follows: Si = expression(ti)/ti. The score is positively correlated with the height of the peak expression value and increases the earlier this value is reached.
To compute the edge weights, we first computed the BLASTP bit score between each pair of protein sequences. We turned the bit scores into a matrix, and set to zero those elements smaller than the 100 (our cutoff). We next computed an approximate sparse inverse of this matrix (Grote and Huckle, 1997) and used it as the weight matrix for the graph.
4.3. Recovering known human immune response genes
To evaluate the performance of our model, we retrieved 642 known human innate immune response genes from Kelley et al. (2005) and used them as our labeled data. We learned the model parameters by threefold cross validation using the labeled data. We compared the performance of GRF, MRF, and the baseline model where genes are ranked by their expression score alone. The MRF model is discussed in detail in Lu et al. (2006). We use the fraction of known immune response genes recovered by a model as the performance measure. Because the set of immune response genes that we used does not have labels indicating the cell types or infection conditions, we treat a gene as “positive” regardless of the cell type and bacteria type. For GRF and MRF models, the genes were ranked by their highest posterior probability (in any of the cell or bacteria types). For the baseline model, the genes are ranked by their expression scores. As we show in Figure 2a, both GRF and MRF models outperform the baseline model. These models are able to infer a better gene's posterior probability by transferring information between the same gene across cell types or from homologous genes across species. For example, for the top 10% ranked genes, MRF is able to recover 28% of known immune response genes, compared with 26% by the baseline model. Encouragingly, GRF leads to the biggest improvement in performance. Of the top 10% high scoring genes based on the posterior computed by GRF, 35% are known immune response genes, a 35% increase compared to the baseline (score only) model.
FIG. 2.

(a) Performance comparison of the Gaussian random field (GRF) with improved weights, the Markov random field (MRF), and a baseline model ranking genes by their expression scores. Using MRF, we were able to recover 18% of the known immune genes in the top 5% of ranked genes. This is a 28% improvement compared with the baseline model (which recovers 14% of the immune genes). The GRF model is able to recover 25% of the known immune genes at the same threshold, a 79% improvement over the baseline method and a 38% improvement over the MRF. (b) Performance comparison of GRF on two different graphs. The first graph contains genes from macrophages and dendritic cells in both human and mouse. The second graph contains genes from human macrophages and dendritic cells, but not from those in mouse. It can be seen that using homology information leads to large improvements.
To study the gain obtained by using cross species analysis we tested the performance of the GRF model when using only the human genes (removing the mouse genes from the graph). As can be seen in Figure 2b, the performance of the GRF when only human genes are included is drastically reduced. The ROC curve when using this data is completely dominated by the curve of the results when using both species, even though the comparison is for recovering known human genes. This indicates that by combining data from both species we can improve the assignment of each species as well.
To determine how sensitive the computed expression cores are to experimental noise, we carried out similar analysis on data that was generated by adding a small Gaussian noise term (mean = 0, variance = m2, where m is the median expression difference between the first two time points) to each time point of the immune response data. For each of the noise added datasets, we determined the score's performance for recovering known human immune response genes as discussed above. We repeated this process 50 times and found that the precision varies by 10% compared to the real data indicating the robustness of the computed scores (Fig. 3).
FIG. 3.

Our expression score function is robust to noise in expression data. We analyzed data that was generated by adding a small Gaussian noise term to each time point of the immune response data. For each of the noise added datasets we compared our method for recovering known human immune response genes to random selection as discussed above. This process was repeated 50 times. The error bars in the figure are based on 50 of these repeats. As can be seen, our method still performs well even with the added noise.
4.4. Identification of common response genes by combined analysis
Based on the learned posterior probabilities, we ranked the genes for each cell type in each species, for both Gram-positive and Gram-negative infections. We identified 57 ortholog pairs for which all nodes for both genes are assigned high posterior. Specifically, we selected ortholog pairs whose posterior probabilities are higher than 0.5 in all cells, bacteria, and species. These genes are commonly induced by all bacteria in both macrophages and dendritic cells across the two species (Fig. 4). As a sanity check, we first compared our list with a separate list of genes commonly induced in human macrophages by various bacteria. This latter list was derived from expression experiments that were not included in our analysis (Nau et al., 2002). The results confirmed the lists we identified. The overlap between the two lists was highly significant with a p-value = 1.70 × 10−25 (p-value computed using hypergeometric distribution).
FIG. 4.

One of the networks of genes commonly induced in both dendritic cells and macrophages when infected by bacteria, in both human and mouse. The network was constructed using Ingenuity Pathway Analysis (www.ingenuity.com). The gray-colored nodes are genes identified by our method. White-colored nodes are genes interacting with commonly induced genes. Note the large fraction of the pathway recovered by our method. Many known immune response genes are present in this network. IL1 is an important mediator of inflammatory response and involved in cell proliferation, differentiation, and apoptosis (Mizutani et al., 1991; Bratt and Palmblad, 1997). ETS2 is an important transcription factor for inflammation. CCL3, CCL4, and CCL5 are chemokines that recruite and activate leucocytes (Wolpe et al., 1988). The profiles for one of these genes, CCL5, are shown in Figure 6.
To reveal the functions of the common response genes we carried out GO enrichment analysis using STEM (Ernst and Bar-Joseph, 2006). The enriched GO categories include many common categories involved in immune responses, including “immune response” (p-value = 3.9 × 10−8, all p-values corrected using Bonferroni), “inflammatory response” (p-value = 2.5 × 10−7), “cell-cell signaling” (p-value = 1.1 × 10−6), “defense response” (p-value = 1.5 × 10−6), and “response to stress” (p-value = 2.4 × 10−5).
Our list recovered many of the classic players of innate immune activation and inflammation. For example, TNF is a pro-inflammatory cytokine and stimulates the acute phase reaction (Lukacs et al., 1995). IL1 is an important mediator of inflammatory response and involved in cell proliferation, differentiation, and apoptosis (Bratt and Palmblad, 1997; Mizutani et al., 1991). The list also includes chemokines that recruit and activate leucocytes (CCL3, CCL4, CCL5, CXCL1) (Wolpe et al., 1988) or attracts T-cells (CXCL9) (Valbuena et al., 2003). Also important to the regulation of inflammation response is IL10, a well-known anti-inflammatory molecule (Lammers et al., 2003). Additionally, ETS2, NFkB, and JUNB are all very important transcription factors that are activated in inflammation (Sun and Andersson, 2002). In addition to recovering genes labeled in the IRIS database (Kelley et al., 2005), which accounts for 21% of our predictions, we also successfully identified many immune response genes that were not included in the labeled dataset. Six out of the top 10 such genes are known to be commonly induced in host response in macrophages and dendritic cells (Jenner and Young, 2005), including PBEF1, an inhibitor of neutrophil apoptosis (Lee and Goodman, 2006), and MMP14, an endopeptidase that degrades various components of the extracellular matrix (Mignon et al., 1995). For complete list, see supporting website: www.cs.cmu.edu/ ∼lyongu/pub/immune/.
To identify the pathways involved in common immune response, we searched for networks enriched by common response genes using Ingenuity Pathway Analysis. One of these networks is shown in Figure 4.
4.5. Immune responses conserved in specific cell types
In addition to genes commonly induced across all dividing factors, we also identified genes that are differentially expressed between the two cell types. We identified 127 genes that are highly induced in dendritic cells in both bacteria types across human and mouse, but are not induced in macrophages (Fig. 5). GO enrichment analysis highlights some of the important characteristics of this set of genes, including “cell communication” (p-value = 1.7 × 10−10) and “signal transduction” (p-value = 1.1 × 10−9) (for the complete lists, see supporting website: www.cs.cmu.edu/∼lyongu/pub/immune/). Many of the genes are known to be associated with functions of dendritic cells, especially antigen processing and presentation. For example, components of the proteosome are prominently represented in the genes determined to be induced in dendritic cells. The proteosome is a necessary first step in MHC class I antigen presentation, a major function of dendritic cells. Peptides generated by the proteosome are then transported from the cytosol to endoplasmic reticulum by TAP, also represented in the gene list, where they are loaded on to MHC I molecules. Antigen presentation by DC is also accomplished through the class II pathway and the DC-specific gene list includes HLA-DRA, a human MHC II (class II) surface molecule. In addition to peptide-MHC complexes, T cell activation during antigen presentation requires a second signal. CD86, identified as a dendritic cell gene by our algorithm is an essential co-stimulatory molecule that delivers this second signal and is also a marker of dendritic cell maturation. Also in this list are TNFSF9 and TNFSF4, two cytokines that play a role in antigen presentation between dendritic cells and T lymphocytes. We searched for pathways enriched by these genes, and one of the enriched networks is shown in Figure 5.
FIG. 5.

One of the networks of genes strongly induced in dendritic cells but less so, unchanged, or suppressed in macrophages. (The legend is the same as in Figure 4.) The network was constructed by Ingenuity Pathway Analysis (www.ingenuity.com). The gray-colored nodes are genes inferred to be expressed at high levels in dendritic cells but not in macrophages, regardless of the bacteria type or species. White-colored nodes are genes interacting with such induced genes. Many known dendritic immune response genes are present in this network. CD86 is an essential costimulatory molecule and is also a marker of dendritic cell maturation. TAP is involved in the transportation of peptides generated by the proteosome from the cytosol to endoplasmic reticulum, which is an important step in MHC class I antigen presentation, a major function of dendritic cells. The profiles of CD86 are shown in Figure 7.
We have also identified 157 genes that are more likely to be induced in macrophages than in dendritic cells. Among these genes, FNGR1 is important for macrophages to detect interferon-gamma (also known as type II interferon), a key activating cytokine of macrophages. HMGB1, a chromatin structural protein, is believed to be involved in inflammation and sepsis. Another interesting gene is ADAM12, which is from a family of proteinases that are likely involved in tissue remodeling/wound healing by macrophages.
5. Conclusion
By combining expression experiments across species, cell types, and bacteria type, we were able to obtain a core set of innate immune response genes. The set we identified contained many of the known key players in this response. These show similar expression patterns across species, cell types and pathogens (Fig. 6). We have also identified unique signatures for macrophages and dendritic cells (Fig. 7) leading to insights regarding the set of processes activated in each of these cell types as part of the response.
FIG. 6.

Expression profiles of CCL5 identified by our method as a common immune response gene. Time (x-axis) is in minutes following infection. (a, b) Expression profiles for human CCL5 in dendritic cells and macrophages. (c, d) Expression profiles for mouse CCL5 in dendritic cells and macrophages. Expression of both genes is strongly induced following infection.
FIG. 7.

Expression profiles of CD86 identified to be activated only in dendritic cells. (a, b) Expression profiles for human CD86 in dendritic cells and macrophages. (c, d) Expression profiles for mouse CD86 in dendritic cells and macrophages. For both species, the expression of CD86 is induced in dendritic cells, but unchanged following infection in macrophages (and only mildly induced at the end of the time course).
While our method assumes that homologous genes share similar functions, it is still sensitive to the observed expression profiles. Thus, if two homologs display different expression patterns they would be assigned different labels. Still, homology information is a very useful feature for most genes. Relying on homology information, we were able to drastically improve the recovery of the correct set of genes.
While we have focused here on immune response, our method is general and can be applied to other diseases or conditions. We would like to further explore the lists derived by our method to determine the interactions and mechanisms leading to the activation of these genes in the cells they were assigned to. We would also like to expand our method so that it can better utilize the temporal information available in the microarray data. An additional area to explore is to incorporate other sources of information in the construction of the weight matrix. For example, it would be interesting to consider protein domains in addition to sequence similarity when creating the weight matrix.
Acknowledgments
This work was supported in part by the NIH (grants 1RO1 GM085022, NO1 AI50018 to Z.B.J.) and the NSF (Career Award 0448453 to Z.B.J.).
Disclosure Statement
No competing financial interests exist.
References
- Altschul S.F. Gish W. Miller W., et al. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay S. Sharan R. Ideker T. Systematic identification of functional orthologs based on protein network comparison. Genome Res. 2006;16:428–435. doi: 10.1101/gr.4526006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop C.M. Pattern Recognition and Machine Learning. Springer; New York: 2006. [Google Scholar]
- Bratt J. Palmblad J. Cytokine-induced neutrophil-mediated injury of human endothelial cells. J. Immunol. 1997;159:912–918. [PubMed] [Google Scholar]
- Chaussabel D. Semnani R.T. McDowell M.A., et al. Unique gene expression profiles of human macrophages and dendritic cells to phylogenetically distinct parasites. Blood. 2003;202:672–681. doi: 10.1182/blood-2002-10-3232. [DOI] [PubMed] [Google Scholar]
- Deng M. Chen T. Sun F. Integrated probabilistic model for functional prediction proteins. J. Comput. Biol. 2004;11:463–435. doi: 10.1089/1066527041410346. [DOI] [PubMed] [Google Scholar]
- Detweiler C.S. Cunanan D.B. Falkow S. Host microarray analysis reveals a role for the Salmonella response regulator phoP in human macrophage cell death. Proc. Natl. Acad. Sci. USA. 2001;98:5850–5855. doi: 10.1073/pnas.091110098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper D.W. Bethea H.N. He Y.W. Toll-like receptor 2-dependent and -independent activation of macrophages by group B streptococci. Immunol. Lett. 2006;102:202–214. doi: 10.1016/j.imlet.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Enright A.J. van Dongen S. Ouzounis C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eppig J.T. Bult C.J. Kadin J.A., et al. The Mouse Genome Database (MGD): from genes to mice—a community resource for mouse biology. Nucleic Acids Res. 2005;33:D471–D475. doi: 10.1093/nar/gki113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J. Bar-Joseph Z. STEM: a tool for the analysis of short time series gene expression data. BMC Bioinform. 2006;7:191. doi: 10.1186/1471-2105-7-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon D.T. Locksley R.M. The instructive role of innate immunity in the acquired immune response. Science. 1996;272:50–54. doi: 10.1126/science.272.5258.50. [DOI] [PubMed] [Google Scholar]
- Granucci F. Vizzardelli C. Pavelka N., et al. Inducible il-2 production by dendritic cells revealed by global gene expression analysis. Nat. Immunol. 2001;2:882–888. doi: 10.1038/ni0901-882. [DOI] [PubMed] [Google Scholar]
- Grote M.J. Huckle T. Parallel preconditioning with sparse approximate inverses. SIAM J. Sci. Comput. 1997;18:838–853. [Google Scholar]
- Hoffmann J.A. Kafatos F.C. Janeway C.A., Jr., et al. Phylogenetic perspectives in innate immunity. Science. 1999;284:1313–1318. doi: 10.1126/science.284.5418.1313. [DOI] [PubMed] [Google Scholar]
- Hoffmann R. van Erp K. Trulzsch K., et al. Transcriptional responses of murine macrophages to infection with yersinia enterocolitica. Cell Microbiol. 2004;6:377–390. doi: 10.1111/j.1462-5822.2004.00365.x. [DOI] [PubMed] [Google Scholar]
- Huang Q. Liu D. Majewski P., et al. The plasticity of dendritic cell responses to pathogens and their components. Science. 2001;294:870–875. doi: 10.1126/science.294.5543.870. [DOI] [PubMed] [Google Scholar]
- Jenner R.G. Young R.A. Insights into host responses against pathogens from transcriptional profiling. Nat. Rev. Microbiol. 2005;3:281–294. doi: 10.1038/nrmicro1126. [DOI] [PubMed] [Google Scholar]
- Keller G. Snodgrass R. Life span of multipotential hematopoietic stem cells in vivo. J. Exp. Med. 1990;171:1407–1418. doi: 10.1084/jem.171.5.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley J. Bono B.D. Trowsdale J. IRIS: a database surveying known human immune system genes. Genomics. 2005;85:503–11. doi: 10.1016/j.ygeno.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Lammers K.M. Brigidi P. Vitali B., et al. Immunomodulatory effects of probiotic bacteria DNA: IL-1 and IL-10 response in human peripheral blood mononuclear cells. FEMS Immunol. Med. Microbiol. 2003;22:165–72. doi: 10.1016/S0928-8244(03)00144-5. [DOI] [PubMed] [Google Scholar]
- Lang R. Patel D. Morris J.J., et al. Shaping gene expression in activated and resting primary macrophages by IL-10. J. Immunol. 2002;169:2253–2263. doi: 10.4049/jimmunol.169.5.2253. [DOI] [PubMed] [Google Scholar]
- Lee H.C. Goodman J.L. Anaplasma phagocytophilum causes global induction of antiapoptosis in human neutrophils. Genomics. 2006;88:496–503. doi: 10.1016/j.ygeno.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Letovsky S. Kasif S. Predicting protein function from protein/protein interaction data: a probabilistic approach. Bioinformatics. 2003;19(Suppl 1):i197–i204. doi: 10.1093/bioinformatics/btg1026. [DOI] [PubMed] [Google Scholar]
- Liu M. Liberzon A. Kong S.W., et al. Network-based analysis of affected biological processes in type 2 diabetes models. PLoS Genet. 2007;3:e96. doi: 10.1371/journal.pgen.0030096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y. Rosenfeld R. Bar-Joseph Z. Identifying cycling genes by combining sequence homology and expression data. Bioinformatics. 2006;22:e314–e322. doi: 10.1093/bioinformatics/btl229. [DOI] [PubMed] [Google Scholar]
- Lu Y. Mahony S. Benos P.V., et al. Combined analysis reveals a core set of cycling genes. Genome Biol. 2007;8:R146. doi: 10.1186/gb-2007-8-7-r146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs N.W. Strieter R.M. Chensue S.W., et al. TNF-alpha mediates recruitment of neutrophils and eosinophils during airway inflammation. J. Immunol. 1995;154:5411–5417. [PubMed] [Google Scholar]
- McCaffrey R.L. Fawcett P. O'Riordan M., et al. A specific gene expression program triggered by Gram-positive bacteria in the cytosol. Proc. Natl. Acad. Sci. USA. 2004;101:11386–11391. doi: 10.1073/pnas.0403215101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignon C. Okada A. Mattei M.G., et al. Assignment of the human membrane-type matrix metalloproteinase (MMP14) gene to 14q11-q12 by in situ hybridization. Genomics. 1995;28:360–361. doi: 10.1006/geno.1995.1159. [DOI] [PubMed] [Google Scholar]
- Mizutani H. Schechter N. Lazarus G., et al. Rapid and specific conversion of precursor interleukin 1 beta (IL-1 beta) to an active IL-1 species by human mast cell chymase. J. Exp. Med. 1991;174:821–825. doi: 10.1084/jem.174.4.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nau G.J. Richmond J.F.L. Schlesinger A., et al. Human macrophage activation programs induced by bacterial pathogens. Proc. Natl. Acad. Sci. USA. 2002;99:1503–1508. doi: 10.1073/pnas.022649799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelder J.A. Mead R. A simplex method for function minimization. Comput. J. 1965;7:308–313. [Google Scholar]
- Sun Z. Andersson R. NF-kappaB activation and inhibition: a review. Shock. 2002;18:99–106. doi: 10.1097/00024382-200208000-00001. [DOI] [PubMed] [Google Scholar]
- Valbuena G. Bradford W. Walker D.H. Expression analysis of the T-cell-targeting chemokines CXCL9 and CXCL10 in mice and humans with endothelial infections caused by rickettsiae of the spotted fever group. Am. J. Pathol. 2003;163:1357–1369. doi: 10.1016/S0002-9440(10)63494-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Erp K. Dach K. Koch I., et al. Role of strain differences on host resistance and the transcriptional response of macrophages to infection with Yersinia enterocolitica. Physiol. Genomics. 2006;25:75–84. doi: 10.1152/physiolgenomics.00188.2005. [DOI] [PubMed] [Google Scholar]
- Wolpe S.D. Davatelis G. Sherry B., et al. Macrophages secrete a novel heparin-binding protein with inflammatory and neutrophil chemokinetic properties. J. Exp. Med. 1988;167:570–581. doi: 10.1084/jem.167.2.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yedidia J.S. Freeman W.T. Weiss Y. In Exploring Artificial Intelligence in the New Millennium. Morgan Kaufmann Publishers Inc.; New York: 2003. Understanding belief propagation and its generalizations, 239–236. [Google Scholar]
- Zhu X. Carnegie Mellon University; Pittsburgh: 2005. Semi-supervised learning with graphs [Ph.D. dissertation] [Google Scholar]