

Abstract
Insulin resistance is a characteristic of late pregnancy, and adipose tissue is one of the tissues that most actively contributes to the reduced maternal insulin sensitivity. There is evidence that pregnancy is a condition of moderate inflammation, although the physiological role of this low-grade inflammation remains unclear. The present study was designed to validate whether low-grade inflammation plays a role in the development of insulin resistance in adipose tissue during late pregnancy. To this end, we analyzed proinflammatory adipokines and kinases in lumbar adipose tissue of nonpregnant and late pregnant rats at d 18 and 20 of gestation. We found that circulating and tissue levels of adipokines, such as IL-1β, plasminogen activator inhibitor-1, and TNF-α, were increased at late pregnancy, which correlated with insulin resistance. The observed increase in adipokines coincided with an enhanced activation of p38 MAPK in adipose tissue. Treatment of pregnant rats with the p38 MAPK inhibitor SB 202190 increased insulin-stimulated tyrosine phosphorylation of the insulin receptor (IR) and IR substrate-1 in adipose tissue, which was paralleled by a reduction of IR substrate-1 serine phosphorylation and an enhancement of the metabolic actions of insulin. These results indicate that activation of p38 MAPK in adipose tissue contributes to adipose tissue insulin resistance at late pregnancy. Furthermore, the results of the present study support the hypothesis that physiological low-grade inflammation in the maternal organism is relevant to the development of pregnancy-associated insulin resistance.
In the last decade, the role of the adipose tissue in the development of insulin resistance has been extensively studied. It is well established that adipose tissue acts as an endocrine organ, secreting biologically active molecules in response to external stimuli or lipid overloading (1) in both an autocrine and paracrine fashion. These adipose tissue-derived signaling molecules include adipokines, such as adiponectin, resistin, and leptin, as well as cytokines/chemokines, such as TNF-α, monocyte chemotactic protein (MCP)-1, IL-1β, and IL-6, and also acute phase reactants, such as plasminogen activator inhibitor (PAI)-1 and C reactive protein (2). Some of these adipokines have been linked with insulin resistance in metabolic disorders, such as obesity (3) and type 2 diabetes (4), whereas adiponectin and visfatin have been related to insulin sensitivity (5, 6). The mechanisms that underlie the physiological effects of these molecules are still incompletely understood. In some cases, they implicate activation of nuclear factor-κB in insulin-sensitive tissues (7) and, in response to proinflammatory stimuli, activation of diverse stress kinase pathways. Accordingly, 3T3-L1 adipocytes show an impaired insulin response when they are treated with IL-1β, which is dependent on p38 MAPK-ERK-1/2 (8). Furthermore, activation of p38 was found to link visceral adiposity to whole-body insulin resistance (9), and ERK-1/2 attenuation was shown to mitigate inflammatory oxidative stress in white adipose tissue during exercise (10).
Insulin resistance is defined as a state, in which more insulin is required to obtain the biological effects that are induced by insulin in the normal condition. Thus, virtually any defect in the insulin signaling cascade can cause insulin resistance. Insulin signaling is initiated upon binding of insulin to the insulin receptor (IR), activating the intrinsic tyrosine kinase activity of the receptor β-subunit. This event initiates a cascade of cell-signaling responses, including auto-tyrosine phosphorylation of IR and phosphorylation of IR substrate (IRS) proteins (11) that act as docking proteins for a number of downstream effector molecules, such as phosphatidylinositol 3-kinase or growth factor receptor-bound protein 2 (12). IR and IRS proteins are susceptible to serine phosphorylation, an event that attenuates insulin signaling in different conditions (13–17). IRS-1 serine phosphorylation (pSer-IRS-1) has been linked to the activation of several Ser/Thr kinases, such as inhibitor of κ light polypeptide gene enhancer in B-cells, kinase β (IKK), mammalian target of rapamycin (mTOR), protein kinase C, protein kinase B, or glycogen synthase kinase-3. Most of these effectors respond to free fatty acids, cytokines, or oxidative stress, whereas others constitute a negative feedback mechanism and become activated by insulin (15).
Pregnancy is characterized by modifications in maternal adiposity, starting with an increase in adipose tissue mass during the earlier phase of gestation and followed by a decrease of fat mass during the late phase (18). During the late phase of pregnancy, insulin resistance eventually develops both in human (19–21) and rat (22, 23). There is evidence that pregnancy is a condition of moderate inflammation (24, 25), in which adipose tissue, together with the placenta, contributes to the local and systemic increase of inflammatory molecules (26). The physiological role of this low-grade inflammation remains unclear. It has been proposed that the activation of inflammatory pathways is implicated in the development of insulin resistance at late pregnancy and, when this physiological adaptation is decompensated, the mother is exposed to an increased risk of developing gestational diabetes (27). In fact, misregulation of cytokine networks can lead to adverse pregnancy outcomes, including preterm labor, preeclampsia, and intrauterine growth restriction (28). Although it has been shown that in human pregnancy circulating TNF-α is significantly correlated with insulin resistance (29), there is no direct evidence for the role of inflammation in insulin signaling during pregnancy. In this context, we have recently shown that adiponectin and pSer-IRS-1 are implicated in adipose tissue insulin resistance in late pregnant rats (30), suggesting that activation of Ser/Thr kinases by inflammatory molecules, including stimulation of MAPK/stress-activated protein kinases pathways, may contribute to the development of insulin resistance. Based on these previous findings, the present study was designed to validate whether low-grade inflammation plays a role in the development of the insulin resistance in adipose tissue during late pregnancy.
Materials and Methods
Animals and sample collection
Female Sprague Dawley rats were housed at 22–24 C with 12-h light, 12-h dark cycles, from 0800 to 2000 h, and free access to water and to a chow diet (Panlab, Barcelona, Spain). Animals were mated when weighting between 180 and 210 g; d 0 of pregnancy was determined by the presence of spermatozoids in vaginal smears. Experimental groups were composed by virgin and 18- and 20-d-pregnant rats. Subsequent to CO2 anesthesia, animals were decapitated, lumbar adipose pads were rapidly dissected, snap frozen in liquid nitrogen, and stored at −80 C until processed for further analysis. Blood was collected from the neck wound in EDTA-tubes for immediate separation of plasma at 4 C that was stored at −30 C until analysis. Experimental protocols were approved by the Animal Research Committee of the Faculty of Pharmacy, Universidad San Pablo-CEU.
Plasma analysis
Enzymatic colorimetric tests were used to determine in EDTA-plasma samples glucose (GOD-PAP, from Roche Diagnostics, Barcelona, Spain), triglycerides (LPL/GPO-Trinder from Roche Diagnostics), and nonesterified fatty acids (NEFA) (ACS-ACOD from Wako Chemicals GmbH, Neuss, Germany). Insulin was determined in plasma samples using a specific ELISA kit for rats (Mercodia, Uppsala, Denmark).
Protein extraction from adipose tissue
Frozen lumbar adipose tissue (100 mg) was powdered in liquid nitrogen in a mortar precooled to −80 C, and lysed for 30 min in an ice-cold 30 mm HEPES buffer (pH 7.4), containing 5 mm EDTA, 1% Triton X-100, 0.5% sodium deoxycholate, 8 mm Na3VO4, 1 mm NaF, and 2 mm protease inhibitor (Pefablock, Roche, Barcelona, Spain). Cellular debris was removed by centrifugation at 17,000 × g for 30 min at 4 C, and protein concentration was determined in the supernatant by the bicinchoninic acid protein assay (Pierce, Rockford, IL).
Adipokine analysis
Adipokine levels in plasma and in adipose tissue homogenates, prepared as indicated above, were determined by multiplexing in a Bioplex system (xMAP Technology, Bio-Rad, Madrid, Spain) using specific kits from Linco, Inc. (St. Charles, MO). Concentrations of PAI-1 (active) and PAI-1 (total), IL-1β, IL-6, MCP-1, C reactive protein, and TNF-α were calculated by interpolatation using recombinant protein standards supplied by the manufacturer.
Calculation of insulin sensitivity indexes
Plasma glucose and insulin values in 6-h fasting animals were used to calculate homeostasis model assessment of insulin resistance (HOMA-IR), quantitative insulin sensitivity check index (QUICKI), and fasting glucose to insulin ratio (FGIR) as estimates of insulin sensitivity. Recently, we have validated the use of these indexes in rats by comparison with the hyperinsulinemic-euglycemic clamp technique (31). Briefly, HOMA-IR was calculated as the product of the fasting plasma glucose (FPG) and fasting plasma insulin (FPI) levels divided by a constant. The equation was as follows: HOMA-IR = (FPG × FPI)/2430, where FPI was in μU/ml and FPG in mg/dl. QUICKI was calculated according to the original formula (32) as the inverse log sum of fasting insulin in μU/ml and fasting glucose in mg/dl. QUICKI = 1/[log(FPG) + log(FPI)]. Finally, FGIR was calculated as the ratio of FPG divided by FPI levels. FGIR = FPG/FPI, where FPG was in mg/dl and FPI in μU/ml.
SB 202190 treatment
The p38 MAPK inhibitor SB 202190 (Sigma-Aldrich, Madrid, Spain) was suspended in a solution of 20% dimethylsulfoxide in 0.9% NaCl. From d 18 to 20 of gestation, rats were treated with a single daily dose of 0.8 mg SB 202190/kg body weight or vehicle (20% dimethylsulfoxide in 0.9% NaCl). In the morning of d 20 of pregnancy, and 2 h after the last SB 202190 dose, rats were killed as described above.
Insulin treatment for insulin signaling studies
For in vivo stimulation of the insulin signaling cascade, 16-h-fasted 20-d-pregnant rats were anesthetized with sodium pentobarbital (40 mg/kg body weight, ip). The abdominal cavity was opened, and saline (0.9% NaCl) with or without insulin (4 IU Humulin NPH/kg body weight; Lilly, Madrid, Spain) was injected via the portal vein. After 90 sec, lumbar adipose tissue was removed, snap frozen in liquid nitrogen, and stored at −80 C.
Isolation of adipocytes and in vitro studies of p38 MAPK inhibition
Adipocytes were prepared from white lumbar adipose tissue of fasted 20-d-pregnant rats as described previously (33). Briefly, freshly isolated adipose pads were digested with collagenase A (1 mg/ml) (activity 0.21 U/mg; Roche Diagnostics) in Krebs-Ringer bicarbonate (KRB) buffer (pH 7.4) containing 4% (wt/vol) BSA (fatty acids free, fraction V; Sigma-Aldrich) and 5.5 mm glucose (KRB buffer), for 30 min at 37 C in an O2/CO2 atmosphere (19:1, vol/vol) under constant shaking (60 cycles/min). After filtering and washing with KRB buffer to eliminate collagenase, cells were resuspended in the same buffer at a concentration of about 0.5 × 106 cells/ml. Then the adipocyte suspension was incubated for 30 min at 37 C in KRB buffer containing 1 U/ml of adenosine deaminase (Sigma-Aldrich) in the absence and presence of 15 μm of SB 202190. Subsequently, cells were incubated for further 30 min in the absence or presence of 90 nm insulin. Subsequently, incubation tubes were placed on ice, and infranatants were removed for the enzymatic determination of glycerol (GPO-Trinder; Sigma-Aldrich). The adipose cells were lysed in an ice-cold lysis buffer and processed, as described in the Protein extraction from adipose tissue section, for the analysis of serine phosphorylation of acetyl-coenzyme A carboxylase (ACC) by Western blot analysis. Glycerol released into the incubation medium as taken as an index of lipolytic activity, and phosphorylation state of ACC, was used as an estimate of activation of lipogenesis.
Immunoblotting
Twenty-five micrograms of tissue or cell protein from each experimental condition were subjected to SDS-PAGE and electrophoretically transferred to polyvinylidene difluoride membranes. Membranes were blocked for 1 h at room temperature with 5% nonfat dry milk and then incubated at 4 C overnight with one of the following antibodies: antiphosphotyrosine-IR (Y972) and anti-IR-ß from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); antiphosphotyrosine-IRS-1 (Y612) from Sigma-Aldrich; antiphosphoserine-IRS-1 (S307) and anti-IRS-1 from Upstate Biotechnology (Lake Placid, NY); antiphospho-p38 MAPK (T180 and Y182), anti-p38 MAPK, antiphospho-c-Jun-N-terminal kinase (JNK)-1/2 (T183 and Y185), and anti-JNK-1/2 from Biosource International (Camarillo, CA); antiphospho-mTOR (S2448), anti-mTOR, and antiphosphoserine (S79)-ACC were from Cell Signaling (Beverly, MA). Membranes were washed and incubated at room temperature with either antimouse or antirabbit horseradish peroxidase-conjugated secondary antibodies as appropriate. Immunoreactive bands were visualized using the enhanced chemiluminescence system and quantified by densitometry; the intensity of each protein band was normalized using β-actin.
Detection of macrophage infiltration in adipose tissue by immunohistochemistry
Lumbar adipose tissue sections were cut off longitudinally, fixed in 4% formalin, and embedded in paraffin. Endogenous peroxidase was neutralized by treatment of deparaffinized sections with 3% hydrogen peroxide for 10 min. Antigen unmasking was performed by incubating samples in a pH 9 buffer at 98 C for 45 min. Nonspecific reactions were blocked by incubation for 30 min with normal swine serum. Sections were incubated with a primary monoclonal antibody against the rat homologue to human CD68 (clone ED1, from DakoCytomation, Glostrup, Denmark), a marker for monocytes and inflammatory macrophages, for 30 min at room temperature. Then, a biotinylated secondary antibody was applied for 30 min, followed by incubation with the streptavidin-horseradish peroxidase conjugate for 15 min; both reagents were obtained from Vector Laboratories (Burlingame, CA). Peroxidase was developed using the LSAB+ kit, from Dako, according to the instructions of the manufacturer. Sections were counterstained using hematoxylin Gill II (Sigma, St. Louis, MO). Digitized images were obtained using a Nikon E600 microscope equipped with a DXM200 camera (Nikon, Melville, NY). Six to 12 images were taken from each slide at a low magnification (×10) to obtain a wide section with significant labeling. Quantitative image analysis was performed using the ImageJ software (version 1.35q) from National Institutes of Health (Bethesda, MD).
Presentation of results
Results are expressed as mean ± sem of 4–10 animals per group. When data were not normally distributed, logarithmic transformation was applied, and the statistical analyses were done on the logarithm of the parameter. As indicated in table and figure legends, statistical comparisons between two groups were made using the Student′s t test; comparisons between three or more groups were based on ANOVA with 95% confidence limits, followed by Student-Newman Keuls, post hoc test, using the GraphPad Prism program (version 5; GraphPad, San Diego, CA).
Results
Metabolic changes at late pregnancy are associated with increased cytokine levels
Here, we used the animal model of the late pregnant rat to explore the potential implication of low-level inflammation in the development of insulin resistance during gestation. As shown in Table 1, both lumbar adipose tissue and maternal body weight increased at late pregnancy. Expectedly, late pregnant rats had significantly lower plasma glucose levels in the presence of hyperinsulinemia (Table 1). In addition, plasma concentration of triglycerides and NEFA in the 18- and 20-d-pregnant rats were significantly increased compared with rats at d 0 of gestation (Table 1). Late pregnancy was further associated with a significant increase of HOMA-IR, an index of insulin resistance, as well as a significant decease of QUICKI and FGIR, indexes of insulin sensitivity, as shown by a comparison of 18- and 20-d-pregnant animals with nonpregnant rats (Table 1). Together, these data reflect on the characteristic metabolic changes at late pregnancy, such as hyperlipemia, an overall increase of lipolytic activity, and insulin resistance.
Table 1.
Effect of late pregnancy on body and adipose tissue weight and on biochemical parameters and insulin sensitivity indexes in rats
| 0 | 18 | 20 | |
|---|---|---|---|
| Body weight (g) | 235 ± 4.76a | 376 ± 19.6b | 394.0 ± 21.7b |
| (n = 9) | (n = 5) | (n = 7) | |
| Lumbar adipose tissue weight (g) | 1.09 ± 0.09a | 2.56 ± 0.15b | 2.35 ± 0.09b |
| (n = 7) | (n = 5) | (n = 5) | |
| Plasma glucose (mg/dl) | 134.0 ± 5.12a | 113.9 ± 6.0b | 106.4 ± 3.2b |
| (n = 6) | (n = 5) | (n = 7) | |
| Plasma insulin (μg/liter) | 1.07 ± 0.07a | 1.99 ± 0.15b | 2.03 ± 0.15b |
| (n = 9) | (n = 5) | (n = 6) | |
| Plasma triglycerides (mg/dl) | 131.0 ± 10.0a | 302.1 ± 44.3b | 685.5 ± 81.7c |
| (n = 5) | (n = 5) | (n = 7) | |
| Plasma NEFA (μm) | 648.6 ± 49.3a | 960.6 ± 74.0b | 1951.5 ± 158.3c |
| (n = 6) | (n = 5) | (n = 5) | |
| HOMA-IR | 1.40 ± 0.11a | 2.35 ± 0.25b | 2.24 ± 0.18b |
| (n = 6) | (n = 5) | (n = 6) | |
| QUICKI | 0.284 ± 0.002a | 0.267 ± 0.003b | 0.268 ± 0.003b |
| (n = 6) | (n = 5) | (n = 6) | |
| FGIR (mg/10–4 U) | 5.53 ± 0.73a | 2.32 ± 0.17b | 2.14 ± 0.14b |
| (n = 6) | (n = 5) | (n = 6) |
Enzymatic colorimetric tests were used to determine in EDTA-plasma samples glucose, triglycerides, and NEFA. Insulin was determined in plasma samples using a specific ELISA kit for rats. Insulin sensitivity indexes, HOMA-IR, QUICKI, and FGIR, were calculated from short-term fasting plasma glucose and insulin values as described in Materials and Methods. Data are mean values ± sem. Values of plasma insulin were log transformed to equalize the variance between conditions. Comparisons were made ANOVA followed by Student-Newman-Keuls as post hoc. Different (a–c)letters represent statistical differences between groups (P < 0.05).
Our previous work points to a possible role of inflammatory processes in whole-body insulin resistance at late pregnancy (30). To corroborate this hypothesis, we measured circulating levels of proinflammatory cytokines including TNF-α, MCP-1, IL-1β, IL-6, and active PAI-1. We found a pronounced increase of IL-1β (7-fold) (Fig. 1A) and active PAI-1 (>30-fold) (Fig. 1B) at both 18 and 20 d of gestation; MCP-1 (Fig. 1C) showed a modest (1.8-fold) but significant increase in 20-d-pregnant rats compared with virgin and 18-d-pregnant animals. Under our experimental conditions, TNF-α and IL-6 levels were undetectable in the rats (data not shown).
Fig. 1.
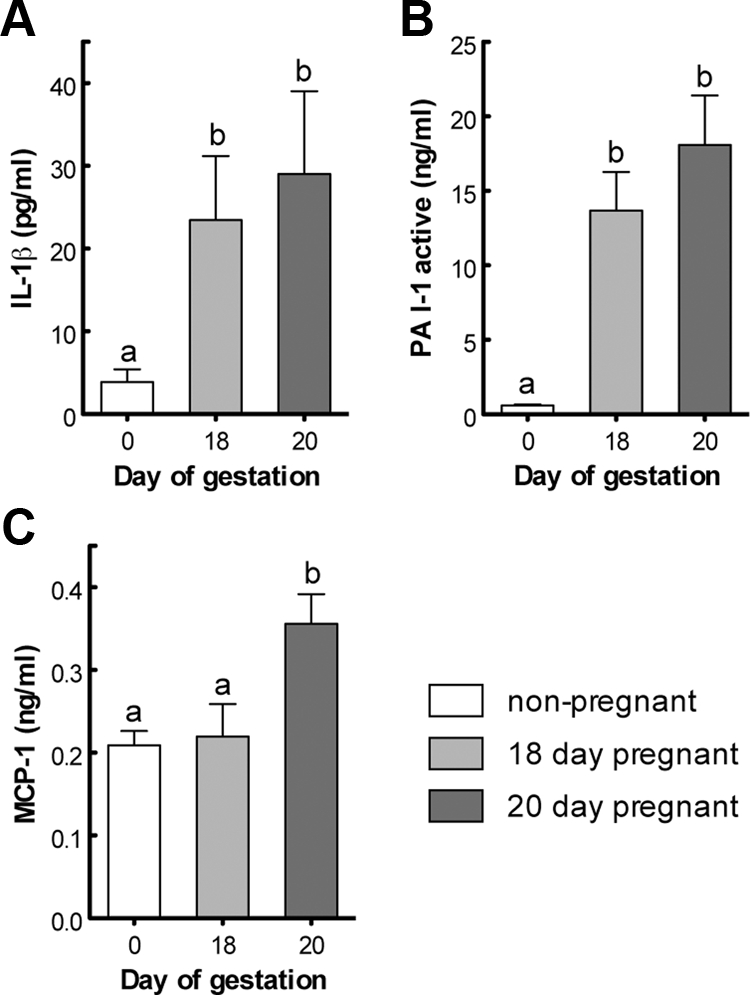
Comparison of circulating concentrations of adipokines in nonpregnant (0) and late pregnant rats (18 and 20 d of gestation). IL-1β (A), PAI-1 (active) (B), and MCP-1 (C).
To explore the impact of pregnancy on cytokine levels in adipose tissue, we measured concentrations of IL-6, total PAI-1, and TNF-α in lumbar adipose tissue from 18- and 20-d-pregnant and virgin rats. We did not detect any significant change of IL-6 values when the three experimental groups were compared (data not shown). PAI-1 concentrations increased progressively and were significantly higher (1.7-fold) in 20-d-pregnant rats compared with nonpregnant controls (Fig. 2A); TNF-α was significantly increased (>2-fold) in both 18- and 20-d-pregnant rats compared with virgin animals (Fig. 2B). The comparably low levels of ED1 immunoreactivity in virgin and 20-d-pregnant animals exclude macrophage infiltration into lumbar adipose tissue as a major source of inflammatory mediators during late pregnancy (Fig. 2C).
Fig. 2.
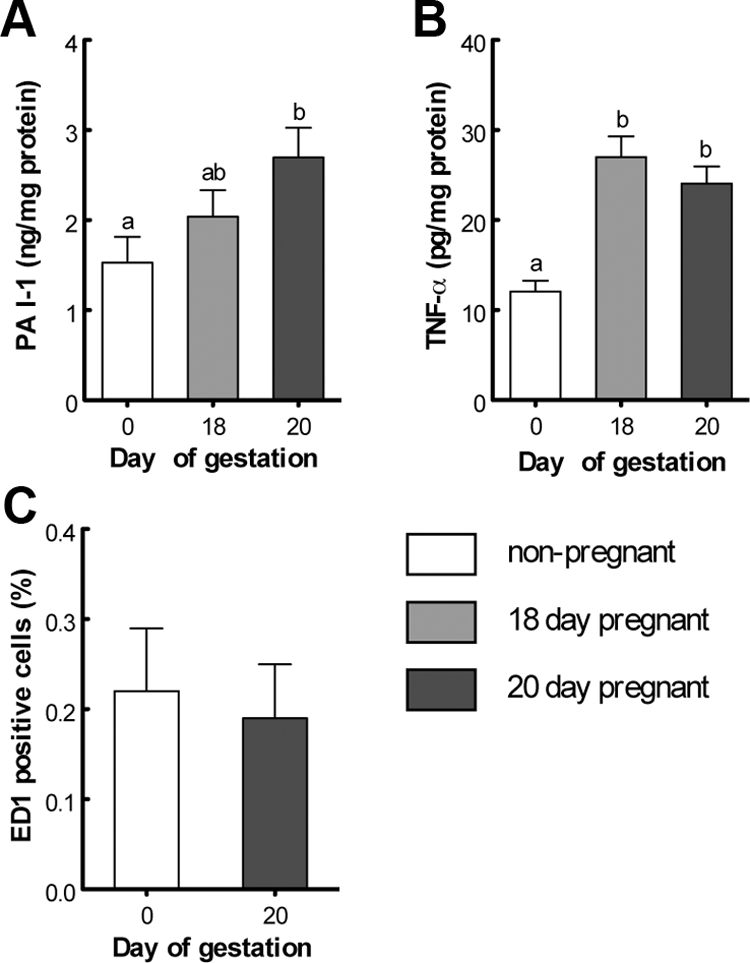
Concentrations of adipokines and immunodetection of ED1, a marker for macrophage infiltration, in lumbar adipose tissue of nonpregnant (0) and late pregnant rats (18 and 20 d of gestation). PAI-1 (total) (A), TNF-α (B), and percentage of ED1-positive cells (C).
To test for an association between increased cytokine levels and insulin resistance at late pregnancy, we performed correlation analysis between these variables in virgin and pregnant rats at d 18 and 20 of gestation. The most significant inverse correlations were obtained for circulating concentrations of active PAI-1 (Fig. 3A) and adipose tissue TNF-α (Fig. 3B) vs. both QUICKI and log(FGIR), indicating that insulin resistance at late pregnancy is associated with an inflammatory response. Correlations were also significant when the values obtained for nonpregnant, 18-, and 20-d-pregnant rats were studied separately.
Fig. 3.
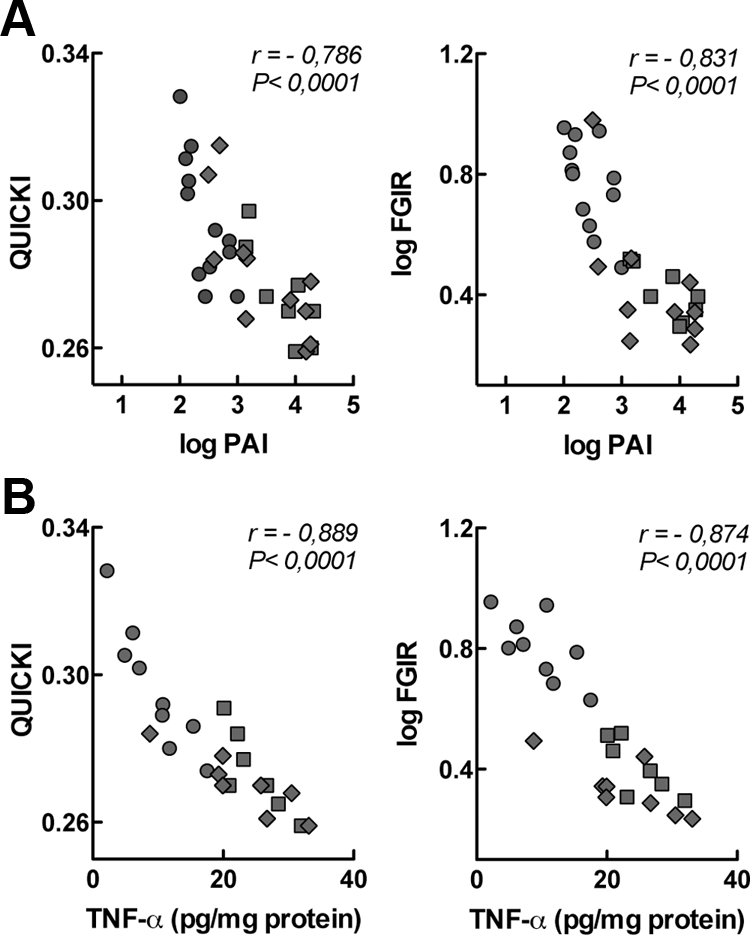
Correlation analysis between adipokine levels and insulin sensitivity in nonpregnant and pregnant rats at d 18 and 20 of gestation. A, Correlation between concentrations of circulating PAI-1 (active) and QUICKI or FGIR. B, Correlation between concentrations of TNF-α in lumbar adipose tissue and QUICKI or FGIR. Circles, Nonpregnant rats; squares, 18-d-pregnant rats; and diamonds, 20-d-pregnant rats.
Activation of p38 MAPK plays a role in insulin resistance at late pregnancy
Inflammatory mediators, such as TNF-α, contribute to the development of insulin resistance in obesity and diabetes through activation of stress-activated serine/threonine kinases, which results in impaired insulin signaling at various levels, including inhibitory serine phosphorylation of IRS-1 (34). The presence of proinflammatory cytokines in lumbar adipose tissue (see Fig. 2) of late pregnant rats prompted us to investigate the status of selected serine/threonine kinases and IRS-1 in these animals.
We did not observe any effect of pregnancy on the basal phosphorylation of mTOR [mTOR/total mTOR, 0.823 ± 0.320 vs. 0.608 ± 0.160 (n = 7); P = 0.56 for nonpregnant and pregnant rats, respectively] and JNK-1/2 [pJNK/total JNK, 3.79 ± 1.23 vs. 2.99 ± 1.16 (n = 6); P = 0.64 for nonpregnant and pregnant rats, respectively]. We found, however, a significant activation of p38 MAPK, assessed as threonine/tyrosine phosphorylation of p38 MAPK, in 20-d-pregnant rats compared with nonpregnant animals (Fig. 4). To further confirm the functional implication of p38 MAPK in the modulation of insulin responsiveness during pregnancy, we investigated whether insulin signaling in adipose tissue of pregnant rats could be restored in vivo upon treatment with SB 202190, a specific inhibitor of p38 MAPK. First, we validated that SB 202190 completely blocked p38 MAPK in adipose tissue of late pregnant rats (Fig. 5A). Pharmacological blockade of p38 MAPK in pregnant rats was paralleled by a more than 15-fold decrease of pSer-IRS-1 (Fig. 5B), confirming a role of p38 MAPK activation in the regulation of IRS-1 at late gestation.
Fig. 4.
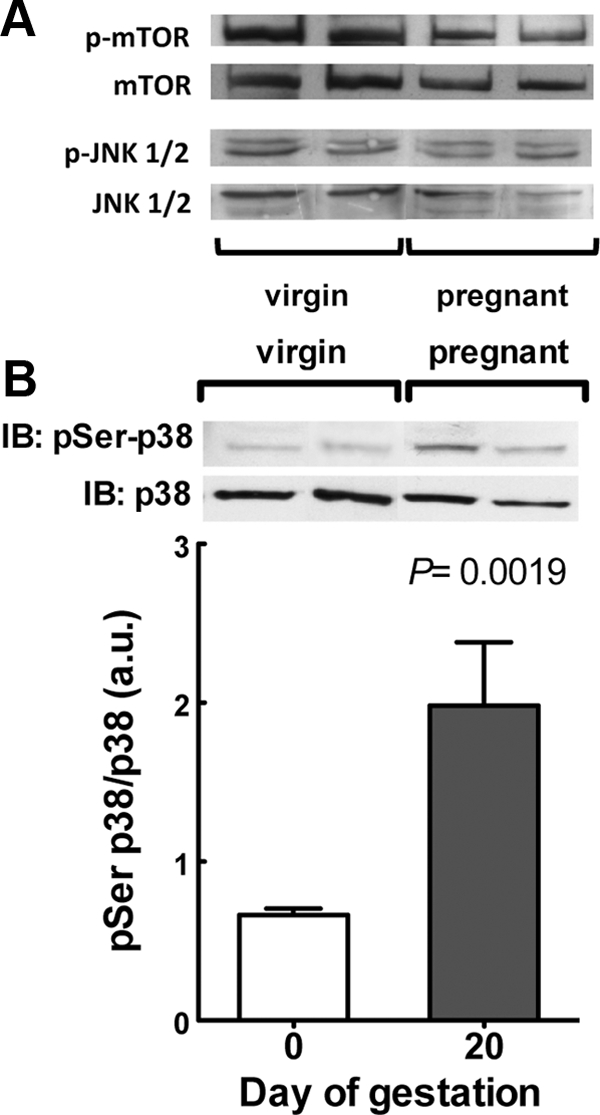
p38 MAPK activation is increased in adipose tissue at late gestation. A, Phosphorylation of mTOR and JNK1/2. Representative immunoblots for phosphorylated and total proteins of two independent experiments. B, Phosphorylation of p38 MAPK. The graph shows the levels of phosphorylated p38 MAPK, normalized to the total amount of p38 MAPK protein, of six independent experiments. A representative immunoblot (IB) for phosphorylated and total protein is shown in the graph.
Fig. 5.

Inhibition of p38 MAPK at late pregnancy increases insulin signaling in adipose tissue. Starting at d 18 of gestation, rats were treated from d 18 to 20 of gestation, with vehicle or a daily dose of the specific p38 MAPK inhibitor SB 202190. On the morning of d 20 of pregnancy, and 2 h after the last SB 202190 dose, rats were killed, and adipose tissue was used for the analysis of p38 MAPK phosphorylation and pSer IRS-1. A, SB 202190 blocks phosphorylation of p38 MAPK at late pregnancy. The graph shows the levels of phosphorylated p38 MAPK, normalized to the total amount of p38 MAPK protein, of four independent experiments. A representative immunoblot for phosphorylated and total protein is shown in the graph. B, Inhibition of p38 MAPK decreases IRS-1 S307 phosphorylation in adipose tissue of late pregnant rats. The graph shows the levels of Ser-phosphorylated IRS-1, normalized to the total amount of IRS-1 in each sample. For in vivo stimulation of the insulin signaling cascade, after the SB 202190 treatment, some animals were anesthetized, and saline with or without insulin was injected via the portal vein. The graphs show the fold stimulation by insulin of tyrosine phosphorylated IR (C) and IRS-1 (D), normalized to the total amount of IR and IRS-1 protein. Representative immunoblots (IB) for phosphorylated and total IRS-1 are shown above each graph.
Based on our previous observation that serine phosphorylation of IRS-1 is involved in the modulation of insulin resistance in adipose tissue at late pregnancy (30), we next analyzed the impact of p38 MAPK inhibition on the activation of both IR and IRS-1 by insulin. We found that treatment of pregnant rats from d 18 to 20 of gestation with the p38 MAPK inhibitor SB 202190 significantly increased insulin-stimulated tyrosine phosphorylation of both IR (∼6-fold higher in pregnant than in virgin rats) (Fig. 5C) and IRS-1 (∼2.5-fold higher in pregnant than in virgin rats) (Fig. 5D). Furthermore, treatment of pregnant rats with the p38 MAPK inhibitor (control vs. SB 202190-treated pregnant rats, n = 4–8 animals per group for the following comparisons) was associated with a significant decrease of lumbar adipose tissue TNF-α (15.70 ± 2.19 vs. 7.97 ± 1.55 pg/mg protein, P < 0.05), which was accompanied by a significant decrease of the insulin resistance index HOMA-IR (1.52 ± 0.23 vs. 0.89 ± 0.08, P < 0.05), as well as a significant increase of the insulin sensitivity indexes QUICKI (0.284 ± 0.007 vs. 0.300 ± 0.004, P < 0.05) and FGIR (2.39 ± 0.25 vs. 5.27 ± 0.32 mg/10−4 U, P < 0.001).
Finally, we analyzed the impact of p38 MAPK inhibition on the metabolic actions of insulin. Regarding circulating metabolites, we observed in control vs. SB 202190-treated pregnant rats (n = 4–8 animals per group) a significant decrease both in plasma triglycerides (366.8 ± 59.1 vs. 212.4 ± 23.8 mg/dl, P < 0.05), NEFA (1468.0 ± 225.0 vs. 962.9 ± 63.2 μm, P < 0.05), and glycerol (498.4 ± 55.1 vs. 356.6 ± 22.8 μm, P < 0.05), whereas glucose levels were not altered (108.5 ± 5.38 vs. 106.6 ± 3.52 mg/dl, P = 0.779) and insulin tended to decrease (1.48 ± 0.26 vs. 0.83 ± 0.14 μg/liter, P = 0.054). The obtained decrease in circulating NEFA and glycerol, regarded as indexes of lipolytic activity, indicates that the lipolytic activity of adipose tissue, and consequently tissue sensitivity to insulin, was higher in the SB 202190-treated animals. To confirm this hypothesis, we performed in vitro experiments incubating isolated adipocytes with SB 202190 before treatment with insulin. We observed that in vitro pretreatment of adipocytes from 20-d-pregnant rats with the p38 MAPK inhibitor SB 202190 significantly increased the antilipolytic effect of insulin (Fig. 6A). Furthermore, incubation with SB 202190 increased dephosphorylation of ACC by insulin compared with adipocytes that had not been incubated with the inhibitor, suggesting that inhibition of p38 MAPK favors the lipogenic activity of insulin (Fig. 6B). Together, these data support the notion that activation of p38 MAPK plays a role in insulin resistance during late pregnancy.
Fig. 6.
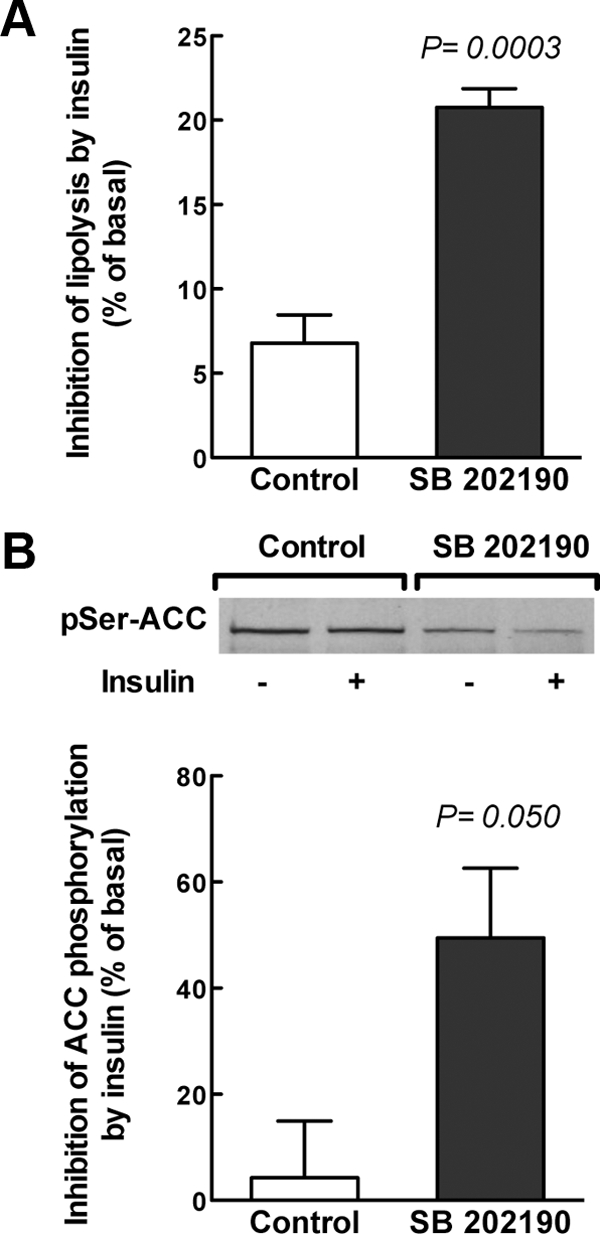
Inhibition of p38 MAPK in adipose cell of late pregnant rats increases the metabolic actions of insulin. Isolated adipocytes from fasted 20-d-pregnant rats were treated for 30 min in the absence (control) and presence of 15 μm of the specific p38 MAPK inhibitor SB 202190 before incubation for further 30 min in the absence (basal) or presence of 90 nm insulin. A, Incubation supernatants were used for glycerol determination to measure the effect of SB 202190 on the antilipolytic action of insulin. Data represent independent experiments with at least five different rats and are expressed as percentage of decrease of lipolytic activity by insulin as compared with the basal condition (i.e. in the absence of insulin). Glycerol determinations were performed in duplicate. B, Adipocytes were harvested to measure the effect of SB 202190 on the lipogenic action of insulin. Western blot analysis of ACC-Ser79 phosphorylation was used as estimate of lipogenesis. Data represent four independent experiments and are expressed as percentage of decrease of ACC phosphorylation by insulin compared with the basal condition (i.e. in the absence of insulin). A representative immunoblot for phosphorylated ACC (pSer-ACC) is shown in the graph.
Discussion
Elevated levels of circulating cytokines reflect on low-grade inflammation at late pregnancy
Late pregnancy is characterized by increased fat depots and insulin resistance, both in human (19–21) and rat (22, 23). Recent work from our group suggests that activation of inflammatory signaling pathways may contribute to the development of insulin resistance in the mother (30). Here, we show that late pregnancy in the rat is associated with low-grade inflammation, as evidenced by an increase of circulating levels of cytokines and acute phase proteins, such as IL-1β, MCP-1, and PAI-1. In accordance with previous studies on pregnancy in humans (35), concentrations of circulating IL-6 and TNF-α were undetectable.
The observed increase in circulating IL-1β concentrations at late pregnancy fits well with previous work showing that this cytokine increases in a model of obesity-related insulin resistance (36), which points to a role of IL-1β in the insulin resistance state of the mother. Furthermore, long-term treatment of adipocytes with IL-1β abrogates insulin signaling and diminishes insulin-mediated glucose uptake (37). It has also been demonstrated that IL-1β represses adiponectin gene expression in adipose tissue (38) and profoundly affects lipid metabolism by decreasing tissue uptake of lipids through inhibition of lipoprotein lipase activity (39) and activation of peroxisome proliferator-activated receptor (PPAR) γ-induced adipogenesis (40). This metabolic effect of IL-1β may contribute to the hypertriglyceridemia and hypoadiponectinemia that we observed in the late pregnant rat (30).
Similarly, the augmented circulating MCP-1 concentrations found in the present study in 20-d-pregnant rats agrees with several observations associating increased MCP-1 levels with insulin resistance, including the increase of circulating MCP-1 levels in diabetic individuals (41), the capacity of MCP-1 to recruit macrophages into adipose tissue (42), and the negative impact of this chemokine on insulin action in skeletal muscle (42), an organ that develops pronounced insulin resistance at late pregnancy (23). We further found that circulating levels of active PAI-1, an acute phase protein that is elevated in the early stages of the inflammatory response, were elevated in late pregnant rats. This observation fits well with the pronounced inhibition of fribrinolysis seen in pregnancy (43) and increased plasma PAI-1 levels in pregnant women (44). In the model of the late pregnant rat, we found a strong correlation between the circulating levels of PAI-1 and insulin resistance at late pregnancy. Several studies relate PAI-1 with insulin resistance. In a model of diet-induced obesity, pharmacological inhibition of PAI-1 has been reported to mitigate the development of insulin resistance, including a reduction in fat and adipocyte volume (45). Another study proposes that PAI-1 levels may reflect on situations characterized by oxidative stress and fat redistribution (46), in which insulin resistance is a common feature, such as late pregnancy. Taken together, these findings lend support to the development of low-grade inflammation at late pregnancy and relate this inflammatory condition to pregnancy-associated insulin resistance in the rat.
Adipose tissue contributes to low-grade inflammation at late pregnancy
The observed accumulation of circulating levels of cytokines and acute phase proteins at late pregnancy raises the question on the origin of this inflammatory response. Adipose tissue may release factors that stimulate the production of inflammatory markers by other organs, such as the liver or, alternatively, adipose tissue itself, may be a relevant source of inflammatory molecules (47). We addressed this question by measuring cytokine levels in lumbar adipose tissue and show that adipose tissue is a relevant source of PAI-1, IL-6, and TNF-α at late gestation.
Regarding PAI-1, it has been suggested that increases in circulating PAI-1 are accompanied by an augmented level of this protein in visceral adipose tissue (48). This is also the case in late pregnant rats, because not only the circulating protein but also tissular PAI-1 expression are significantly increased at late gestation. In rodent models, elevated local expression of PAI-1 has been associated with larger fat pads and hyperthropic adipocytes (49). Thus, the elevated expression of PAI-1 that we observed during gestation may be implicated in the accretion of fat of the mother during pregnancy.
IL-6 and TNF-α are well-known inducers of insulin resistance in several animal models of obesity, type 2 diabetes, and gestational diabetes. We observed comparable amounts of IL-6 in lumbar adipose tissue of late pregnant and virgin rats, whereas other groups reported increases of IL-6 in sc adipose tissue in the last part of gestation (50). IL-6 has a strong impact on insulin signaling due to its ability to reduce IRS-1 and glucose transporter type 4 mRNA levels in 3T3-L1 adipocytes, but it does neither impair tyrosine phosphorylation of the IR nor induce serine phosphorylation of IRS-1 (51). Thus, our results regarding IL-6 suggest that this cytokine does not play a significant role in the augmented serine phosphorylation of IRS-1 that we found in lumbar adipose tissue of late pregnant rats (30). Although TNF-α was undetectable in plasma, we found increased amounts of TNF-α in lumbar adipose tissue of 18- and 20-d-pregnant rats compared with nonpregnant animals. This finding fits well with observations on increased TNF-α levels during pregnancy in women (44, 52), the placenta being the primary source of this cytokine (29). Although IL-6 appears to be released systemically by adipose tissue (53), the extent to which TNF-α, locally produced in white adipose tissue, is released into the circulation is a matter of debate (47). Accordingly, despite low circulating concentrations of this cytokine, locally produced TNF-α may act within adipose tissue as a potent autocrine and paracrine regulator of diverse metabolic processes (47). In fact, TNF-α seems to play a pivotal role in the synthesis of other cytokines within white adipose tissue (53), including PA1–1 (54).
Together, our findings indicate that an increased local production of proinflammatory mediators, such as TNF-α, may contribute to low-grade inflammation at late pregnancy. In obesity, white adipose tissue becomes infiltrated by macrophages, suggesting that nonadipose cells may be a significant component of the inflammatory state within adipose tissue (55). The increment of circulating MCP-1 in late pregnant rats would also be compatible with the recruitment of macrophages to adipose tissue (56). In our model of late pregnant rats, however, we did not find any evidence for the infiltration of lumbar adipose tissue by macrophages, which supports the conclusion that adipocytes account for the observed increment of PAI-1 and TNF-α levels in adipose tissue at late pregnancy and, consequently, contribute to the low-grade inflammatory state of this tissue.
Activation of MAPK contributes to pregnancy-associated insulin resistance in adipose tissue
TNF-α is an important mediator of insulin resistance in obesity and diabetes. In this context, relevant downstream effects of TNF-α include increased serine phosphorylation of IRS-1 (57), through the activation of different serine/threonine kinases, including p38 MAPK, JNK, ERK, mTOR, and IKK, in muscle and adipose tissue (reviewed in Refs. 58, 59). There is evidence that activation of MAPK signaling induces insulin resistance in 3T3-L1 adipocytes (60). Other cytokines have also been reported to impair insulin signaling through activation of stress kinases. For example, there is evidence that the impaired response of 3T3-L1 adipocytes to insulin upon treatment with IL-1β involves p38 MAPK-ERK-1/2 (8) and that adipocytes exposed to TNF-α and IL-1β exhibit a reduced insulin response that can be restored by inhibiting ERK-1/2 and p38 MAPK (61). In obese humans, increased phosphorylation of JNK-1/2 and p38 MAPK has been observed in omental adipose tissue compared with their relative lean controls (9). Furthermore, activation of p38 MAPK has been proposed to functionally link visceral adiposity with whole-body insulin resistance (9). Similarly, ERK-1/2 attenuation has been correlated with exercise-induced improvement of inflammatory-oxidative stress in white adipose tissue (10).
We did not detect any effect of pregnancy on the basal phosphorylation of mTOR or JNK-1/2. In the present study, we found a significant increase of p38 MAPK activity and, as previously published, a modest increase in ERK activity (30) in lumbar adipose tissue of late pregnant rats compared with virgin animals. This observation indicates a functional link between MAPK activation in adipose tissue, probably involving locally produced TNF-α, and pregnancy-associated insulin resistance. To confirm the role of p38 MAPK in the impaired insulin response in lumbar adipose tissue that develops at late pregnancy, we treated pregnant rats with SB 202190, a well-established specific inhibitor of p38 MAPK. Activation of p38 is dependent on dual phosphorylation on Thr180 and Tyr182 (62). Treatment of pregnant rats with SB 202190 completely blocked p38 MAPK phosphorylation and, consequently, its activation. Inhibition of p38 MAPK was paralleled by a significant decrease of adipose tissue TNF-α and, as evidenced by increased IR and IRS-1 tyrosine phosphorylation in response to insulin, a pronounced improvement of insulin signaling in lumbar adipose tissue of late pregnant rats. In addition, we found a significant decrease in pSer-IRS-1, which reflects on the role of p38 MAPK in the physiological deterioration of insulin sensitivity during pregnancy. It remains to be established whether the effect of p38 MAPK inhibition on pSer-IRS-1 is mediated by p38 MAPK itself or by IKK, a downstream effector of p38 MAPK, a mechanism that may be relevant to insulin signaling in adipose cells (63, 64). Furthermore, we observed a higher antilipolitic and lipogenic action of insulin in cells that were pretreated with the p38 inhibitor, suggesting that inhibition of p38 MAPK restored not only the insulin signaling cascade but also the metabolic effects of the hormone in adipose cells at late pregnancy. Such improved insulin sensitivity of adipose tissue contributes to an overall decrease of insulin resistance in late pregnant rats. In previous work, we have shown that treatment of pregnant rats with the peroxisome proliferator-activated receptor γ ligand englitazone restored not only insulin sensitivity in adipose tissue but also serum adiponectin concentrations to levels found in virgin animals (30). Interestingly, rosiglitazone is able to reverse TNF-α-induced insulin resistance in rat brown adipocytes by blocking the activation of ERK-1/2 and p38 MAPK (65). Although we cannot definitely conclude that TNF-α is responsible for p38 MAPK activation at late pregnancy, the present results together with previously published data on the role TNF-α on p38 MAPK allow us to suggest that TNF-α, locally synthesized in fat depots at late pregnancy, may mediate pSer-IRS-1 through p38 MAPK activation and, thus, contribute to the development of insulin resistance at late pregnancy. TNF-α has been reported to increase expression of PAI-1 through mechanisms that include, among others, activation of p38 MAPK (66). Because TNF-α that is synthesized in fat depots at late pregnancy is not secreted, as indicated by its undetectable circulating levels, TNF-α-induced p38 MAPK activation may, therefore, further exacerbate the inflammatory state of the mother by incrementing PAI-1 levels in adipose tissue.
It remains to be established which are the pregnancy-related factor/s that drive adipocyte-associated inflammation. Cortisol and placental-derived hormones, including placental lactogen and estrogen, have been proposed as mediators of insulin resistance at late pregnancy (67). There is also evidence that female sex hormones are involved in the regulation of the immune response. In this context, it is noteworthy that some of the immunomodulatory actions of estrogen are mediated through the local synthesis of cytokines. For example, estradiol increases TNF-α in vitro (68) and in vivo, amplifying the response to lipopolysaccharide challenge at several points critical to the regulation of the progression of the proinflammatory cascade (69). A recent study with healthy premenopausal women with and without intake of contraceptives reported a correlation between the concentration of circulating estradiol with the release of TNF-α and IL-6 during the luteal phase (70). Furthermore, female sex steroids, such as estradiol and progesterone, are likely to affect insulin sensitivity through modulation of adiponectin and body fat (71). Thus, it is tempting to speculate that gestational hormones, such as estradiol, could be one of the initial factors promoting maternal inflammation through modulation of both antiinflammatory factors, such as adiponectin, and/or proinflammatory cytokines, such as TNF-α.
In conclusion, to our knowledge, this is the first study providing evidence that p38 MAPK activation in adipose tissue contributes to adipose tissue insulin resistance at late pregnancy. Our findings lend support to the hypothesis that physiological low-grade inflammation in the maternal organism is relevant to the development of pregnancy-associated insulin resistance.
Acknowledgments
We thank the veterinarian and animal house technicians of the Universidad San Pablo-CEU for helpful assistance with the animals.
This work was supported by Spanish Ministry of Science and Innovation Grants SAF2007-64881 and SAF2010-19603, the University San Pablo-CEU Grant PC15-10), and the National Institutes of Health Grant HD 057236.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ACC
- Acetyl-coenzyme A carboxylase
- FGIR
- fasting glucose to insulin ratio
- FPG
- fasting plasma glucose
- FPI
- fasting plasma insulin
- HOMA-IR
- homeostasis model assessment of insulin resistance
- IKK
- inhibitor of κ light polypeptide gene enhancer in B-cells, kinase β
- IR
- insulin receptor
- IRS
- IR substrate
- JNK
- c-Jun-N-terminal kinase
- KRB
- Krebs-Ringer bicarbonate
- MCP
- monocyte chemotactic protein
- mTOR
- mammalian target of rapamycin
- NEFA
- nonesterified fatty acid
- PAI
- plasminogen activator inhibitor
- pSer-IRS-1
- IRS-1 serine phosphorylation
- QUICKI
- quantitative insulin sensitivity check index.
References
- 1. Flier JS. 1995. The adipocyte: storage depot or node on the energy information superhighway? Cell 80:15–18 [DOI] [PubMed] [Google Scholar]
- 2. Tilg H, Moschen AR. 2008. Inflammatory mechanisms in the regulation of insulin resistance. Mol Med 14:222–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shoelson SE, Lee J, Goldfine AB. 2006. Inflammation and insulin resistance. J Clin Invest 116:1793–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sjöholm A, Nyström T. 2006. Inflammation and the etiology of type 2 diabetes. Diabetes Metab Res Rev 22:4–10 [DOI] [PubMed] [Google Scholar]
- 5. Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, Kishimoto K, Matsuki Y, Murakami M, Ichisaka T, Murakami H, Watanabe E, Takagi T, Akiyoshi M, Ohtsubo T, Kihara S, Yamashita S, Makishima M, Funahashi T, Yamanaka S, Hiramatsu R, Matsuzawa Y, Shimomura I. 2005. Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science 307:426–430 [DOI] [PubMed] [Google Scholar]
- 6. Tschritter O, Fritsche A, Thamer C, Haap M, Shirkavand F, Rahe S, Staiger H, Maerker E, Häring H, Stumvoll M. 2003. Plasma adiponectin concentrations predict insulin sensitivity of both glucose and lipid metabolism. Diabetes 52:239–243 [DOI] [PubMed] [Google Scholar]
- 7. Bastard JP, Maachi M, Lagathu C, Kim MJ, Caron M, Vidal H, Capeau J, Feve B. 2006. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur Cytokine Netw 17:4–12 [PubMed] [Google Scholar]
- 8. Jager J, Grémeaux T, Cormont M, Le Marchand-Brustel Y, Tanti JF. 2007. Interleukin-1β-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology 148:241–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blüher M, Bashan N, Shai I, Harman-Boehm I, Tarnovscki T, Avinaoch E, Stumvoll M, Dietrich A, Klöting N, Rudich A. 2009. Activated Ask1-MKK4–p38MAPK/JNK stress signaling pathway in human omental fat tissue may link macrophage infiltration to whole-body Insulin sensitivity. J Clin Endocrinol Metab 94:2507–2515 [DOI] [PubMed] [Google Scholar]
- 10. Sakurai T, Izawa T, Kizaki T, Ogasawara JE, Shirato K, Imaizumi K, Takahashi K, Ishida H, Ohno H. 2009. Exercise training decreases expression of inflammation-related adipokines through reduction of oxidative stress in rat white adipose tissue. Biochem Biophys Res Commun 379:605–609 [DOI] [PubMed] [Google Scholar]
- 11. Virkamäki A, Ueki K, Kahn CR. 1999. Protein-protein interaction in insulin signaling and the molecular mechanisms of insulin resistance. J Clin Invest 103:931–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Saltiel AR, Kahn CR. 2001. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414:799–806 [DOI] [PubMed] [Google Scholar]
- 13. Le Roith D, Zick Y. 2001. Recent advances in our understanding of insulin action and insulin resistance. Diabetes Care 24:588–597 [DOI] [PubMed] [Google Scholar]
- 14. Paz K, Hemi R, LeRoith D, Karasik A, Elhanany E, Kanety H, Zick Y. 1997. A molecular basis for insulin resistance. Elevated serine/threonine phosphorylation of IRS-1 and IRS-2 inhibits their binding to the juxtamembrane region of the insulin receptor and impairs their ability to undergo insulin-induced tyrosine phosphorylation. J Biol Chem 272:29911–29918 [DOI] [PubMed] [Google Scholar]
- 15. Zick Y. 2001. Insulin resistance: a phosphorylation-based uncoupling of insulin signaling. Trends Cell Biol 11:437–441 [DOI] [PubMed] [Google Scholar]
- 16. Rui L, Aguirre V, Kim JK, Shulman GI, Lee A, Corbould A, Dunaif A, White MF. 2001. Insulin/IGF-1 and TNF-α stimulate phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. J Clin Invest 107:181–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Delahaye L, Mothe-Satney I, Myers MG, White MF, Van Obberghen E. 1998. Interaction of insulin receptor substrate-1 (IRS-1) with phosphatidylinositol 3-kinase: effect of substitution of serine for alanine in potential IRS-1 serine phosphorylation sites. Endocrinology 139:4911–4919 [DOI] [PubMed] [Google Scholar]
- 18. López-Luna P, Maier I, Herrera E. 1991. Carcass and tissue fat content in the pregnant rat. Biol Neonate 60:29–38 [DOI] [PubMed] [Google Scholar]
- 19. Ryan EA, O'Sullivan MJ, Skyler JS. 1985. Insulin action during pregnancy. Studies with the euglycemic clamp technique. Diabetes 34:380–389 [DOI] [PubMed] [Google Scholar]
- 20. Catalano PM, Tyzbir ED, Wolfe RR, Calles J, Roman NM, Amini SB, Sims EA. 1993. Carbohydrate metabolism during pregnancy in control subjects and women with gestational diabetes. Am J Physiol 264:E60–E67 [DOI] [PubMed] [Google Scholar]
- 21. Buchanan TA, Metzger BE, Freinkel N, Bergman RN. 1990. Insulin sensitivity and B-cell responsiveness to glucose during late pregnancy in lean and moderately obese women with normal glucose tolerance or mild gestational diabetes. Am J Obstet Gynecol 162:1008–1014 [DOI] [PubMed] [Google Scholar]
- 22. Leturque A, Ferre P, Burnol AF, Kande J, Maulard P, Girard J. 1986. Glucose utilization rates and insulin sensitivity in vivo in tissues of virgin and pregnant rats. Diabetes 35:172–177 [DOI] [PubMed] [Google Scholar]
- 23. Ramos P, Herrera E. 1995. Reversion of insulin resistance in the rat during late pregnancy by 72-h glucose infusion. Am J Physiol 269:E858–E863 [DOI] [PubMed] [Google Scholar]
- 24. Faas MM, Schuiling GA, Linton EA, Sargent IL, Redman CW. 2000. Activation of peripheral leukocytes in rat pregnancy and experimental preeclampsia. Am J Obstet Gynecol 182:351–357 [DOI] [PubMed] [Google Scholar]
- 25. von Dadelszen P, Wilkins T, Redman CW. 1999. Maternal peripheral blood leukocytes in normal and pre-eclamptic pregnancies. Br J Obstet Gynaecol 106:576–581 [DOI] [PubMed] [Google Scholar]
- 26. Hauguel-de Mouzon S, Guerre-Millo M. 2006. The placenta cytokine network and inflammatory signals. Placenta 27:794–798 [DOI] [PubMed] [Google Scholar]
- 27. Di Benedetto A, Russo GT, Corrado F, Di Cesare E, Alessi E, Nicocia G, D'Anna R, Cucinotta D. 2005. Inflammatory markers in women with a recent history of gestational diabetes mellitus. J Endocrinol Invest 28:34–38 [DOI] [PubMed] [Google Scholar]
- 28. Orsi NM, Tribe RM. 2008. Cytokine networks and the regulation of uterine function in pregnancy and parturition. J Neuroendocrinol 20:462–469 [DOI] [PubMed] [Google Scholar]
- 29. Kirwan JP, Hauguel-De Mouzon S, Lepercq J, Challier JC, Huston-Presley L, Friedman JE, Kalhan SC, Catalano PM. 2002. TNF-α is a predictor of insulin resistance in human pregnancy. Diabetes 51:2207–2213 [DOI] [PubMed] [Google Scholar]
- 30. Sevillano J, de Castro J, Bocos C, Herrera E, Ramos MP. 2007. Role of insulin receptor substrate-1 serine 307 phosphorylation and adiponectin in adipose tissue insulin resistance in late pregnancy. Endocrinology 148:5933–5942 [DOI] [PubMed] [Google Scholar]
- 31. Cacho J, Sevillano J, de Castro J, Herrera E, Ramos MP. 2008. Validation of simple indexes to assess insulin sensitivity during pregnancy in Wistar and Sprague-Dawley rats. Am J Physiol Endocrinol Metab 295:E1269–E1276 [DOI] [PubMed] [Google Scholar]
- 32. Katz A, Nambi SS, Mather K, Baron AD, Follmann DA, Sullivan G, Quon MJ. 2000. Quantitative insulin sensitivity check index: a simple, accurate method for assessing insulin sensitivity in humans. J Clin Endocrinol Metab 85:2402–2410 [DOI] [PubMed] [Google Scholar]
- 33. Ramos MP, Crespo-Solans MD, del Campo S, Cacho J, Herrera E. 2003. Fat accumulation in the rat during early pregnancy is modulated by enhanced insulin responsiveness. Am J Physiol Endocrinol Metab 285:E318–E328 [DOI] [PubMed] [Google Scholar]
- 34. Zick Y. 2004. Uncoupling insulin signalling by serine/threonine phosphorylation: a molecular basis for insulin resistance. Biochem Soc Trans 32:812–816 [DOI] [PubMed] [Google Scholar]
- 35. Bernardi F, Guolo F, Bortolin T, Petronilho F, Dal-Pizzol F. 2008. Oxidative stress and inflammatory markers in normal pregnancy and preeclampsia. J Obstet Gynaecol Res 34:948–951 [DOI] [PubMed] [Google Scholar]
- 36. Maury E, Ehala-Aleksejev K, Guiot Y, Detry R, Vandenhooft A, Brichard SM. 2007. Adipokines oversecreted by omental adipose tissue in human obesity. Am J Physiol Endocrinol Metab 293:E656–E665 [DOI] [PubMed] [Google Scholar]
- 37. Lagathu C, Yvan-Charvet L, Bastard JP, Maachi M, Quignard-Boulangé A, Capeau J, Caron M. 2006. Long-term treatment with interleukin-1β induces insulin resistance in murine and human adipocytes. Diabetologia 49:2162–2173 [DOI] [PubMed] [Google Scholar]
- 38. Lihn AS, Bruun JM, He G, Pedersen SB, Jensen PF, Richelsen B. 2004. Lower expression of adiponectin mRNA in visceral adipose tissue in lean and obese subjects. Mol Cell Endocrinol 219:9–15 [DOI] [PubMed] [Google Scholar]
- 39. Argilés JM, Lopez-Soriano FJ, Evans RD, Williamson DH. 1989. Interleukin-1 and lipid metabolism in the rat. Biochem J 259:673–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Suzawa M, Takada I, Yanagisawa J, Ohtake F, Ogawa S, Yamauchi T, Kadowaki T, Takeuchi Y, Shibuya H, Gotoh Y, Matsumoto K, Kato S. 2003. Cytokines suppress adipogenesis and PPAR-γ function through the TAK1/TAB1/NIK cascade. Nat Cell Biol 5:224–230 [DOI] [PubMed] [Google Scholar]
- 41. Nomura S, Shouzu A, Omoto S, Nishikawa M, Fukuhara S. 2000. Significance of chemokines and activated platelets in patients with diabetes. Clin Exp Immunol 121:437–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sell H, Dietze-Schroeder D, Kaiser U, Eckel J. 2006. Monocyte chemotactic protein-1 is a potential player in the negative cross-talk between adipose tissue and skeletal muscle. Endocrinology 147:2458–2467 [DOI] [PubMed] [Google Scholar]
- 43. Cerneca F, Ricci G, Simeone R, Malisano M, Alberico S, Guaschino S. 1997. Coagulation and fibrinolysis changes in normal pregnancy. Increased levels of procoagulants and reduced levels of inhibitors during pregnancy induce a hypercoagulable state, combined with a reactive fibrinolysis. Eur J Obstet Gynecol Reprod Biol 73:31–36 [DOI] [PubMed] [Google Scholar]
- 44. Stewart FM, Freeman DJ, Ramsay JE, Greer IA, Caslake M, Ferrell WR. 2007. Longitudinal assessment of maternal endothelial function and markers of inflammation and placental function throughout pregnancy in lean and obese mothers. J Clin Endocrinol Metab 92:969–975 [DOI] [PubMed] [Google Scholar]
- 45. Crandall DL, Quinet EM, El Ayachi S, Hreha AL, Leik CE, Savio DA, Juhan-Vague I, Alessi MC. 2006. Modulation of adipose tissue development by pharmacological inhibition of PAI-1. Arterioscler Thromb Vasc Biol 26:2209–2215 [DOI] [PubMed] [Google Scholar]
- 46. Alessi MC, Juhan-Vague I. 2006. PAI-1 and the metabolic syndrome: links, causes, and consequences. Arterioscler Thromb Vasc Biol 26:2200–2207 [DOI] [PubMed] [Google Scholar]
- 47. Trayhurn P, Wood IS. 2004. Adipokines: inflammation and the pleiotropic role of white adipose tissue. Br J Nutr 92:347–355 [DOI] [PubMed] [Google Scholar]
- 48. Shimomura I, Funahashi T, Takahashi M, Maeda K, Kotani K, Nakamura T, Yamashita S, Miura M, Fukuda Y, Takemura K, Tokunaga K, Matsuzawa Y. 1996. Enhanced expression of PAI-1 in visceral fat: possible contributor to vascular disease in obesity. Nat Med 2:800–803 [DOI] [PubMed] [Google Scholar]
- 49. Scroyen I, Jacobs F, Cosemans L, De Geest B, Lijnen HR. 2009. Effect of plasminogen activator inhibitor-1 on adipogenesis in vivo. Thromb Haemost 101:388–393 [PubMed] [Google Scholar]
- 50. Caja S, Puerta M. 2008. White adipose tissue production and release of IL-6 and TNF-α do not parallel circulating and cerebrospinal fluid concentrations in pregnant rats. Horm Metab Res 40:375–380 [DOI] [PubMed] [Google Scholar]
- 51. Rotter V, Nagaev I, Smith U. 2003. Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-α, overexpressed in human fat cells from insulin-resistant subjects. J Biol Chem 278:45777–45784 [DOI] [PubMed] [Google Scholar]
- 52. Melczer Z, Bánhidy F, Csömör S, Kovács M, Siklós P, Winkler G, Cseh K. 2002. Role of tumour necrosis factor-α in insulin resistance during normal pregnancy. Eur J Obstet Gynecol Reprod Biol 105:7–10 [DOI] [PubMed] [Google Scholar]
- 53. Coppack SW. 2001. Pro-inflammatory cytokines and adipose tissue. Proc Nutr Soc 60:349–356 [DOI] [PubMed] [Google Scholar]
- 54. Plomgaard P, Keller P, Keller C, Pedersen BK. 2005. TNF-α, but not IL-6, stimulates plasminogen activator inhibitor-1 expression in human subcutaneous adipose tissue. J Appl Physiol 98:2019–2023 [DOI] [PubMed] [Google Scholar]
- 55. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr 2003. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112:1796–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashira K, Kasuga M. 2006. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest 116:1494–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. 1996. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-α- and obesity-induced insulin resistance. Science 271:665–668 [DOI] [PubMed] [Google Scholar]
- 58. Nieto-Vazquez I, Fernández-Veledo S, Krämer DK, Vila-Bedmar R, Garcia-Guerra L, Lorenzo M. 2008. Insulin resistance associated to obesity: the link TNF-α. Arch Physiol Biochem 114:183–194 [DOI] [PubMed] [Google Scholar]
- 59. Draznin B. 2006. Molecular mechanisms of insulin resistance: serine phosphorylation of insulin receptor substrate-1 and increased expression of p85α: the two sides of a coin. Diabetes 55:2392–2397 [DOI] [PubMed] [Google Scholar]
- 60. Fujishiro M, Gotoh Y, Katagiri H, Sakoda H, Ogihara T, Anai M, Onishi Y, Ono H, Abe M, Shojima N, Fukushima Y, Kikuchi M, Oka Y, Asano T. 2003. Three mitogen-activated protein kinases inhibit insulin signaling by different mechanisms in 3T3-L1 adipocytes. Mol Endocrinol 17:487–497 [DOI] [PubMed] [Google Scholar]
- 61. Kobashi C, Asamizu S, Ishiki M, Iwata M, Usui I, Yamazaki K, Tobe K, Kobayashi M, Urakaze M. 2009. Inhibitory effect of IL-8 on insulin action in human adipocytes via MAP kinase pathway. J Inflamm 6:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ. 1995. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem 270:7420–7426 [DOI] [PubMed] [Google Scholar]
- 63. Herschkovitz A, Liu YF, Ilan E, Ronen D, Boura-Halfon S, Zick Y. 2007. Common inhibitory serine sites phosphorylated by IRS-1 kinases, triggered by insulin and inducers of insulin resistance. J Biol Chem 282:18018–18027 [DOI] [PubMed] [Google Scholar]
- 64. Gao Z, Zhang X, Zuberi A, Hwang D, Quon MJ, Lefevre M, Ye J. 2004. Inhibition of insulin sensitivity by free fatty acids requires activation of multiple serine kinases in 3T3-L1 adipocytes. Mol Endocrinol 18:2024–2034 [DOI] [PubMed] [Google Scholar]
- 65. Hernandez R, Teruel T, Lorenzo M. 2003. Rosiglitazone produces insulin sensitisation by increasing expression of the insulin receptor and its tyrosine kinase activity in brown adipocytes. Diabetologia 46:1618–1628 [DOI] [PubMed] [Google Scholar]
- 66. Pandey M, Loskutoff DJ, Samad F. 2005. Molecular mechanisms of tumor necrosis factor-α-mediated plasminogen activator inhibitor-1 expression in adipocytes. FASEB J 19:1317–1319 [DOI] [PubMed] [Google Scholar]
- 67. Ryan EA. 2003. Hormones and insulin resistance during pregnancy. Lancet 362:1777–1778 [DOI] [PubMed] [Google Scholar]
- 68. D'Agostino P, Milano S, Barbera C, Di Bella G, La Rosa M, Ferlazzo V, Farruggio R, Miceli DM, Miele M, Castagnetta L, Cillari E. 1999. Sex hormones modulate inflammatory mediators produced by macrophages. Ann NY Acad Sci 876:426–429 [DOI] [PubMed] [Google Scholar]
- 69. Kahl S, Elsasser TH, Li CJ. 2011. Modeling the effects of estradiol and progesterone on the acute phase proinflammatory axis: variability in tumor necrosis factor-α, nitric oxide, and xanthine oxidase responses to endotoxin challenge in steers. Domest Anim Endocrinol 40:213–221 [DOI] [PubMed] [Google Scholar]
- 70. Schwarz E, Schäfer C, Bode JC, Bode C. 2000. Influence of the menstrual cycle on the LPS-induced cytokine response of monocytes. Cytokine 12:413–416 [DOI] [PubMed] [Google Scholar]
- 71. Leung KC, Xu A, Craig ME, Martin A, Lam KS, O'Sullivan AJ. 2009. Adiponectin isoform distribution in women–relationship to female sex steroids and insulin sensitivity. Metabolism 58:239–245 [DOI] [PubMed] [Google Scholar]