

Abstract
The sarcomeric Z-disc defines the lateral borders of the sarcomere and has primarily been seen as a structure important for mechanical stability. This view has changed dramatically within the last one or two decades. A multitude of novel Z-disc proteins and their interacting partners have been identified, which has led to the identification of additional functions and which have now been assigned to this structure. This includes its importance for intracellular signalling, for mechanosensation and mechanotransduction in particular, an emerging importance for protein turnover and autophagy, as well as its molecular links to the t-tubular system and the sarcoplasmic reticulum. Moreover, the discovery of mutations in a wide variety of Z-disc proteins, which lead to perturbations of several of the above-mentioned systems, gives rise to a diverse group of diseases which can be termed Z-discopathies. This paper provides a brief overview of these novel aspects as well as points to future research directions.
1. Introduction
Z-discs (Z-disk, Z-line, Z-band) delineate the lateral borders of sarcomeres and are the smallest functional units in striated muscle. The core of a Z-disc consists of actin filaments coming from adjacent sarcomeres which are crosslinked by α actinin molecules [1]. Mature Z-discs are probably composed of hundreds of different proteins, and one regards them as one of the most complex macromolecular structures in biology [2]. Z-discs, which are difficult to detect in conventional light microscopy, appear in the longitudinal view of electron microscopy as electron dense bands with varying sizes, ranging between 30 and 50 nm in fast muscle and between 100 and 140 nm in slow muscle and cardiac myocytes. In the transverse view, they appear as a “basketweave” or as “small square lattices” [3]. F-actin filaments, titin, and the nebulin/nebulette system directly attach to the Z-disc, and, because of their importance for sarcomere mechanics, the Z-disc has initially been regarded as important for mechanical stability only.
However, within the last decade, our knowledge with regard to this structure increased steadily, and it is now clear that the sarcomeric Z-disc serves as a nodal point for signalling in general and mechanosensation and mechanotransduction in particular. The Z-disc also links to the t-tubular system, the sarcoplasmic reticulum, and several E3 ubiquitin ligases localized to the Z-disc and link this structure to protein turnover and probably autophagy.
While excellent reviews with regard to the structural aspects of the Z-disc [4, 5] and with emphasis on its role in signalling [6–9] have been published recently, we will focus more on the role of the Z-disc in disease and will highlight recent developments in the exciting field of Z-disc biology.
Although we use the term “Z-disc protein” in this paper, such a protein is difficult to define. This is because (i) a protein at the Z-disc may not always be confined to the disc—depending on developmental stage and physiological circumstances; (ii) it may be found at other locations within the sarcomere, such as the I- and M-bands (i.e., FHL1 and 2); (iii) these proteins can also be in multiple localizations, including the nucleus (such as muscle LIM protein, (MLP)), costamere (such as integrin linked kinase, (ILK)), cytoplasm, intercalated disc, sarcoplasmic reticulum, and t-tubules. Another example is desmin, which is present at the sarcomeric Z-disc but as an intermediate filament (IF); it connects not only this structure with the desmosomes but has other links to other compartments, linking these to the nucleus as well. Other examples of Z-disc proteins include the giant protein titin, where only the amino-terminus is located to the Z-disc, and nebulin/nebulette, where only the carboxy-terminus is localized to the Z-disc and/or F-actin. In this context, an NCBI online search (http://www.ncbi.nlm.nih.gov/guide/) revealed that currently about 227 gene products or proteins are associated with the Z disc, and at least about 50 human and about 60 mouse gene products have been assigned to it. Due to space restrictions, this paper cannot discuss all of these proteins, but will focus on some significant recent developments and point out some important and particularly disease-associated proteins and their genes.
2. Structural Aspects
Z-discs can be defined as plate-like structures in sarcomeres to which the plus ends of actin filaments are localized, or they can be defined as the centre of the I-band. As pointed out above, they are probably composed of hundreds of different proteins and are regarded as one of the most complex macromolecular structures in biology [2]. Major Z-disc proteins include cardiac actin which is crosslinked by α actinin and “capped” by CapZ, titin, which spans a complete half sarcomere, and nebulin/nebulette which runs along actin filaments.
Z-discs, which are difficult to detect in conventional light microscopy, appear in the longitudinal view of electron microscopy as dense zigzag bands with varying but myofibre-specific sizes, ranging between 30 to 50 nm in fast muscle and between 100 to 140 nm in slow muscle and cardiac myocytes. Z-disc widths are determined by the layers of α actinin, which can range from two to six or even more molecules [10, 11]. It is interesting to find the thinnest Z-discs in the fastest muscle fibres, which, functionally, produce the highest sarcomere-shortening velocities, and the widest Z-discs in slow muscle. It is difficult to give an explanation for this phenomenon, but, if the Z disc contributes to series elasticity, perhaps via its connection to titin, thinner discs may provide less elasticity (=stiffer cell). As the contractile apparatus is activated, it has to stretch the discs in series before force transmits—so cellular force transmission throughout the muscle would have a shorter delay and be faster. Also, may be via z-disc/titin/actomyosin interaction crossbridge that cycling may be faster. In the transverse view, Z-discs appear as “basketweaves” or “small square lattices” [3], and it has been proposed by Goldstein and coworkers [12–14] as well as by Yamaguchi et al. [15], that the Z-disc changes its structure, from small square lattice in resting muscle to a wider basketweave form in activated muscle [13]. A study of α actinin binding to actin within the Z-disc revealed that the angles between both proteins varied preferentially between 60 and 120 degrees [16]. Both the conformational change of the Z-disc during contraction as well as the changes of the angles between α actinin and actin could well be part of a stress-sensing mechanism.
3. Z-Disc-Related Proteins
3.1. α Actinin
α actinin belongs to the spectrin gene superfamily which includes a wide variety of different cytoskeletal components including dystrophin, utrophin, fimbrin, and alpha and beta spectrins. The skeletal and cardiac muscle isoforms are localized to the sarcomeric Z-discs and are important for actin localization. Various splice variants have been reported, the largest of which contains 892 amino acids. The mature protein is about 35 nm in length and occurs as an antiparallel homodimer. The single protein consists of four spectrin repeats at its centre as well as of an actin-binding domain (ABD) at its amino-terminus and two EF hands (calmodulin homology domains) at the carboxy-terminus, which enable α actinin to crosslink actin filaments within the Z-disc. A single mutation in α actinin (Q9R) has been found in an individual affected by DCM [17], and via linkage analysis the gene for α actinin (ACTN2) has been linked recently to HCM [18, 19] (please see also Table 1 and Figure 1). The α actinin Q9R mutation in particular disrupts the interaction with MLP and seems to affect cellular differentiation.
Table 1.
Z-disc proteins involved in human disease.
| Name of gene | Type of disease |
|---|---|
| α actinin (central Z-disc) | Cardiomyopathy |
| Muscle LIM protein (MLP) | Cardiomyopathy |
| Telethonin (TCAP) | Cardiomyopathy, LGMD2G |
| Actin | Cardiomyopathy |
| Integrin-linked kinase (ILK) | DCM |
| Calsarcin1 (Myozenin, ZASP) | Cardiomyopathy, myofibrillar myopathy (MFM) zaspopathies |
| FHL1 (four and a half LIM domain protein) | X-linked myopathy with postural muscle atrophy (XMPMA), scapuloperoneal (SP), reducing body myopathy (RBM) |
| Cypher (Zasp, Oracle) | Cardiomyopathy, MFM |
| Desmin (Z-disc related) | Multiple |
| Nebulin/Nebulette | Nemaline myopathy |
| Myotilin | LGMD, MFM, myotilinopathy |
| BAG3 | Cardiomyopathy, MFM |
| Titin (Z-disc)* | Cardiomyopathy (titinopathies) |
Z-disc protein mutations cause a wide spectrum of diseases, which may be called “Z-discopathies”. Titin mutation itself cause a broad spectrum of diseases (here we refer only to mutations affecting the Z-disc). The term cardiomyopathy includes at least HCM and DCM, other types of heart failure such as arrhythmogenic right ventricular myopathy.
Figure 1.
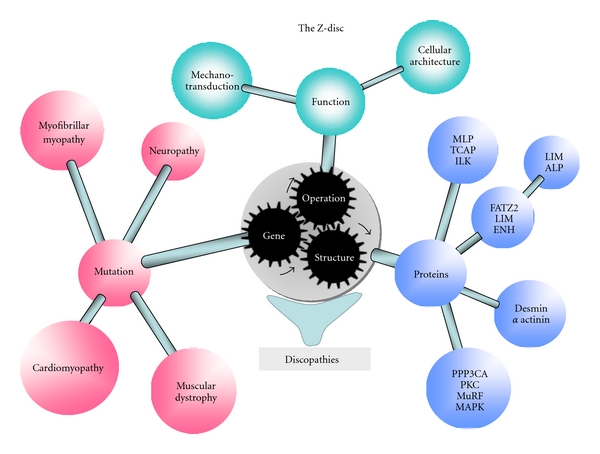
The sarcomeric Z-disc—multiple functions and links to maladaptation. The figure links various functions and structural aspects to maladaptation and disease, such as mutations in Z-disc proteins which are linked to different diseases. Defects in mechanosensation and mechanotransduction are linked to Z-disc mediated autophagy and UPS which in turn are linked to skeletal and cardiac muscle disease.
3.2. Muscle LIM Protein (MLP)
Muscle LIM protein (for Lin11, Isl 1, and Mec 3 (LIM)) is a 194 amino acid LIM-only protein localized not only to the sarcomeric Z-disc, where it interacts most probably with α actinin [20], calcineurin [21], and telethonin [22, 23], but is also present in the nucleus where this protein is able to interact with myoD, myogenin, and MRF4 [24], as well as at the intercalated discs where it interacts with NRAP [25], and at the costameres where it interacts with zyxin [20], integrin linked kinase (ILK) [26], and β1 spectrin [27]. MLP-deficient animals develop a severe form of heart failure, and patients with mutations in this gene develop forms of cardiomyopathy—either dilated cardiomyopathy DCM (dilated, enlarged ventricular chambers and inadequate systolic function) or hypertrophic cardiomyopathy HCM (increase in myocardial wall thicknesses which may or may not be associated with fatal arrhythmias) [28, 29]. MLP interacts with a variety of additional proteins such as cofilin 2 [30] and actin [31] and is involved in actin polymerization. Moreover, the protein has been implicated in various signal transduction cascades, including mechanosensation and mechanotransduction, calcium metabolism, and myofibrillogenesis. Of note, MLP has also been reported to be deacetylated by histone deacetylase 4 (HDAC4) and acetylated by the histone acetylase (HAT) PCAM and that its acetylation is linked to an increase myofibrillar calcium sensitivity [32]. Although little is known about the role of HDACs and HATs with regard to their effects on calcium sensitivity, a possible link between these molecules, the Z-disc, and contractility is of great interest because a direct link between the Z-disc and calcium sensitivity has never been shown before. However, future research will have to elucidate this link in much more detail (for a recent review on MLP, see [33]).
3.3. Telethonin (TCAP)
Telethonin (titin cap, t-cap, TCAP) is a small 167 amino acid protein (molecular weight ~19 kDa) striated-muscle-specific protein with a unique β-sheet structure. This protein, with unknown homologue genes, binds in an antiparallel (2 : 1) sandwich complex at the periphery of the sarcomeric Z-disc to the titin Z1-Z2 domains and essentially “glues” together the N-termini of two adjacent titin molecules [2]. This interaction represents also the strongest protein-protein interaction observed to date [34]. In this regard, it will be interesting to analyze telethonin's function during biomechanical stress. Telethonin is an in vitro substrate of titin kinase, an interaction thought to be critical during myofibril growth [35], and the protein also can be phosphorylated by protein kinase D (PKD) which might link telethonin to the regulation of G-protein-coupled receptor (GPCR) signalling [36].
Telethonin interacts also with a wide variety of additional and very different proteins, such as ankyrin repeat protein 2, small ankyrin 1 (a transmembrane protein of the sarcoplasmic reticulum) [37], minK, a potassium channel β-subunit, as well as with sodium channels such as Nav1.5 [38, 39]. Interestingly, the interaction with mink indicates a link between telethonin and the t-tubular system, a connection also observed in a telethonin-deficient zebrafish model [40]. However, the precise interplay between telethonin and the t-tubular system needs to be elucidated.
In addition, telethonin interacts with MDM2 and muscle ring fingers 1/2 (MuRF1/2) [41, 42]—E3 ubiquitin ligases strongly impacting cardiac protein turnover. It remains to be elucidated which telethonin domains are involved in the interaction with MuRF 1/2 and whether mutations affect the degradation process of telethonin. It might well be that some telethonin mutations may lead to forms of proteinopathies, which are characterized by aggresome formation, autophagy, and apoptosis (see next chapter).
Moreover, telethonin may participate in the regulation of myocardial hypertrophy by interacting with calsarcin-1 (also known as FATZ-2 or myozenin-2) [43], which is a mediator of calcineurin activity. Telethonin interacts with, as well as regulates, the secretion of myostatin, an important negative regulator of muscle growth [44], and interacts with BMP10, a positive regulator of hypertrophy [45]. Therefore, telethonin is well suited to influence the delicate balance between atrophy and hypertrophy or myocardial remodelling [28]. However, this link needs to be analyzed in more detail. Telethonin may also affect cell survival by interacting with the proapoptotic protein Siva [46] and is involved in cardiac mechanosensation [22].
Nonsense mutations in the telethonin gene are associated with limb-girdle muscular dystrophy type 2G (LGMD 2G) [47–49], heterozygous missense mutations, with dilated and hypertrophic forms of cardiomyopathy [18, 22, 50], as well as with intestinal pseudoobstruction [39]. However, a recently discovered telethonin mutation (R76C), found in an individual affected by intestinal pseudoobstruction, affected the Nav1.5 (SCN5A) channel when coexpressed in HEK 293 cells altering steady-state activation kinetics [39]. Whether this effect can also be observed in the heart or whether other telethonin mutations also affect ion channels requires elucidation.
To date it is not at all clear why and how telethonin mutations cause these broad-spectrum diseases. Ion channels, E3 ubiquitin ligases, autophagy, and apoptosis could well play a role. In this regard, telethonin deficiency in a genetically altered mouse model is not associated with a spontaneous cardiac phenotype but leads to severe maladaptation and heart failure following biomechanical stress. Telethonin interacts with the proapoptotic protein p53 and facilitates its degradation via MDM2. Loss of telethonin in the presence of biomechanical stress increases p53 as well as apoptotic cell death [51]. This novel link might help to explain some of the pathologies observed in patients affected by mutations in this gene.
3.4. Integrin-Linked kinase (ILK)
ILK has been identified as an integrin-binding protein and has also been localized to the sarcomeric Z-disc, but the functionality of its kinase domain in vivo has recently been questioned [52]. The ternary complex of ILK, Pinch and Parvin (IPP) interacts with the cytoplasmic tail of β integrin and couples them to the actin cytoskeleton. ILK is found at costameres and has also been localized to the Z-disc area where this protein interacts with a host of different proteins, including MLP [26] or c-Src [53]. It has also been implicated in Akt-mediated survival signalling. An ILK nonsense mutation has been identified in zebrafish which leads to a “lost contact” phenotype indicating that cells, particularly endothelial cells and cardiac myocytes, lose contact with their surrounding cells. In addition, a human ILK mutation (A262V) has been described in a DCM patient. This mutation, when injected into an ILK-deficient zebrafish, did not rescue the phenotype, while wildtype ILK does. Among other effects, ILK A262V was also associated with a decrease in capillary density in human myocardial tissue. By affecting both, cardiac myocytes and endothelial cells, ILK A262V causes cardiomyopathy and associated heart failure via a novel molecular mechanism [54].
3.5. Melusin
Melusin is a muscle-specific protein located at costameres near the Z-disc, where it binds to the cytoplasmic domain of β1 integrin [55]. Homozygous deletion of this gene is not associated with a spontaneous phenotype, but leads to a reduced left ventricular hypertrophy and a transition to DCM following transverse aortic constriction (TAC). More interestingly, deficiency of this protein is not associated with any loss of sensitivity to humoral factors such as angiotensin II or phenylephrine in terms of a myocardial hypertrophic response. Further analysis revealed that following pressure overload glycogen synthase kinase 3β (GSK 3β) and Akt phosphorylation were blunted in melusin deficient hearts [56]. Conversely, melusin overexpression is protective after TAC [57]. In addition, a search for melusin mutations in human heart failure patients revealed the presence of three different genetic variations, but a clear genotype-phenotype relationship could not be established [58].
Thus, melusin is an element of the integrin-dependent cardiac mechanosensor machinery, it may be indispensable for adaptive cardiac remodelling, and melusin might serve as a therapeutic target in heart failure.
3.6. Calsarcin 1 (FATZ2, Myozenin2)
The calsarcins have initially been cloned via their ability to interact with the protein phosphatase calcineurin, and they are localized to the Z-disc [59]. Further research showed that calsarcin 1 knock-out animals do not have any spontaneous phenotype, but, when subjected to biomechanical stress, for example, TAC, they respond with increased calcineurin activity, massive hypertrophy, and heart failure, which suggests that calsarcin1 inhibits calcineurin function [60]. Recently HCM-associated mutations have been identified in this gene, making it another candidate gene for cardiomyopathy [61].
3.7. Four-and-Half-Domain-LIM Proteins 1/2/3 (FHL1/2/3)
This group of proteins contains an amino-terminal zinc finger/half LIM domain followed by four complete LIM domains. This arrangement has led to the designation of this class of proteins as four and a half LIM domain proteins (FHL). It encompasses currently five different members, of which FHL 1, 2, and 3 are expressed in striated muscle where they localize to the sarcomeric Z-disc/I-band area. But they can also sometimes be found in the nucleus [62]. FHL1 knock-out animals subjected to TAC which undergo protective remodelling—a favourable outcome. It has been suggested that this protein plays an important role in the mechanism of pathological hypertrophy by sensing biomechanical stress responses via the N2B titin stretch sensor domain. This initiates changes in the titin- and MAPK-mediated responses [63]. FHL1, 2, and 3 interact with extracellular-regulated kinases (Erk), with, particularly, FHL2 as an inhibitor of Erk-mediated transcriptional events [62].
Human mutations in FHL1 have been identified in patients affected by X linked myopathy with postural muscle atrophy (XMPMA, probably a form of Emery Dreyfuss Muscular Dystrophy) [64] and scapuloperoneal (SP) syndrome [65] as well as in patients with reducing body myopathy (RBM). The latter is characterized by progressive muscle weakness and the presence of cytoplasmic aggregates (“aggresomes”) [66–68]. Aggresomes are inclusion bodies formed when the cellular degradation machinery, usually the ubiquitin proteasome system (UPS), is impaired or overwhelmed. This leads to an accumulation of proteins for disposal. The aggresomal response can be interpreted as protective in the presence of a high load of damaged, abnormal or misfolded protein within the cytoplasm which fails to be degraded by the UPS and before autophagy is fully engaged. A careful analysis of these aggregates in RBM show that they contain a number of different proteins, such as desmin, ubiquitin, the lysosome-associated protein LAMP1, and the endoplasmic reticulum chaperone GRP78 [68]. How mutations in FHL1 are precisely linked to these aggresomes and may be to autophagy remains unclear, but the presence of E3 ubiquitin ligases at the sarcomeric Z-disc and the defects observed in this system in RBM link both together.
3.8. Enigma/Enigma Homologue (ENH)/Cypher Family and Actinin-Binding LIM Proteins (ALP)
The enigma family of proteins consists of enigma, ENH, and cypher (mouse) (Zasp (human isoform), Oracle) and is defined by an amino-terminal PDZ domain as well as by one to three carboxy-terminal LIM domains [69]. All of them are expressed in striated muscle and localize to the Z-disc, including enigma [69], ENH [70], and cypher [71].
The ALP family consists of four proteins, namely, ALP (PDLIM3), CLP36 (CLIM1, Elfin, PDLIM1), RIL (PDLIM4), and Mystique (SLIM, PDLIM2), which are all expressed in the heart. Each member of this family contains an N-terminal PDZ domain and a C-terminal LIM domain, and actinin-associated LIM protein (ALP) is also present at the Z-disc [72].
ENH and enigma [70] and cypher [71] interact with protein kinase C and influences this important pathway. In contrast, ALP is important for actin filament organization [73]. Most of the genes in these two groups have been knocked out with striking different phenotypes. For example, the ALP knock-out mouse develops a pronounced right ventricular cardiomyopathy, where a developmental pathway is responsible for this phenotype [72]. In this context, ENH-deficient animals have recently been generated and develop a DCM-like phenotype [74]. Cypher is expressed in at least six different isoforms and plays an essential role in striated muscle structure and function. Mice homozygous null for cypher die during the first week because of congenital myopathy with symptoms that include decreased milk intake, limb muscle weakness, cyanosis, and cardiomyopathy [71].
Electron microscopy in these mice revealed severely disorganized skeletal and cardiac muscle with discontinuous/punctuate Z-discs. These findings are somewhat more severe but similar to defects found in MLP-deficient mice [22, 75]. The fact that homozygous deletion of this gene in animals leads to neonatal death implies that, although cypher is not necessary for myofibrillogenesis, it stabilizes Z-discs after the organism starts to sustain its own cardiovascular system. Conditional cypher knock-out animals support this conclusion, where deletion of this gene in adult animals also leads to a severe cardiomyopathy and heart failure phenotype, with Z-disc defects [76].
Knocking in two different skeletal-muscle-specific isoforms to the cypher locus produces partial cardiac rescue, with animals surviving up to one year [77]. In addition, human mutations have also been identified in the cypher (ZASP) gene which produces dilated and or hypertrophic cardiomyopathy, in left ventricular noncompaction [78–80]. Another set of human mutations has been found in patients affected by myofibrillar myopathy (MFM), a disorder in which disintegration of the Z-disc and then of the myofibrils is followed by abnormal accumulation of multiple proteins such as desmin, αB crystalline, and dystrophin. Clinically the disease is characterized by progressive muscle weakness, often starting or involving distal muscles, but limb girdle and scapuloperoneal distributions may also occur. Cardiomyopathy and peripheral neuropathy are frequently present. To date all MFM-causing mutations are present in Z-disk proteins: Zasp, BAG3 (please see next chapter), desmin, αB crystalline, myotilin, and filamin C [81].
The disease causing molecular mechanism for all of the observed phenotypes, in the knock-out animals or in the patients, is not well defined. However, it is possible that the D626N ZASP mutation, which changes the affinity to PKC, might cause the phenotype via alterations in this pathway [78]. Beside skeletal muscle and cardiac phenotypes, ZASP mutations also produce neuropathy, and; hence, the spectrum of phenotypes caused by mutations in this gene has been called zaspopathies [82]. (For an excellent review on this topic, please see [83].)
3.9. Bcl2-Associated Athanogene (BAG3)
Bcl2-associated athanogene (BAG) family proteins belong to an evolutionary conserved group of Hsp70/HSC70 binding cochaperones with up to six different members. They all contain a conserved carboxy-terminal Hsp70/HSC70-binding domain but differ significantly in their amino-terminal architecture, which points to different functions of different family members in cell biology. BAG3 is localized to the sarcomeric Z-disc, and BAG3-deficient animals are born in the expected Mendelian ratios, undistinguishable from wild-type littermates within the first days after birth but develop, during the first and second weeks, severe skeletal and cardiac muscle pathologies characterized by myofibrillar degeneration and apoptosis which leads to death at the age of 4 weeks [84]. In addition, human mutations in BAG3 have been found in patients affected by MFM (please see above) [85], and probably, most importantly, BAG3 recently has been identified as a novel significant candidate gene for human cardiomyopathy and associated heart failure in large genome-wide association studies (GWA) [86, 87].
3.10. Myotilin
Myotilin is a 57 kDa cytoskeletal protein, localized to the sarcomeric Z-disc and important for the stability of thin filaments during muscle contraction. It binds F-actin, crosslinks actin filaments, and prevents latrunculin A-induced filament disassembly. Mutations in this gene have been associated with limb-girdle muscular dystrophy and myofibrillar myopathies (“myotilinopathies”). Von Nandelstadh and coworkers [88] analyzed the molecular mechanisms underlying myotilinopathies and found at least two different calpain cleavage sites in myotilin and that the protein is degraded by the UPS. It turned out that proteolysis inhibitor induced reduction in myotilin protein turnover, which results in formation of intracellular myotilin and actin-containing aggregates which are similar to the aggregates observed in MFM. Moreover, myotilinopathy causing-mutations prevent or at least significantly slow down myotilin protein degradation.
3.11. Alpha-B Crystalline and HSPB7
Alpha crystallins are composed of two gene products: alpha-A and alpha-B (for acidic and basic). Alpha crystallins can be induced by heat shock and are members of the small heat shock protein (sHSP or HSP20) family. They act as molecular chaperones, but they do not renature proteins and release them in the fashion of a true chaperone, instead they hold them in large soluble aggregates, and posttranslational modifications decrease their ability to chaperone. Two additional functions of alpha crystallins are an autokinase activity and participation in the intracellular architecture. Alpha-A and alpha-B gene products are differentially expressed: alpha-A is preferentially restricted to the lens, and alpha-B is expressed widely in many tissues and organs and particularly in the heart. Alpha-B crystallin interacts with desmin, vimentin, and actin and is localized to the Z-disc [89, 90]. Moreover, elevated expression of alpha-B crystallin occurs in many neurological diseases and a missense mutation cosegregated in a family with a desmin-related myopathy [91]. Particularly, the R120G alpha-B crystallin mutation has been analyzed in great detail and increases autophagy in a transgenic mouse model. Autophagy is likely to be an adaptive response to an increase in misfolded proteins, and p62 promotes aggresome formation and autophagy which protects cardiac myocytes against proteotoxic stress [92].
HSPB7 or cardiovascular heat shock protein (cvHSP) is a 170 amino acid protein with high expression in myocardium. The mRNA is induced in monocrotaline-induced right ventricular hypertrophy but downregulated in left ventricular hypertrophy following TAC. Moreover, the protein interacts with alpha filamin and with actin-binding protein 280 [93] and has been found to colocalize with alpha-B crystallin [94]. However, recently a variety of different GWAs pointed to HSPB7 as an important novel candidate gene for DCM [87, 95–97].
3.12. Desmin
Desmin, which is muscle-specific expressed, belongs together with vimentin and lamin A/C to the intermediate filaments (IF), a group of related proteins that share common structural and sequence features, such as the central alpha helical rod and amino-terminal as well as carboxy-terminal globular domains. Desmin is a major IF and present in almost every cell type. In cardiac myocytes desmin connects desmosomes not only with the Z-disc but also with other organelles. Mutations in this gene cause a variety of cardiac diseases (beside the above-discussed MFM). These include desmin-related myopathy [98, 99], limb-girdle muscular dystrophy [100]. They also cause several cardiomyopathies such as dilated cardiomyopathy (DCM) [101], arrhythmogenic right-ventricular cardiomyopathy (ARVC) [102], cardiomyopathy with advanced AV block and arrhythmia [103], familial restrictive cardiomyopathy [104], and DCM with conduction system defects [105]. Desmin interacts with a variety of proteins, and, because it is elastic, it can sense deformation in a cellular structure. It has been linked directly to mechanosensation [106] and might well have a function via the “tensegration” of the intracellular cytoskeletal network; that is, its elasticity may cause a conformational change in response to any type of mechanical stimulation.
3.13. Nebulin/Nebulette
Nebulin is a giant 600–900 kDa filamentous protein present in skeletal muscle whereas the much shorter 100 kDa-related nebulette is present in cardiac tissue. Nebulin/nebulette run along actin filaments, and patients with mutations in nebulin develop nemaline myopathy, which is characterized by weakness, hypotonia, and depressed or absent deep tendon reflexes. Interestingly two different nebulin knock-out mouse models recapitulate all major features of this disease and are available for further analysis [107, 108]. (For an elegant recent review on nebulin, please see [109].)
3.14. Phosphatases (Calcineurin/PPP3CA): Protein Kinases (PKC)
Calcineurin is a heterodimer of a 58 to 64 kDa catalytic subunit, comprising calcineurin A and a 19 kDa regulatory subunit, calcineurin B, and is well known to be one of the major four Ser/Thr phosphatases found in eukaryotic cells implicated in the control of cardiac hypertrophy as well as associated heart failure [110]. Numerous articles have pointed to the importance of this phosphatase, which dephosphorylates nuclear factor activated T-cells (NFAT), enables nuclear translocation of this transcription factor, and initiates changes in gene expression, which in the majority of cases leads to hypertrophy and associated heart failure. Within the scope of this paper it is impossible to review completely the importance of calcineurin; however, it colocalizes with MLP at the sarcomeric Z-disc, and MLP is necessary for calcineurin activation [21].
Protein kinase C (PKC) belongs to a large family of serine/threonine kinases with at least 12 different members and plays major roles in cardiac physiology in mediating hypertrophy and remodelling. They are classically subdivided into three groups: (i) classical calcium sensitive (a, β I, β II, γ), (ii) novel isoforms which lack the calcium sensitive C2 domain (e, d, h, θ), (iii) atypical isoforms (ζ, λ murine-ι in human) which cannot be activated by calcium, diacylglycerol, or by phorbol esters (reviewed in [111]). In particular, PKCε is localized to the Z-disc [112] and it translocates to the nucleus upon biomechanical stress (on TAC) [113], where this kinase most probably elicits a physiological type of hypertrophy or at least exerts protective functions [114]. This view is supported by transgenic over-expression of a constitutive active PKCε in a mouse model where concentric hypertrophy but not heart failure was observed [115].
In this context, protein kinase D (PKD) translocates to the Z-disc in response to GPCR stimulation via norepinephrine, angiotensin II, and endothelin 1 and is activated via forming a complex with PKCε—an effect particularly important after α adrenergic receptor triggered hypertrophy, where PKCε-mediated PKD activation was essential [116]. This type of hypertrophy might be antagonized by the MuRF1, which is present at the sarcomeric Z-disc and which inhibits PKCε [117].
3.15. Protein Degradation (Muscle Ring Fingers—MuRF): Calpain
Protein degradations via the UPS and via different autophagy pathways are important processes in cardiac biology. The UPS recognizes specific proteins and places polyubiquitin chains on them for subsequent destruction by the proteasome. This process is important not only for misfolded and damaged proteins, but also in regulating a variety of different cell signalling pathways involved in proliferation, adaptation to stress, regulation of atrophy/hypertrophy-myocardial remodelling, sarcopenia, and cell survival. However, the UPS may also exert protective functions during ischemia/reperfusion or during myocardial remodelling. Some ubiquitinated proteins aggregate and cannot be degraded via the UPS, in which case cytosolic receptors such as p62, NBR, and histone deacetylase 6 recognize aggregated ubiquitinated proteins and target them for autophagy (for excellent recent reviews, please see [118–120]). Here, because of limited space, we just point out that E3 ubiquitin ligases MuRF1 and MuRF3 are localized to the sarcomeric Z-disc [121, 122] in addition to atrogin 1, which mediates calcineurin degradation and might also have major implications for cardiac function [123]. Interestingly, MuRFs interact with serum response factor and seem to play a major role in the initiation of myocardial hypertrophy [124], and particularly MuRF1 may have additional functions in muscle fatigue and twitch potentiation [125].
In addition, calpain 1, which is an important protease particularly for the selective degradation of cell cycle components and for the regulation of apoptosis (together with caspases) and for the cleavage of cell membrane-cytoskeleton components, has also been shown to colocalize with myotilin at the sarcomeric Z-disc [126].
3.16. Transcription Factors and Mitogen-Activated Protein Kinases (MAPK)
The sarcomeric Z-disc harbours a number of important transcription factors, such as NFAT, which is a target of the phosphatase calcineurin and which has been implicated in the onset of maladaptive myocardial hypertrophy. Another example is clock, a helix loop helix transcription factor involved in the circadian regulation of myocardial function. The protein localizes to the Z-disc and increases its expression as well as translocating into the nucleus upon an increase in contractility. It also decreases its expression on loss of contractility. It has been suggested that this protein links glucose and fatty acid metabolism to myofilament crossbridge activity [127]. In addition, MLP, telethonin and, FHL proteins shuttle into the nucleus, where they function as cofactors of transcription [62, 128, 129] and/or affect p53 [51]. It has also become clear that a variety of MAPKs such as Erk2 and p38 converge at the sarcomeric Z-disc/I-band area and interact with FHL1 and 2 among others [62, 63, 130].
In conclusion, the sarcomeric Z-disc may well act as a central structure linking myofilament activity, mechanosensation, mechanotransduction, and transcriptional activity, an effect which may be called Z-disc mechanotranscriptional coupling.
4. Summary
The sarcomeric Z-disc, barely detectable in conventional light microscopy, is a far more complex structure than initially recognised. Novel protein/protein interaction discovery strategies have led to the identification of a variety of novel Z-disc proteins which also led to the discovery of novel functional aspects. The Z-disc is not only important for mechanical stability and force transmission but also for signalling, mechanosensation, mechanotransduction, apoptosis, and cell survival. The sarcomeric Z-disc might also be important for protein turnover via the UPS and autophagy and has molecular links to the t-tubular system as well as to the sarcoplasmic reticulum. Links to energy turnover point to additional important but less well-studied aspects. It is important to stress that proteins localized at the sarcomeric Z-disc might also be present at other locations such as the nucleus, and, for a variety of different Z-disc proteins, nuclear shuttling has been demonstrated (for an excellent review see: [131]). Mutations in Z-disc genes give rise to Z-discopathies, a heterogenous group of diseases encompassing various cardiomyopathies such as DCM, HCM, ARVC, titinopathies, zaspopathies, myotilinopathies, myofibrillar myopathy, and muscular dystrophies such as the LGMD and XMPMA (please see table and figure). Next generation sequencing is expected to significantly speed up the mutation discovery process, and we can expect that within the next few years a variety of novel mutations located in Z-disc genes will be discovered. This will probably lead to the identification of novel and so far unknown Z-discopathies and may also lead to a better understanding of the underlying molecular mechanism as well as to the development of novel therapeutic strategies.
Moreover, most probably because of the numerous different functions carried by various genetic Z-disc protein mutations, this could lead to perturbations in a vast array of different systems, and the sarcomeric Z-disc is becoming a “hotspot” for cardiomyopathy-causing mutations [18].
It is possible to define central or core Z-disc proteins such as α actinin which serve probably primarily a mechanical function. Another set of proteins may be called transversal or partial Z-disc proteins such as titin, nebulin, or actin which may serve a mechanical function as well but which may have additional implications, for example, in signal transduction, contractility, relaxation, and passive elasticity. In addition, peripheral Z-disc proteins such as telethonin, calsarcin, calcineurin, and MLP may be important for cell signalling, communication, and survival. These proteins can be found at various locations such as the nucleus, costameres, intercalated discs, M-lines, I-bands, and the cytoplasm.
4.1. Future Directions
Future research needs to focus on the link between the sarcomeric Z-disc (probably via MLP), acetylation/deacetylation, and calcium sensitivity. To date a clear genotype-phenotype relationship is not possible for cardiomyopathy and heart-failure-causing mutations. More effort has to be invested in analyzing the relationship between a mutation, its effects on protein function (i.e., poison peptide?), and its effects on protein metabolism in general (i.e., proteinopathy). The link between mechanosensation, mechanotransduction, and apoptosis as well as autophagy needs more investigation. In addition, telethonin might link the sarcomeric Z-disc to the t-tubular system, to the sarcoplasmic reticulum, and directly to apoptosis—these links should be exploited for development of novel therapeutic strategies to combat heart failure, an otherwise lethal condition.
The link between the sarcomeric Z-disc, gene expression, and cell survival, which can be called Z-disc transcriptional coupling, as well as the link between mechanical activity and apoptosis (mechanoptosis) can probably be exploited for novel therapeutic strategies [51].
Finally, Z-disc biology is an exciting newly emerging field and will certainly provide important stimuli for biology and physiology.
Acknowledgments
The authors are grateful to Dr. Pradeep Luther for helpful comments and in-depth discussions on the structural aspects of the sarcomeric Z-disc. R. Knöll is supported by the British Heart Foundation (PG/11/34/28793), the Deutsche Forschungsgemeinschaft (Kn 448/9-1 and Kn 448/10-1), FP7-PEOPLE-2011-IRSES Proposal No. 291834—Acronym: SarcoSi and Fritz Thyssen Foundation.
Abbreviations
- ARVC:
Arrhythmogenic right ventricular cardiomyopathy
- DCM:
Dilated cardiomyopathy
- GPCR:
G-protein-coupled receptor
- HCM:
Hypertrophic cardiomyopathy
- IF:
Intermediate filament
- LGMD:
Limb-girdle muscular dystrophy
- LIM:
Lin11, Isl 1, and Mec 3 motive
- PKC/D:
Protein kinase C/D
- RBM:
Reducing body myopathy
- SP:
Scapuloperoneal
- TAC:
Transverse aortic constriction
- UPS:
Ubiquitin proteasome system
- XMPMA:
X-linked myopathy with postural muscle atrophy.
References
- 1.Luther PK. Three-dimensional reconstruction of a simple Z-band in fish muscle. Journal of Cell Biology. 1991;113(5):1043–1055. doi: 10.1083/jcb.113.5.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zou P, Pinotsis N, Lange S, et al. Palindromic assembly of the giant muscle protein titin in the sarcomeric Z-disk. Nature. 2006;439(7073):229–233. doi: 10.1038/nature04343. [DOI] [PubMed] [Google Scholar]
- 3.Luther PK, Barry JS, Squire JM. The three-dimensional structure of a vertebrate wide (slow muscle) Z-band: lessons on Z-band assembly. Journal of Molecular Biology. 2002;315(1):9–20. doi: 10.1006/jmbi.2001.5217. [DOI] [PubMed] [Google Scholar]
- 4.Luther PK. The vertebrate muscle Z-disc: sarcomere anchor for structure and signalling. Journal of Muscle Research and Cell Motility. 2009;30(5-6):171–185. doi: 10.1007/s10974-009-9189-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Squire JM, Al-Khayat HA, Knupp C, Luther PK. Molecular architecture in muscle contractile assemblies. Advances in Protein Chemistry. 2005;71:17–87. doi: 10.1016/S0065-3233(04)71002-5. [DOI] [PubMed] [Google Scholar]
- 6.Clark KA, McElhinny AS, Beckerle MC, Gregorio CC. Striated muscle cytoarchitecture: an intricate web of form and function. Annual Review of Cell and Developmental Biology. 2002;18:637–706. doi: 10.1146/annurev.cellbio.18.012502.105840. [DOI] [PubMed] [Google Scholar]
- 7.Frank D, Frey N. Cardiac Z-disc signaling network. The Journal of Biological Chemistry. 2011;286:9897–9904. doi: 10.1074/jbc.R110.174268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frank D, Kuhn C, Katus HA, Frey N. The sarcomeric Z-disc: a nodal point in signalling and disease. Journal of Molecular Medicine. 2006;84(6):446–468. doi: 10.1007/s00109-005-0033-1. [DOI] [PubMed] [Google Scholar]
- 9.Sheikh F, Bang ML, Lange S, Chen J. ‘Z’eroing in on the role of Cypher in striated muscle function, signaling, and human disease. Trends in Cardiovascular Medicine. 2007;17(8):258–262. doi: 10.1016/j.tcm.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luther PK, Padron R, Ritter S, Craig R, Squire JM. Heterogeneity of Z-band structure within a single muscle sarcomere: implications for sarcomere assembly. Journal of Molecular Biology. 2003;332(1):161–169. doi: 10.1016/s0022-2836(03)00883-0. [DOI] [PubMed] [Google Scholar]
- 11.Rowe RW. The ultrastructure of Z disks from white, intermediate, and red fibers of mammalian striated muscles. Journal of Cell Biology. 1973;57(2):261–277. doi: 10.1083/jcb.57.2.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldstein MA, Michael LH, Schroeter JP, Sass RL. Z band dynamics as a function of sarcomere length and the contractile state of muscle. FASEB Journal. 1987;1(2):133–142. doi: 10.1096/fasebj.1.2.3609610. [DOI] [PubMed] [Google Scholar]
- 13.Goldstein MA, Michael LH, Schroeter JP, Sass RL. Structural states in the Z band of skeletal muscle correlate with states of active and passive tension. Journal of General Physiology. 1988;92(1):113–119. doi: 10.1085/jgp.92.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldstein MA, Schoeter JP, Sass RL. Two structural states of the vertebrate Z band. Electron Microscopy Reviews. 1990;3(2):227–248. doi: 10.1016/0892-0354(90)90003-b. [DOI] [PubMed] [Google Scholar]
- 15.Yamaguchi M, Izumimoto M, Robson RM, Stromer MH. Fine structure of wide and narrow vertebrate muscle Z-lines. A proposed model and computer simulation of Z-line architecture. Journal of Molecular Biology. 1985;184(4):621–643. doi: 10.1016/0022-2836(85)90308-0. [DOI] [PubMed] [Google Scholar]
- 16.Hampton CM, Taylor DW, Taylor KA. Novel structures for alpha-actinin:F-actin interactions and their implications for actin-membrane attachment and tension sensing in the cytoskeleton. Journal of Molecular Biology. 2007;368(1):92–104. doi: 10.1016/j.jmb.2007.01.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mohapatra B, Jimenez S, Lin JH, et al. Mutations in the muscle LIM protein and alpha-actinin-2 genes in dilated cardiomyopathy and endocardial fibroelastosis. Molecular Genetics and Metabolism. 2003;80(1-2):207–215. doi: 10.1016/s1096-7192(03)00142-2. [DOI] [PubMed] [Google Scholar]
- 18.Bos JM, Ackerman MJ. Z-disc genes in hypertrophic cardiomyopathy: stretching the cardiomyopathies? Journal of the American College of Cardiology. 2010;55:1136–1138. doi: 10.1016/j.jacc.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 19.Chiu C, Bagnall RD, Ingles J, et al. Mutations in alpha-actinin-2 cause hypertrophic cardiomyopathy: a genome-wide analysis. Journal of the American College of Cardiology. 2010;55(11):1127–1135. doi: 10.1016/j.jacc.2009.11.016. [DOI] [PubMed] [Google Scholar]
- 20.Louis HA, Pino JD, Schmeichel KL, Pomies P, Beckerle MC. Comparison of three members of the cysteine-rich protein family reveals functional conservation and divergent patterns of gene expression. Journal of Biological Chemistry. 1997;272(43):27484–27491. doi: 10.1074/jbc.272.43.27484. [DOI] [PubMed] [Google Scholar]
- 21.Heineke J, Ruetten H, Willenbockel C, et al. Attenuation of cardiac remodeling after myocardial infarction by muscle LIM protein-calcineurin signaling at the sarcomeric Z-disc. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(5):1655–1660. doi: 10.1073/pnas.0405488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knöll R, Hoshijima M, Hoffman HM, et al. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell. 2002;111(7):943–955. doi: 10.1016/s0092-8674(02)01226-6. [DOI] [PubMed] [Google Scholar]
- 23.Knöll R, Kostin S, Klede S, et al. A common MLP (muscle LIM protein) variant is associated with cardiomyopathy. Circulation Research. 2010;106:695–704. doi: 10.1161/CIRCRESAHA.109.206243. [DOI] [PubMed] [Google Scholar]
- 24.Kong Y, Flick MJ, Kudla AJ, Konieczny SF. Muscle LIM protein promotes myogenesis by enhancing the activity of MyoD. Molecular and Cellular Biology. 1997;17(8):4750–4760. doi: 10.1128/mcb.17.8.4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ehler E, Horowits R, Zuppinger C, et al. Alterations at the intercalated disk associated with the absence of muscle LIM protein. Journal of Cell Biology. 2001;153(4):763–772. doi: 10.1083/jcb.153.4.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Postel R, Vakeel P, Topczewski J, Knöll R, Bakkers J. Zebrafish integrin-linked kinase is required in skeletal muscles for strengthening the integrin-ECM adhesion complex. Developmental Biology. 2008;318(1):92–101. doi: 10.1016/j.ydbio.2008.03.024. [DOI] [PubMed] [Google Scholar]
- 27.Flick MJ, Konieczny SF. The muscle regulatory and structural protein MLP is a cytoskeletal binding partner of betaI-spectrin. Journal of Cell Science. 2000;113(9):1553–1564. doi: 10.1242/jcs.113.9.1553. [DOI] [PubMed] [Google Scholar]
- 28.Knöll R, Iaccarino G, Tarone G, et al. Towards a re-definition of “cardiac hypertrophy” through a rational characterization of left ventricular phenotypes: a position paper of the Working Group “Myocardial Function” of the ESC. European Journal of Heart Failure. 2011;13:811–819. doi: 10.1093/eurjhf/hfr071. [DOI] [PubMed] [Google Scholar]
- 29.Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies: an American heart association scientific statement from the council on clinical cardiology, heart failure and transplantation committee; quality of care and outcomes research and functional genomics and translational biology interdisciplinary working groups; and council on epidemiology and prevention. Circulation. 2006;113(14):1807–1816. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- 30.Papalouka V, Arvanitis DA, Vafiadaki E, et al. Muscle Lim protein interacts with cofilin 2 and regulates F-actin dynamics in cardiac and skeletal muscle. Molecular and Cellular Biology. 2009;29(22):6046–6058. doi: 10.1128/MCB.00654-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arber S, Caroni P. Specificity of single LIM motifs in targeting and LIM/LIM interactions in situ. Genes and Development. 1996;10(3):289–300. doi: 10.1101/gad.10.3.289. [DOI] [PubMed] [Google Scholar]
- 32.Gupta MP, Samant SA, Smith SH, Shroff SG. HDAC4 and PCAF bind to cardiac sarcomeres and play a role in regulating myofilament contractile activity. Journal of Biological Chemistry. 2008;283(15):10135–10146. doi: 10.1074/jbc.M710277200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buyandelger B, Ng KE, Miocic S, et al. MLP (muscle LIM protein) as a stress sensor in the heart. Pflügers Archiv. 2011;462(1):135–142. doi: 10.1007/s00424-011-0961-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bertz M, Wilmanns M, Rief M. The titin-telethonin complex is a directed, superstable molecular bond in the muscle Z-disk. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(32):13307–13310. doi: 10.1073/pnas.0902312106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mayans O, van der Ven PFM, Wilm M, et al. Structural basis for activation of the titin kinase domain during myofibrillogenesis. Nature. 1998;395(863):p. 869. doi: 10.1038/27603. [DOI] [PubMed] [Google Scholar]
- 36.Haworth RS, Cuello F, Herron TJ, et al. Protein kinase D is a novel mediator of cardiac troponin I phosphorylation and regulates myofilament function. Circulation Research. 2004;95(11):1091–1099. doi: 10.1161/01.RES.0000149299.34793.3c. [DOI] [PubMed] [Google Scholar]
- 37.Kontrogianni-Konstantopoulos A, Jones EM, Van Rossum DB, Bloch RJ. Obscurin is a ligand for small ankyrin 1 in skeletal muscle. Molecular Biology of the Cell. 2003;14(3):1138–1148. doi: 10.1091/mbc.E02-07-0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Furukawa T, Ono Y, Tsuchiya H, et al. Specific interaction of the potassium channel beta-subunit minK with the sarcomeric protein T-cap suggests a T-tubule-myofibril linking system. Journal of Molecular Biology. 2001;313(4):775–784. doi: 10.1006/jmbi.2001.5053. [DOI] [PubMed] [Google Scholar]
- 39.Mazzone A, Strege PR, Tester DJ, et al. A mutation in telethonin alters Nav1.5 function. Journal of Biological Chemistry. 2008;283(24):16537–16544. doi: 10.1074/jbc.M801744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang R, Yang J, Zhu J, Xu X. Depletion of zebrafish Tcap leads to muscular dystrophy via disrupting sarcomere-membrane interaction, not sarcomere assembly. Human Molecular Genetics. 2009;18(21):4130–4140. doi: 10.1093/hmg/ddp362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tian LF, Li HY, Jin BF, et al. MDM2 interacts with and downregulates a sarcomeric protein, TCAP. Biochemical and Biophysical Research Communications. 2006;345(1):355–361. doi: 10.1016/j.bbrc.2006.04.108. [DOI] [PubMed] [Google Scholar]
- 42.Witt SH, Granzier H, Witt CC, Labeit S. MURF-1 and MURF-2 target a specific subset of myofibrillar proteins redundantly: towards understanding MURF-dependent muscle ubiquitination. Journal of Molecular Biology. 2005;350(4):713–722. doi: 10.1016/j.jmb.2005.05.021. [DOI] [PubMed] [Google Scholar]
- 43.Frey N, Olson EN. Calsarcin-3, a novel skeletal muscle-specific member of the calsarcin family, interacts with multiple Z-disc proteins. Journal of Biological Chemistry. 2002;277(16):13998–14004. doi: 10.1074/jbc.M200712200. [DOI] [PubMed] [Google Scholar]
- 44.Nicholas G, Thomas M, Langley B, et al. Titin-cap associates with, and regulates secretion of, Myostatin. Journal of Cellular Physiology. 2002;193(1):120–131. doi: 10.1002/jcp.10158. [DOI] [PubMed] [Google Scholar]
- 45.Nakano N, Hori H, Abe M, et al. Interaction of BMP10 with Tcap may modulate the course of hypertensive cardiac hypertrophy. American Journal of Physiology. 2007;293(6):H3396–H3403. doi: 10.1152/ajpheart.00311.2007. [DOI] [PubMed] [Google Scholar]
- 46.Mihatsch K, Nestler M, Saluz HP, Henke A, Munder T. Proapoptotic protein Siva binds to the muscle protein telethonin in cardiomyocytes during coxsackieviral infection. Cardiovascular Research. 2009;81(1):108–115. doi: 10.1093/cvr/cvn276. [DOI] [PubMed] [Google Scholar]
- 47.Ferreiro A, Mezmezian M, Olive M, et al. Telethonin-deficiency initially presenting as a congenital muscular dystrophy. Neuromuscular Disorders. 2011;21(6):433–438. doi: 10.1016/j.nmd.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 48.Moreira ES, Wiltshire TJ, Faulkner G, et al. Limb-girdle muscular dystrophy type 2G is caused by mutations in the gene encoding the sarcomeric protein telethonin. Nature Genetics. 2000;24(2):163–166. doi: 10.1038/72822. [DOI] [PubMed] [Google Scholar]
- 49.Olive M, Shatunov A, Gonzalez L, et al. Transcription-terminating mutation in telethonin causing autosomal recessive muscular dystrophy type 2G in a European patient. Neuromuscular Disorders. 2008;18(12):929–933. doi: 10.1016/j.nmd.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hayashi T, Arimura T, Itoh-Satoh M, et al. Tcap gene mutations in hypertrophic cardiomyopathy and dilated cardiomyopathy. Journal of the American College of Cardiology. 2004;44(11):2192–2201. doi: 10.1016/j.jacc.2004.08.058. [DOI] [PubMed] [Google Scholar]
- 51.Knöll R, Linke WA, Zou P, et al. Telethonin deficiency is associated with maladaptation to biomechanical stress in the mammalian heart. Circulation Research. 2011;109(7):758–769. doi: 10.1161/CIRCRESAHA.111.245787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lange A, Wickstrom SA, Jakobson M, Zent R, Sainio K, Fassler R. Integrin-linked kinase is an adaptor with essential functions during mouse development. Nature. 2009;461(7266):1002–1006. doi: 10.1038/nature08468. [DOI] [PubMed] [Google Scholar]
- 53.Kim YB, Choi S, Choi MC, et al. Cell adhesion-dependent cofilin serine 3 phosphorylation by the integrin-linked kinase·c-Src complex. Journal of Biological Chemistry. 2008;283(15):10089–10096. doi: 10.1074/jbc.M708300200. [DOI] [PubMed] [Google Scholar]
- 54.Knöll R, Postel R, Wang J, et al. Laminin-alpha4 and integrin-linked kinase mutations cause human cardiomyopathy via simultaneous defects in cardiomyocytes and endothelial cells. Circulation. 2007;116(5):515–525. doi: 10.1161/CIRCULATIONAHA.107.689984. [DOI] [PubMed] [Google Scholar]
- 55.Brancaccio M, Guazzone S, Menini N, et al. Melusin is a new muscle-specific interactor for beta(1) integrin cytoplasmic domain. Journal of Biological Chemistry. 1999;274(41):29282–29288. doi: 10.1074/jbc.274.41.29282. [DOI] [PubMed] [Google Scholar]
- 56.Brancaccio M, Fratta L, Notte A, et al. Melusin, a muscle-specific integrin beta(1)-interacting protein, is required to prevent cardiac failure in response to chronic pressure overload. Nature Medicine. 2003;9(1):68–75. doi: 10.1038/nm805. [DOI] [PubMed] [Google Scholar]
- 57.De Acetis M, Notte A, Accornero F, et al. Cardiac overexpression of melusin protects from dilated cardiomyopathy due to long-standing pressure overload. Circulation Research. 2005;96(10):1087–1094. doi: 10.1161/01.RES.0000168028.36081.e0. [DOI] [PubMed] [Google Scholar]
- 58.Palumbo V, Segat L, Padovan L, et al. Melusin gene (ITGB1BP2) nucleotide variations study in hypertensive and cardiopathic patients. BMC Medical Genetics. 2009;10, article 140 doi: 10.1186/1471-2350-10-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Frey N, Richardson JA, Olson EN. Calsarcins, a novel family of sarcomeric calcineurin-binding proteins. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(26):14632–14637. doi: 10.1073/pnas.260501097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Frey N, Barrientos T, Shelton JM, et al. Mice lacking calsarcin-1 are sensitized to calcineurin signaling and show accelerated cardiomyopathy in response to pathological biomechanical stress. Nature Medicine. 2004;10(12):1336–1343. doi: 10.1038/nm1132. [DOI] [PubMed] [Google Scholar]
- 61.Osio A, Tan L, Chen SN, et al. Myozenin 2 is a novel gene for human hypertrophic cardiomyopathy. Circulation Research. 2007;100(6):766–768. doi: 10.1161/01.RES.0000263008.66799.aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Purcell NH, Darwis D, Bueno OF, Muller JM, Schule R, Molkentin JD. Extracellular signal-regulated kinase 2 interacts with and is negatively regulated by the LIM-only protein FHL2 in cardiomyocytes. Molecular and Cellular Biology. 2004;24(3):1081–1095. doi: 10.1128/MCB.24.3.1081-1095.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sheikh F, Raskin A, Chu PH, et al. An FHL1-containing complex within the cardiomyocyte sarcomere mediates hypertrophic biomechanical stress responses in mice. Journal of Clinical Investigation. 2008;118(12):3870–3880. doi: 10.1172/JCI34472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Windpassinger C, Schoser B, Straub V, et al. An X-linked myopathy with postural muscle atrophy and generalized hypertrophy, termed XMPMA, is caused by mutations in FHL1. American Journal of Human Genetics. 2008;82(1):88–99. doi: 10.1016/j.ajhg.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Quinzii CM, Vu TH, Min KC, et al. X-linked dominant scapuloperoneal myopathy is due to a mutation in the gene encoding four-and-a-half-LIM protein 1. American Journal of Human Genetics. 2008;82(1):208–213. doi: 10.1016/j.ajhg.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schessl J, Columbus A, Hu Y, et al. Familial reducing body myopathy with cytoplasmic bodies and rigid spine revisited: identification of a second LIM domain mutation in FHL1. Neuropediatrics. 2010;41(1):43–46. doi: 10.1055/s-0030-1254101. [DOI] [PubMed] [Google Scholar]
- 67.Schessl J, Taratuto AL, Sewry C, et al. Clinical, histological and genetic characterization of reducing body myopathy caused by mutations in FHL1. Brain. 2009;132(2):452–464. doi: 10.1093/brain/awn325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schessl J, Zou Y, McGrath MJ, et al. Proteomic identification of FHL1 as the protein mutated in human reducing body myopathy. Journal of Clinical Investigation. 2008;118(3):904–912. doi: 10.1172/JCI34450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guy PM, Kenny DA, Gill GN. The PDZ domain of the LIM protein enigma binds to beta-tropomyosin. Molecular Biology of the Cell. 1999;10(6):1973–1984. doi: 10.1091/mbc.10.6.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kuroda S, Tokunaga C, Kiyohara Y, et al. Protein-protein interaction of zinc finger LIM domains with protein kinase C. Journal of Biological Chemistry. 1996;271(49):31029–31032. doi: 10.1074/jbc.271.49.31029. [DOI] [PubMed] [Google Scholar]
- 71.Zhou Q, Chu PH, Huang C, et al. Ablation of Cypher, a PDZ-LIM domain Z-line protein, causes a severe form of congenital myopathy. Journal of Cell Biology. 2001;155(3):605–612. doi: 10.1083/jcb.200107092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pashmforoush M, Pomies P, Peterson KL, et al. Adult mice deficient in actinin-associated LIM-domain protein reveal a developmental pathway for right ventricular cardiomyopathy. Nature Medicine. 2001;7(5):591–597. doi: 10.1038/87920. [DOI] [PubMed] [Google Scholar]
- 73.Han HF, Beckerle MC. The ALP-enigma protein ALP-1 functions in actin filament organization to promote muscle structural integrity in caenorhabditis elegans. Molecular Biology of the Cell. 2009;20(9):2361–2370. doi: 10.1091/mbc.E08-06-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cheng H, Kimura K, Peter AK, et al. Loss of enigma homolog protein results in dilated cardiomyopathy. Circulation Research. 2010;107(3):348–356. doi: 10.1161/CIRCRESAHA.110.218735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Arber S, Hunter JJ, Ross J, Jr., et al. MLP-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy, and heart failure. Cell. 1997;88(3):393–403. doi: 10.1016/s0092-8674(00)81878-4. [DOI] [PubMed] [Google Scholar]
- 76.Zheng M, Cheng H, Li X, et al. Cardiac-specific ablation of Cypher leads to a severe form of dilated cardiomyopathy with premature death. Human Molecular Genetics. 2009;18(4):701–713. doi: 10.1093/hmg/ddn400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang C, Zhou Q, Liang P, et al. Characterization and in vivo functional analysis of splice variants of cypher. Journal of Biological Chemistry. 2003;278(9):7360–7365. doi: 10.1074/jbc.M211875200. [DOI] [PubMed] [Google Scholar]
- 78.Arimura T, Hayashi T, Terada H, et al. A Cypher/ZASP mutation associated with dilated cardiomyopathy alters the binding affinity to protein kinase C. Journal of Biological Chemistry. 2004;279(8):6746–6752. doi: 10.1074/jbc.M311849200. [DOI] [PubMed] [Google Scholar]
- 79.Vatta M, Mohapatra B, Jimenez S, et al. Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction. Journal of the American College of Cardiology. 2003;42(11):2014–2027. doi: 10.1016/j.jacc.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 80.Xing Y, Ichida F, Matsuoka T, et al. Genetic analysis in patients with left ventricular noncompaction and evidence for genetic heterogeneity. Molecular Genetics and Metabolism. 2006;88(1):71–77. doi: 10.1016/j.ymgme.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 81.Selcen D, Engel AG. Myofibrillar myopathies. Handbook of Clinical Neurology. 2011;101:143–154. doi: 10.1016/B978-0-08-045031-5.00011-6. [DOI] [PubMed] [Google Scholar]
- 82.Selcen D, Engel AG. Mutations in ZASP define a novel form of muscular dystrophy in humans. Annals of Neurology. 2005;57(2):269–276. doi: 10.1002/ana.20376. [DOI] [PubMed] [Google Scholar]
- 83.Zheng M, Cheng H, Banerjee I, Chen J. ALP/Enigma PDZ-LIM domain proteins in the heart. Journal of Molecular Cell Biology. 2010;2(2):96–102. doi: 10.1093/jmcb/mjp038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Homma S, Iwasaki M, Shelton GD, Engvall E, Reed JC, Takayama S. BAG3 deficiency results in fulminant myopathy and early lethality. American Journal of Pathology. 2006;169(3):761–773. doi: 10.2353/ajpath.2006.060250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Selcen D, Muntoni F, Burton BK, et al. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Annals of Neurology. 2009;65(1):83–89. doi: 10.1002/ana.21553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Norton N, Li D, Rieder MJ, et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. American Journal of Human Genetics. 2011;88(3):273–282. doi: 10.1016/j.ajhg.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Villard E, Perret C, Gary F, et al. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. European Heart Journal. 2011;32:1065–1076. doi: 10.1093/eurheartj/ehr105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.von Nandelstadh P, Souymani R, Baumann M, Carpen O. Analysis of myotilin turnover provides mechanistic insight into the role of myotilinopathy-causing mutations. Biochemical Journal. 2011;436(1):113–121. doi: 10.1042/BJ20101672. [DOI] [PubMed] [Google Scholar]
- 89.Bennardini F, Wrzosek A, Chiesi M. Alpha B-Crystallin in cardiac tissue: association with actin and desmin filaments. Circulation Research. 1992;71(2):288–294. doi: 10.1161/01.res.71.2.288. [DOI] [PubMed] [Google Scholar]
- 90.Nicholl ID, Quinlan RA. Chaperone activity of alpha-crystallins modulates intermediate filament assembly. EMBO Journal. 1994;13(4):945–953. doi: 10.1002/j.1460-2075.1994.tb06339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Vicart P, Caron A, Guicheney P, et al. A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nature Genetics. 1998;20(1):92–95. doi: 10.1038/1765. [DOI] [PubMed] [Google Scholar]
- 92.Zheng Q, Su H, Ranek MJ, Wang X. Autophagy and p62 in cardiac proteinopathy. Circulation Research. 2011;109:296–308. doi: 10.1161/CIRCRESAHA.111.244707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Krief S, Faivre JF, Robert P, et al. Identification and characterization of cvHsp. A novel human small stress protein selectively expressed in cardiovascular and insulin-sensitive tissues. Journal of Biological Chemistry. 1999;274(51):36592–36600. doi: 10.1074/jbc.274.51.36592. [DOI] [PubMed] [Google Scholar]
- 94.Brodehl A, Martin I, Gawlowski T, Stork I, Gummert J, Milting H. The small heat shock proteins cvHSP and α-B-Crystallin are colocalized and interact in human myocardial tissue. Clinical Research in Cardiology. 2010;99(supplement 1):p. 467. [Google Scholar]
- 95.Cappola TP, Li M, He J, et al. Common variants in HSPB7 and FRMD4B associated with advanced heart failure. Circulation. 2010;3(2):147–154. doi: 10.1161/CIRCGENETICS.109.898395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Matkovich SJ, Van Booven DJ, Hindes A, et al. Cardiac signaling genes exhibit unexpected sequence diversity in sporadic cardiomyopathy, revealing HSPB7 polymorphisms associated with disease. Journal of Clinical Investigation. 2010;120(1):280–289. doi: 10.1172/JCI39085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stark K, Esslinger UB, Reinhard W, et al. Genetic association study identifies HSPB7 as a risk gene for idiopathic dilated cardiomyopathy. PLoS Genetics. 2010;6(10, article e1001167) doi: 10.1371/journal.pgen.1001167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dalakas MC, Dagvadorj A, Goudeau B, et al. Progressive skeletal myopathy, a phenotypic variant of desmin myopathy associated with desmin mutations. Neuromuscular Disorders. 2003;13(3):252–258. doi: 10.1016/s0960-8966(02)00271-7. [DOI] [PubMed] [Google Scholar]
- 99.Dalakas MC, Park KY, Semino-Mora C, Lee HS, Sivakumar K, Goldfarb LG. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. The New England Journal of Medicine. 2000;342(11):770–780. doi: 10.1056/NEJM200003163421104. [DOI] [PubMed] [Google Scholar]
- 100.Walter MC, Reilich P, Huebner A, et al. Scapuloperoneal syndrome type Kaeser and a wide phenotypic spectrum of adult-onset, dominant myopathies are associated with the desmin mutation R350P. Brain. 2007;130(6):1485–1496. doi: 10.1093/brain/awm039. [DOI] [PubMed] [Google Scholar]
- 101.Li D, Tapscoft T, Gonzalez O, et al. Desmin mutation responsible for idiopathic dilated cardiomyopathy. Circulation. 1999;100(5):461–464. doi: 10.1161/01.cir.100.5.461. [DOI] [PubMed] [Google Scholar]
- 102.Klauke B, Kossmann S, Gaertner A, et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Human Molecular Genetics. 2010;19(23):4595–4607. doi: 10.1093/hmg/ddq387. [DOI] [PubMed] [Google Scholar]
- 103.Otten E, Asimaki A, Maass A, et al. Desmin mutations as a cause of right ventricular heart failure affect the intercalated disks. Heart Rhythm. 2010;7(8):1058–1064. doi: 10.1016/j.hrthm.2010.04.023. [DOI] [PubMed] [Google Scholar]
- 104.Pruszczyk P, Kostera-Pruszczyk A, Shatunov A, et al. Restrictive cardiomyopathy with atrioventricular conduction block resulting from a desmin mutation. International Journal of Cardiology. 2007;117(2):244–253. doi: 10.1016/j.ijcard.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 105.Taylor MRG, Slavov D, Ku L, et al. Prevalence of desmin mutations in dilated cardiomyopathy. Circulation. 2007;115(10):1244–1251. doi: 10.1161/CIRCULATIONAHA.106.646778. [DOI] [PubMed] [Google Scholar]
- 106.Herrmann H, Bar H, Kreplak L, Strelkov SV, Aebi U. Intermediate filaments: from cell architecture to nanomechanics. Nature Reviews Molecular Cell Biology. 2007;8(7):562–573. doi: 10.1038/nrm2197. [DOI] [PubMed] [Google Scholar]
- 107.Bang ML, Li X, Littlefield R, et al. Nebulin-deficient mice exhibit shorter thin filament lengths and reduced contractile function in skeletal muscle. Journal of Cell Biology. 2006;173(6):905–916. doi: 10.1083/jcb.200603119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Witt CC, Burkart C, Labeit D, et al. Nebulin regulates thin filament length, contractility, and Z-disk structure in vivo. EMBO Journal. 2006;25(16):3843–3855. doi: 10.1038/sj.emboj.7601242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Labeit S, Ottenheijm CAC, Granzier H. Nebulin, a major player in muscle health and disease. FASEB Journal. 2011;25(3):822–829. doi: 10.1096/fj.10-157412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Molkentin JD, Lu JR, Antos CL, et al. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93(2):215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Newton AC. Protein kinase C: poised to signal. American Journal of Physiology. 2010;298(3):395–402. doi: 10.1152/ajpendo.00477.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Robia SL, Ghanta J, Robu VG, Walker JW. Localization and kinetics of protein kinase C-epsilon anchoring in cardiac myocytes. Biophysical Journal. 2001;80(5):2140–2151. doi: 10.1016/S0006-3495(01)76187-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gu X, Bishop SP. Increased protein kinase C and isozyme redistribution in pressure-overload cardiac hypertrophy in the rat. Circulation Research. 1994;75(5):926–931. doi: 10.1161/01.res.75.5.926. [DOI] [PubMed] [Google Scholar]
- 114.Wu G, Toyokawa T, Hahn H, Dorn GW., II Epsilon protein kinase C in pathological myocardial hypertrophy. Analysis by combined transgenic expression of translocation modifiers and Galphaq. The Journal of Biological Chemistry. 2000;275:29927–29930. doi: 10.1074/jbc.C000380200. Article ID 10.1074/jbc.C000380200. [DOI] [PubMed] [Google Scholar]
- 115.Takeishi Y, Ping P, Bolli R, Kirkpatrick DL, Hoit BD, Walsh RA. Transgenic overexpression of constitutively active protein kinase C epsilon causes concentric cardiac hypertrophy. Circulation Research. 2000;86(12):1218–1223. doi: 10.1161/01.res.86.12.1218. [DOI] [PubMed] [Google Scholar]
- 116.Iwata M, Maturana A, Hoshijima M, et al. PKCepsilon-PKD1 signaling complex at Z-discs plays a pivotal role in the cardiac hypertrophy induced by G-protein coupling receptor agonists. Biochemical and Biophysical Research Communications. 2005;327:1105–1113. doi: 10.1016/j.bbrc.2004.12.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Arya R, Kedar V, Hwang JR, et al. Muscle ring finger protein-1 inhibits PKC{epsilon} activation and prevents cardiomyocyte hypertrophy. Journal of Cell Biology. 2004;167(6):1147–1159. doi: 10.1083/jcb.200402033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Arndt V, Dick N, Tawo R, et al. Chaperone-assisted selective autophagy is essential for muscle maintenance. Current Biology. 2010;20(2):143–148. doi: 10.1016/j.cub.2009.11.022. [DOI] [PubMed] [Google Scholar]
- 119.Mearini G, Schlossarek S, Willis MS, Carrier L. The ubiquitin-proteasome system in cardiac dysfunction. Biochimica et Biophysica Acta. 2008;1782(12):749–763. doi: 10.1016/j.bbadis.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 120.Willis MS, Townley-Tilson WHD, Kang EY, Homeister JW, Patterson C. Sent to destroy: the ubiquitin proteasome system regulates cell signaling and protein quality control in cardiovascular development and disease. Circulation Research. 2010;106(3):463–478. doi: 10.1161/CIRCRESAHA.109.208801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Centner T, Yano J, Kimura E, et al. Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. Journal of Molecular Biology. 2001;306(4):717–726. doi: 10.1006/jmbi.2001.4448. [DOI] [PubMed] [Google Scholar]
- 122.Spencer JA, Eliazer S, Ilaria RL, Jr., Richardson JA, Olson EN. Regulation of microtubule dynamics and myogenic differentiation by MURF, a striated muscle RING-finger protein. Journal of Cell Biology. 2000;150(4):771–784. doi: 10.1083/jcb.150.4.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li HH, Kedar V, Zhang C, et al. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. Journal of Clinical Investigation. 2004;114(8):1058–1071. doi: 10.1172/JCI22220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Willis MS, Ike C, Li L, Wang DZ, Glass DJ, Patterson C. Muscle ring finger 1, but not muscle ring finger 2, regulates cardiac hypertrophy in vivo. Circulation Research. 2007;100(4):456–459. doi: 10.1161/01.RES.0000259559.48597.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Labeit S, Kohl CH, Witt CC, Labeit D, Jung J, Granzier H. Modulation of muscle atrophy, fatigue and MLC phosphorylation by MuRF1 as indicated by hindlimb suspension studies on MuRF1-KO mice. Journal of Biomedicine and Biotechnology. 2010;2010:9 pages. doi: 10.1155/2010/693741. Article ID 693741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Raynaud F, Jond-Necand C, Marcilhac A, Furst D, Benyamin Y. Calpain 1-gamma filamin interaction in muscle cells: a possible in situ regulation by PKC-alpha. The International Journal of Biochemistry and Cell Biology. 2006;38(3):404–413. doi: 10.1016/j.biocel.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 127.Qi L, Boateng SY. The circadian protein Clock localizes to the sarcomeric Z-disk and is a sensor of myofilament cross-bridge activity in cardiac myocytes. Biochemical and Biophysical Research Communications. 2006;351(4):1054–1059. doi: 10.1016/j.bbrc.2006.10.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Boateng SY, Belin RJ, Geenen DL, et al. Cardiac dysfunction and heart failure are associated with abnormalities in the subcellular distribution and amounts of oligomeric muscle LIM protein. American Journal of Physiology. 2007;292(1):H259–H269. doi: 10.1152/ajpheart.00766.2006. [DOI] [PubMed] [Google Scholar]
- 129.Boateng SY, Senyo SE, Qi L, Goldspink PH, Russell B. Myocyte remodeling in response to hypertrophic stimuli requires nucleocytoplasmic shuttling of muscle LIM protein. Journal of Molecular and Cellular Cardiology. 2009;47(4):426–435. doi: 10.1016/j.yjmcc.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Vahebi S, Ota A, Li M, et al. p38-MAPK induced dephosphorylation of alpha-tropomyosin is associated with depression of myocardial sarcomeric tension and ATPase activity. Circulation Research. 2007;100(3):408–415. doi: 10.1161/01.RES.0000258116.60404.ad. [DOI] [PubMed] [Google Scholar]
- 131.Lange S, Ehler E, Gautel M. From A to Z and back? Multicompartment proteins in the sarcomere. Trends in Cell Biology. 2006;16(1):11–18. doi: 10.1016/j.tcb.2005.11.007. [DOI] [PubMed] [Google Scholar]